
Epstein–Barr Virus History and Pathogenesis

1
Department of Hematology, The Affiliated Hospital of Nanjing University of Chinese Medicine, Jiangsu Province Hospital of Chinese Medicine, Nanjing 210029, China
2
Departments of Otorhinolaryngology-Head and Neck Surgery, and Microbiology, The Tumor Virology Program, Abramson Cancer Center, Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA 19104, USA
*
Authors to whom correspondence should be addressed.
Viruses 2023, 15(3), 714; https://doi.org/10.3390/v15030714
Submission received: 26 January 2023
/
Revised: 4 March 2023
/
Accepted: 7 March 2023
/
Published: 9 March 2023
(This article belongs to the Special Issue Epstein-Barr Virus Replication and Pathogenesis)
Abstract
:Epstein–Barr virus (EBV) is the first identified human oncogenic virus that can establish asymptomatic life-long persistence. It is associated with a large spectrum of diseases, including benign diseases, a number of lymphoid malignancies, and epithelial cancers. EBV can also transform quiescent B lymphocytes into lymphoblastoid cell lines (LCLs) in vitro. Although EBV molecular biology and EBV-related diseases have been continuously investigated for nearly 60 years, the mechanism of viral-mediated transformation, as well as the precise role of EBV in promoting these diseases, remain a major challenge yet to be completely explored. This review will highlight the history of EBV and current advances in EBV-associated diseases, focusing on how this virus provides a paradigm for exploiting the many insights identified through interplay between EBV and its host during oncogenesis, and other related non-malignant disorders.
1. Introduction
Epstein–Barr virus (EBV or human herpesvirus 4 (HHV-4)), is a member of the γ-herpesviruses family. It is the first human tumorigenic virus to be characterized. It is one of the most successful viruses, infecting up to 95% of the world’s adult human population and sustaining a life-long asymptomatic infection of the B-lymphocyte pool [1,2,3,4]. Natural EBV infection is strikingly ubiquitous in humans [2,5]. Studies have elucidated its role as a causative factor in the development of a diverse spectrum of diseases, including benign diseases (infectious mononucleosis (IM)) [2], oral diseases [6], diseases related to functional abnormalities of immunity, multiple sclerosis (MS) [7], systemic autoimmune diseases (SADs) [8], various malignancies (hematological malignances, epithelial cancers) [3,9], and EBV-associated hemophagocytic lymphohistiocytosis (EBV-HLH).
It is estimated that more than 250,000 cases of cancer every year are induced by EBV, and that approximately 2% of all cancer deaths are due to EBV-attributed malignancies [10]. Improved understanding of pathogenic mechanisms, which drives the many aforementioned diseases, provides clues as to potential opportunities for recognizing and identifying early and effective treatments at different stages in the development of EBV-associated diseases. In addition, the more recent utility of immunotherapy to target EBV-infected cells can potentially overcome viral-mediated immune evasion. Over the years, many vaccine strategies and therapeutic agents have been developed to target the EBV gene products, as well as to promote immune surveillance.
2. EBV History
EBV was discovered by electron microscopy of cells cultured from fresh African Burkitt’s lymphoma (BL) biopsy after outgrowth of the cells in culture in the laboratory of Dr. Anthony Epstein in 1964 [11]. There was almost immediate speculation that the virus might be involved in the development of tumorigenesis. Since then, EBV has been an object of intense interest. In 1966, studies showed that there were high EBV antibody levels in BL and nasopharyngeal carcinomas (NPCs) patients [12], as well as in healthy donors [13]. EBV was shown to be the causative agent of IM in 1968 [14]. The presence of EBV DNA was reported in BL cells, as well as extracts from anaplastic NPC samples, which were derived from patient biopsies in 1970 [15]. It was then further confirmed that EBV-specific nucleic acids were present within NPC cells [16]. In the 1980s, EBV was found to be associated with non-Hodgkin’s lymphoma and oral hairy leukoplakia in patients with acquired immunodeficiency syndrome (AIDS) [17,18]. Since then, EBV DNA has been found in tissues from a wide range of different cancer types. Those include hematological disorders, such as post-transplant lymphoproliferative disorders (PTLDs) [19]; Hodgkin’s lymphoma [20]; T-cell lymphoma [21]; NK cell leukemia and other T-cell, NKT-cell and NK-cell lymphoproliferative diseases [22,23]; and certain epithelial neoplasms, including lymphoepithelioma-like carcinomas occurring in a variety of organs, such as gastric adenocarcinoma [24], breast cancer [25], and liver carcinoma [26]. More recently, several SADs and MS have been shown to correlate with chronically reactivated EBV infection and dysfunctional immune control of the virus [8]. However, the detailed contributions of EBV to the pathogenesis of the majority of the aforementioned diseases remain to be explored. Therefore, a thorough examination of the infection, reactivation, and cell transformation induced by EBV will continue to provide more details of its contribution in driving pathogenesis, which will eventually lead to the development of therapeutic targets, agents, and strategies for clinical interventions.
3. Biology and the Potent Transforming Ability of Oncogenic EBV
The stages of the EBV life cycle include the primary infection, establishment of latency, and reactivation or lytic stage to produce new virions, and depend on the interplay between the virus and the host immune system. The fact that EBV persists for the lifetime of the host indicates that it has evolved successful strategies to obstruct the host immunity. Typically, the majority of primary EBV infections are asymptomatic, which occurs during infancy and early childhood in areas where general hygiene conditions are less strictly enforced. Teens and young adults who have not been exposed to EBV early in life are prone to develop IM, which is also called “kissing disease”, as the major transmission route from the carrier to the naive host occurs orally through direct contact with virus-containing saliva, such as with kissing. It can also be transferred via semen during sexual contact, organ transplantation, and blood transfusion [6]. Following primary infection, EBV displays latent infection in the circulating B-cell pool of peripheral blood, minimizes viral production to evade the immune system, and persists in the host with minimal impact. Acute infection and periodic reactivation, on the other hand, allows the infectious progeny virus to spread to new hosts.
Oropharyngeal epithelial cells and B cells are the typical cell types that have shown strong tropism for EBV [8]. The question of the original cell type (either oropharyngeal epithelial cells or B cells) to first get infected by EBV during salivary transmission remains a debate. It is speculated that EBV first infects the oropharyngeal epithelial cells, where it replicates and releases virions. The newly produced virions then infect the co-localized B cells in the lympho–epithelial structures of Waldeyer’s ring [27]. However, there is an opposite point of view assuming that EBV is transmitted via the saliva, enters the tonsillar crypts, crosses the thin layer of epithelium overlying the bed of lymphocytes below, and infects naive B cells residing in the follicular mantle by a yet to be determined mechanism [28]. This point of view was supported by a prospective study that showed that EBV viral genomes are detected by nested PCR at low levels in peripheral blood about three weeks before any IM symptoms, suggesting that B cells are the major reservoir of the virus before epithelial amplification in the oral cavity [29]. Human primary B lymphocytes, as the major reservoir for EBV, are infected through interaction between the major viral outer envelope glycoprotein gp350/220 and the CD21 receptor on the surface of B cells, together with gp42, which is essential for penetration of B cells by forming a complex along with the histocompatibility complex (MHC)-II [30]. EBV can shuttle between different cell types under certain circumstances, mainly B cells and epithelial cells. However, other lymphocytes, such as T cells, NK-lineage cells, monocytes, smooth muscle cells, follicular dendritic cells (DC), and even neurons, can also become infected, as well [31,32,33,34]. Oropharyngeal epithelial cells, unlike B lymphocytes, are permissive for viral replication [35,36], and it was demonstrated that EBV binds much less efficiently to epithelial cells than B cells. The mechanism of how EBV enters into primary B cells and nasopharyngeal epithelial cells has been largely elucidated. However, further understanding of EBV entry into other different cell types is still somewhat incomplete, requiring further examination (Figure 1).
EBV initiates a distinct dual-life cycle, either latent (nonproductive) infection or lytic (productive) replication. The virus undergoes predominantly lytic replication in epithelial cells in vitro and establishes lifelong latency in circulating memory B lymphocytes, but can reactivate periodically from latency. As a result, EBV can shuttle between different cell types, mainly B cells and epithelial cells. EBV can also have chronic active recurrence or reactivate based on specific triggers that results in switching between the latent and lytic life cycle. Moreover, during latent infection, only a limited number of genes are expressed, which are required for maintenance of the viral genome (as an episome in the nucleus), evasion of the host immune system surveillance, cell growth, and proliferation.
The B-lymphotropic nature of EBV is evidenced by the potent transforming ability of the virus to immortalize normal resting B lymphocytes in vitro, converting them into permanently growing lymphoblastoid cell lines (LCLs) [39,40]. EBV remains the most efficient transforming agent, which rapidly immortalizes B cells during in vitro infection. The complete sequence of an EBV strain (the B95-8 strain) was first reported in 1984 [41], and 84 different EBV strains have been isolated to date [38]. There are two types of EBV subtypes, type 1 and type 2; the major identified differences between these two types are the sequences of the latent infection cycle nuclear antigen genes EBNA2, EBNA-LP, and EBNA3 [42,43] and the small, non-polyadenylated RNAs EBER1 and EBER2 [44]. Type 1 strains are more prevalent worldwide and have greater transforming potential [45]. There are four patterns of EBV latency [46,47], which were defined based on the pattern of EBV proteins and small RNAs expressed in in vitro infections and in EBV-induced tumors [48] (Table 1). The EBV genome within LCLs usually expresses all latent genes, which is also known as the latency Ⅲ (growth program), which includes six Epstein–Barr nuclear antigens (EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNA leading protein (EBNA-LP)), three latent membrane proteins (LMP1, LMP2A, and LMP2B), two small nonpolyadenylated RNAs (EBER-1 and EBER-2), and transcripts from the BamHI-A region (BARTs, also known as miRNAs) [49]. The latency Ⅲ program also occurs in iatrogenic immunodeficiency lymphoma (i.e., post-transplant lymphoproliferative disease (PTLD)), primary CNS lymphoma (HIV-associated), NHLs with primary immune disorders, DLBCL associated with chronic inflammation, and EBV-associated T- and NK-cell lymphomas [32]. Latency Ⅰ is described as a program where EBNA1 is the only viral protein expressed, and is seen in immunocompetent/immunocompromised BL; primary effusion lymphoma (PEL); and plasmablastic lymphoma, oral type, in immunodeficient HIV patients [50]. Latency Ⅱ (also referred to as the default program), in which EBNA1 and LMPs are expressed, is typically displayed in Hodgkin’s lymphoma; EBV-positive DLBCL; lymphomatoid granulomatosis; angioimmunoblastic T-cell lymphoma; extranodal NK/T-cell lymphoma, nasal type; aggressive NK-cell leukemia; NPC; and gastric carcinoma (GC). Latency 0, in which none of the EBV antigens are expressed, avoids immune recognition and is typically observed in circulating memory B cells in healthy persons. The expression of EBERs, as well as miRNAs, is present in all forms of latency. Furthermore, it has been documented that in the normal course of EBV infection in healthy individuals, even latency Ⅲ is very transiently seen. However, owing to fluctuations in immune pressures, these different forms of latency may exist [3,51]. Although the different types of latency programs predominate in different EBV-driven tumors, lytic viral replication has also been shown to have pathogenic importance [52,53,54]. Considering the relatively low incidence of EBV-associated malignances when compared to the high proportion of EBV infection in the worldwide population, a detailed understanding of the mechanism of pathogenesis underlying its contribution to the different associated malignancies needs further investigation.
4. Pathogenesis Associated with EBV Latent and Lytic Infection
4.1. EBV Latent Infection. Contribution of EBV Latent Antigens to the Oncogenic Phenotype Remains the Subject of Intense Study
4.1.1. The EBV-Encoded Nuclear Antigens (Summarized in Inset 1)
EBV-encoded nuclear antigens (EBNAs) can influence both viral and cellular transcription. EBNA1 is the only known EBV protein to be consistently expressed in all EBV-related cancers. Its main identified function is to ensure the replication of the double-stranded DNA (dsDNA) EBV genome as cells multiply [55,56]. It can enhance cell survival by inhibiting apoptosis [57], induce genomic instability via the production of reactive oxygen species (ROS) [58], and induce B-cell neoplasia in transgenic mice [59]. EBNA1 also contributes to transcriptional regulation of the EBNAs (including EBNA1 itself) and LMP1 by interacting with certain viral promoters [4]. The main function of the Gly-Ala repeat sequence domain of EBNA1 serves to prevent the protein from proteasomal degradation so as to avoid presentation as peptides to cytotoxic T lymphocytes (CTLs) [60,61]. EBNA1 can also counteract the stabilization of the host-encoded p53/TP53 and MDM2 by host ubiquitin-specific protease USP7, thereby decreasing apoptosis and increasing host cell survival [62]. High-affinity molecular mimicry between EBNA1 and the central nervous system protein glial cell adhesion molecule (GlialCAM) in MS was recently demonstrated. Meanwhile, structural and in vivo functional evidence for their relevance was also illustrated [63]. Taken together, the function of EBNA1 is clearly not restricted to simply maintaining the viral genome in EBV-infected cells.
EBNA2 is an EBV-encoded nuclear transcription factor that plays an essential role in the transformation process in vitro. It is the essential EBV transactivator that is required for activating cellular and viral genes. It can directly activate the important viral latent genes, which include EBNAs and LMPs genes, along with other cellular genes, such as MYC and CD23 [64,65,66]. EBNA2 was also highlighted in having a critical role in rewiring gene regulatory programs of the host through rearrangement of the chromatin landscape, which suggests that these interactions are potential components of genetic mechanisms that can influence the risk of multiple autoimmune diseases [67,68]. EBNA2 can functionally replace the intracellular region of Notch to interact with RBP-Jk to derepress the transcriptional activity at its responsive binding sites, and has been implicated in the development of EBV-associated T-cell tumors in humans [69,70].
EBNA-LP functions as a transcriptional coactivator by cooperating with EBNA2 in an RBP-Jk-mediated large multi-protein complex in the transcriptional activation of responsive cellular and viral promoters. These activities have been shown to be modulated by the EBNA3 family of proteins, which results in modulation of transcription activation through its interaction with cellular and viral transcription activator complexes [4,71,72]. EBNA-LP may also play a role in evading immune surveillance by suppressing the innate cell response to viral DNA, which allows transcription of the EBV genome and survival of EBV-infected naive B cells [73].
The EBNA3 family of proteins comprises EBNA3A, EBNA3B, and EBNA3C. Both EBNA3A and EBNA3C are considered oncogenic and are required for efficient transformation of B cells [74,75]. EBNA3A and EBNA3C can mediate the inhibition of various cyclin-dependent kinase inhibitors [76,77] and potentiate EBV-mediated B-cell transformation and subsequent viral persistence by inhibiting the apoptotic response to viral infection and cell transformation [50,78,79]. This includes inhibition of the BIM gene (a member of the pro-apoptotic B-lymphoma-2 gene (BCL-2) family), further promoting virus-induced proliferation and tumorigenesis [80]. EBNA3A and EBNA3C help establish a long-term latency and subsequent lymphoma development by blocking B-cell differentiation to plasma cell phenotype through transcriptional activation of the cyclin-dependent kinase inhibitor p18INK4c and the master transcriptional regulator of plasma cell differentiation, B lymphocyte-induced maturation protein-1 (BLIMP-1) [81]. In contrast, EBNA3B functions distinctly from EBNA3A and EBNA3C as a tumor suppressor, whose inactivation promotes immune escape and EBV-driven lymphomagenesis [82,83].

Inset 1. Summary of the functions associated with latent EBV nuclear antigens.
4.1.2. EBV-Encoded Latent Membrane Antigens (Summarized in Inset 2)
LMP1 is the main transforming antigen of EBV; it functions like a constitutively active CD40 in a ligand-independent manner [84]. LMP1 has a large number of pleiotropic effects; it can block p53-mediated apoptosis, by stimulating the expression of the zinc finger protein A20 (A20) gene that promotes cell proliferation, and regulate the inflammatory response [85,86]. LMP1 mimics the activated tumor necrosis factor receptor (TNFR) superfamily member CD40 receptor and recruits important intracellular adaptor molecules, TNFR-associated factor (TRAF) family members TRAF1, TRAF2, and TRAF3, to activate nuclear factor κB (NF-κB)-inducing kinase (NIK) and inhibitory kappa B kinases (IKKs), which phosphorylate IKKα and further promote cell survival by inhibiting apoptosis [84,87]. LMP1 also promotes B-cell proliferation by inducing a STAT1-dependent IFN-γ secretion [88] and upregulates anti-apoptotic proteins (BCL-2, myeloid cell leukemia protein-1, BCL2A1 homology, A20) by inducing the expression of cell surface adhesion molecules CD23, CD40, intercellular adhesion molecule-1, and lymphocyte function-related antigen-3 (LFA-3) to inhibit apoptosis and promote tumorigenesis [89,90]. The oncogenic ability of LMP-1 can also be attributed to its ability to induce the activation and secretion of different matrix metalloproteinases, suggesting an important role for this oncoprotein in both the angiogenic and metastatic process during the onset and development of EBV-associated tumors [91]. LMP1 can utilize the PI3K–AKT–mTOR pathway for inducing CD137 expression to support growth of the Hodgkin and Reed/Sternberg (HRS) cells and escape from immune surveillance [92]; it can also promote tumor immune escape by upregulating tumor cell PD-L1 through the NF–κB pathway in extranodal natural killer/T-cell lymphoma (ENKTL) [93].
The LMP2 proteins, LMP2A and LMP2B, first identified in 1990 [94], are not essential for EBV-induced transformation in vitro [95]. LMP2A can drive the proliferation and survival of B cells in a BCR-deficient context, which indicates that LMP2A is a mimic of BCR [96,97]. LMP2A mimics a subset of BCR signaling events, including tyrosine phosphorylation of the kinase SYK, the calcium initiation complex consisting of BLNK, BTK, and PLCγ2, and its downstream transcription factor NFAT [98]. LMP2A affects apoptosis and cell-cycle checkpoints by dysregulating the expression of apoptotic regulators, such as BCl-xL and the tumor suppressor retinoblastoma-associated protein 1 (RB1) [98]. LMP2A can also transform epithelial cells and enhance their adhesion and motility via the PI3K–Akt pathway [99]. Furthermore, LMP2A attenuates signaling from type I and type II interferon receptors (IFNRs), which circumvents the innate immune response, an effect that is generally exploited in persistent virus infections, but which can, in the context of oncogenic viruses, such as EBV, contribute to tumor development by stimulating cell growth and limiting cellular responses to virus-infected cells [100]. The detailed functions of LMP2B remain obscure, although some studies have suggested that it serves to modulate the effects of LMP2A on BCR function, thereby rendering latently infected B cells susceptible to lytic re-activation [101,102].

Inset 2. Summary of the functions associated with EBV-encoded latent membrane antigens.
4.1.3. EBV-Encoded Functional Small Non-Coding RNAs
Two small non-coding RNAs, EBER1 and EBER2, are expressed in all forms of latency. However, the precise function of EBERs remains to be elucidated. EBERs are not essential for EBV-induced transformation of primary B lymphocytes in vitro [4]. They can increase tumorigenicity, promote cell survival, induce interleukin-10 (IL-10) expression in BL cell lines [103,104,105], and significantly enhance the tumorigenicity of EBV-negative BL cells in SCID mice [106]. The BamH1A right frame 1 (BARF1), a transcript generated from the BamH1A region, shares limited homology with the human colony-stimulating factor 1 receptor (CSF1R), also called the FMS proto-oncogene, and displays oncogenic activity when expressed in rodent fibroblasts and primary monkey epithelial cells [107,108]; it is also capable of activating anti-apoptotic Bcl-2 expression [108]. It is originally identified as an early antigen expressed on induction of the EBV lytic phase, but has been shown to be expressed as a latent protein in EBV-associated NPC and GC [109,110,111].
4.2. EBV Lytic Genes and Their Associated Functions
Latency occurs predominantly in B lymphocytes; however, lytic infection can occur in both lymphocytes and epithelial cells. While EBV tumors are comprised of cells in the different latency programs, multiple studies have pointed out that lytic infection, which leads to high viral load, is a cancer determinant in EBV-induced malignances [54]. It has become increasingly clear that, in certain circumstances, the lytic cycle contributes to EBV-induced oncogenesis [112,113]. Lytically infected cells could release growth factors and immunosuppressive cytokines capable of signaling through many different signaling pathways, leading to tumor growth enhancement [53]. Besides the contribution of the lytic cycle itself to the development of EBV-induced cancers, numerous studies have showed that specific lytic proteins can promote oncogenesis [114,115,116]. The two EBV immediate early transcriptional activators, BZLF1 and BRLF1, can synergistically induce expression of multiple early lytic genes involved in viral DNA amplification, late gene expression, and virion production [38,117]. Expression of BZLF1 (the latent to lytic infection switch protein) is the beginning of lytic infection, which has been found in BL, NPC, and GC samples [118,119,120]. BZLF1 can also activate IL-8 and IL-10 expression, leading to the enhancement of cell growth and survival [121]. Another two EBV-encoded immediate early genes, BHRF1 and BALF1, which were found to be turned off once latent infection is established, are homologues of the cellular, anti-apoptotic gene Bcl2, also named viral Bcl-2 proteins (vBcl-2s) [122,123,124]. Genetic inactivation of both these two vBcl-2 genes disabled EBV’s ability to transform primary resting B lymphocytes and resulted in immediate apoptosis of cells [124], which implied that there is a critical role for the vBcl-2s in enabling cell survival during the early stages of the transformation process. BILF1 encodes a constitutively active G protein-coupled receptor, which is capable of modulating various intracellular signaling pathways, including the NF-κB-mediated signaling pathway [125].
Several EBV lytic genes are also known to induce genome instability. These include BZLF1 (impairs accumulation of host DNA damage proteins) [126], BGLF4 (induces the DNA damage response and chromatin remodeling through TIP60) [127], BGLF5 (mediates DNA damage and repression of DNA repair, subsequently increasing microsatellite instability (MSI) and genetic mutations) [128], BNRF1 (induces centrosome amplification) [129], BPLF1 (interferes with DNA repair through its effects on PCNA and Pol η) [130], and BKRF4 (binds histones and interferes with double-stranded DNA break repair) [131].
In addition, several lytic genes have been shown to contribute to immune evasion, including the innate and adaptive immune response. For example, BPLF1 evades the host immune system through interference with the Toll-like receptor signaling pathway [132]; BZLF1 downregulates MHC class Ⅱ genes [121]; BNLF2a inhibits the transporter of antigen processing [133]; BILF1 induces MHC class I internalization and degradation [134]; BZLF1 and BNLF2a synergistically inhibit the processing and presentation of cytotoxic T-cell epitopes [135]; BGLF5 mediates host shutoff [136]; and LF2 antagonizes type I interferon signaling leading in immune evasion [137]. Understanding the relationship between the EBV virus with its host cell and the interaction between EBV latent with lytic antigens in the context of associated cancers will continue to be an important topic for future studies.
5. EBV-Associated Malignances
5.1. EBV-Associated Lymphomas
5.1.1. EBV-Associated B-Cell Lymphomas
Burkitt’s Lymphoma
Burkitt lymphoma (BL) is a highly aggressive mature B-cell neoplasm and includes the historically recognized three subtypes, which are endemic BL, sporadic BL, and immunodeficiency-associated BL. Endemic BL, with the EBV genome present in >95% of the neoplastic cells, occurs in sub-Saharan Africa and regions endemic for malaria [138]. All BLs have a characteristic chromosome translocation of the MYC gene to one of the Ig loci, which juxtaposes MYC to the downstream of Ig heavy or light chain enhancer, resulting in enhanced and deregulated C-MYC expression [138]. The translocations are thought to occur in the lymph node germinal center, where Ig gene modifications associated with class switching and somatic hypermutation normally happen in B cells [9]. In the context of endemic BLs, EBV and malaria induce activation-induced cytidine deaminase (AID) activity in the lymph node and eventually initiate Ig/MYC translocations [138]. It was also shown that EBNA2 can activate MYC and consequently increase the sensitivity of upstream enhancer regions to AID activity [139]. The fact that EBV, together with malaria, can increase the frequency of BLs is attributed to malaria-related higher EBV load, higher levels of AID, and the production of an increased number of B cells in response to malaria in the germinal center, and lower surveillance of EBV due to immunodeficiency [9]. Emerging evidence suggests a dual mechanism of BL pathogenesis: virus-driven versus mutational burden, which depends on the EBV status, regardless of the epidemiologic context and geographic location. Thus, based on recent insights into BL biology, it is recommended by the 5th edition of the WHO classification of hematolymphoid tumors (WHO-HAEM5) to classify BL into two subtypes, EBV-positive BL and EBV-negative BL [140].
Hodgkin Lymphoma
Classical Hodgkin lymphoma (cHL) is a major type of HL, which accounts for approximately 90% of all HLs and is characterized by the presence of scattered malignant HRS cells in the non-neoplastic inflammatory tumor microenvironment background [141]. EBV is present in all of the HRS cells in about 30% of HL cases, the majority of which are the mixed cellularity type [20]. EBV infection of the HRS cells is considered to play a causal role in the oncogenesis of cHL, which is confirmed by in situ hybridization for detection of the EBV genome in HRS cells [20] and EBERs as a target for the routine detection of EBV in cHL [142]. EBV is consistently retained during disease progression, suggesting it is required for maintenance of the tumor phenotype [143]. EBV-infected HRS cells were shown to express a restricted pattern of EBV latency Ⅱ characterized by the presence of EBNA1, LMP1, and LMP2A, as well as a subset of viral miRNA [144]. Furthermore, the HRS cells are germinal center experienced B cells at various stages of development, which lack surface BCR expression due to the crippled rearrangement of BCR [145]. Interestingly, LMP1 and LMP2A are thought to function as surrogates for BCR function, protecting HRS cells from apoptosis, and allow the survival and proliferation of the HRS cells.
EBV-Positive Diffuse Large B-Cell Lymphoma
EBV-positive diffuse large B-cell lymphoma (DLBCL), which was formerly designated as EBV-positive DLBCL of the elderly, usually occurs in individuals aged >50 years, with an inferior clinical prognosis in comparison to EBV-negative DLBCL [146]. The incidence of EBV-positive DLBCL is less than 5% in Western countries, but that percentage goes up to 10–15% in Asian and Latin American countries [147]. Most EBV-positive DLBCL cases have an activated B-cell (ABC) phenotype, expressing MUM1/IRF4, and are negative for CD10 and BCL6 [147]. Expression of NF-κB and phosphorylated STAT3 are more commonly seen in EBV-positive DLBCL compared with EBV-negative DLBCL [146]. Gene expression profiles revealed that the activation of the NF–κB and/or JAK/STAT3 pathways are characteristics of EBV-positive DLBCL, even when compared with ABC type EBV-negative DLBCL [148,149]. However, the interaction with EBV is still poorly understood [148]. It has been shown that infection with EBV lacking EBNA3B leads to aggressive, immune-evading, monomorphic, DLBCL-like tumors in NOD/SCID/γc-/-mice with reconstituted human immune system components [150]. There were also additional EBNA3B mutations identified when screening EBV-positive DLBCL human samples [150]. Thus, EBNA3B is a virus-encoded tumor suppressor, and its mutations can result in increased lymphomagenesis.
5.1.2. EBV-Associated NK-Cell and T-Cell Lymphomas
Extranodal Natural Killer/T-Cell Lymphoma
Extranodal natural killer (NK)/T-cell lymphoma (ENKTL) is an aggressive lymphoma strongly associated with EBV infection that occurs mainly in Asians and the indigenous populations of Latin American, but rarely in native Europeans and North American populations [151]. The exact role of EBV in ENKTL remains elusive; however, the characteristic geographical distribution suggests that ethnicity plays an important genetic role in regards to the predisposition of these populations to the disease [141,152]. In these cancers, EBV is present as a clonal episomal form with the type Ⅱ latency expression pattern. The EBV type 1 strain is common in ENKTL compared with type 2 [153]. A 30-base pair deletion in the LMP1 gene, C terminus region of the type 2 EBV strain, was common and associated with enhancement of its oncogenic potential in ENKTL [154]. EBV BART9 miRNA modulates LMP1 levels and promotes proliferation of ENKTL cells [155]. Different cytokines (IL-2, IL-9, IL-10, IL-15) are produced by EBV-infected NK cells via autocrine or paracrine loops, and the microenvironment can stimulate cell growth, along with expression of LMP1 [156].
Angioimmunoblastic T-Cell Lymphoma
Angioimmunoblastic T-cell lymphoma (AITL) is a neoplasm of mature T-follicular helper (TFH) cells strongly associated with EBV infection, which is less common in North America or Asia than in Europe [157]. EBV-positive B cells are detected in more than 90% of biopsies. However, the neoplastic T cells are EBV-negative. To date, the role of EBV in this pathology is still unknown. It has been reported that EBV itself can drive the development of AITL by activating TFH cells [158]. It is also proposed that AITL generates a profound immunodeficiency at the origin of EBV reactivation favoring the expansion of TFH and B cells, thus contributing to the development of the tumor microenvironment (Figure 2). Moreover, the co-expression of BNLF2a and BCRF1 (a homologous protein of human interleukin 10, vIL-10) can contribute to the immune escape and survival of infected cells [159].
5.2. EBV-Associated Epithelial Carcinomas
5.2.1. Nasopharyngeal Carcinoma
Undifferentiated (also called anaplastic) nasopharyngeal carcinoma (NPC) has an exceptionally high incidence in the Cantonese population in Southern China (95%), as well as Southeast Asia, Northern Africa, and Eskimos in the Arctic [160]. NPC occurs less commonly in the United States and Europe, with rates of 0.5~2/100,000, about 50-fold less than that seen in Southern China. Undifferentiated NPC virtually always has EBV present in the transformed malignant epithelial cell component, but not in the characteristically infiltrating, substantial population of lymphocytes, which are thought to be important in maintaining cancer by providing cytokines or other signals, and in immune escape.
Besides EBV infection, other risk factors for NPC include male sex, HLA type, and environmental/lifestyle factors [161]. EBV is clonal in high-grade preinvasive lesions (carcinoma in situ or dysplasia), as well as in NPC [162], but not in low-grade preinvasive lesions. Notably, epithelial cells are not as susceptible to transformation as B lymphocytes in vitro, either by co-culture or by transfer infection of EBV from lymphocytes. Only a few NPC cell lines have been generated for use in in vitro studies [163,164]. NPC is believed to be a multistep process by accumulating mutations (overexpression of oncogenes and loss of expression of the suppressor genes), which results in a hyperplastic lesion. Meanwhile, EBV is proposed to act as a trigger by inducing dysplastic lesions and eventually invasive NPC. NPC is described as EBV latency Ⅱ program; however, the expression of LMP1 is often undetectable in many of the cells [165,166]. LMP2A can block STAT3 activation by inhibiting NF-κB, thereby impairing LMP1 expression, which provides a mechanistic basis for the scenario that NPC cells express LMP2A, but very little LMP1 [167]. Fingerprints of Epstein–Barr virus in NPC have revealed the activation of distinct pathways (for example, chromatin modification, ERBB–PI3K signaling, and autophagy machinery) that drive the development of this tumor [168].
5.2.2. Gastric Cancer
EBV was first detected in typical gastric adenocarcinoma tumor cells in 1992 [24]. About 10% of cases of gastric cancer (GC) were confirmed to be EBV-associated GC (EBV-GC), which manifests unique genomic aberrations, significant clinicopathological features, and a good prognosis compared to EBV-negative GC [169]. EBV-GCs also display viral latency Ⅱ program, similar to that of NPC [56]. However, LMP1 is absent in some cases, while BART miRNAs are highly expressed [116]. The precise contribution of EBV to the development of EBV-GC is unknown, but it is shown that EBV-GC development is also linked to several other pathogenic factors, including genetic susceptible background, dietary habits, and metal dust environmental cofactors exposure [170]. The lack of p53 mutation in EBV-GC has led to the speculation that EBV can in some way bypass the need for p53 mutation, which seems essential for most types of carcinomas [9]. Other notable genetic differences between EBV-positive GC and EBV-negative GC include high CpG methylation of host genes upon EBV infection, inactivation of tumor suppressor genes, loss of cyclin-dependent kinase inhibitor p16INK4A expression, and relatively frequent PI3K mutations, to name a few [9,38].
6. Summary and Future Perspectives
Since EBV was discovered in 1964, our understanding of EBV has continuously grown. EBV has now been accepted, with its current leading role as a prime example of a human tumor virus that is etiologically linked to an unexpectedly diverse range of diseases, from benign infectious diseases, SADs, to EBV-associated malignancies. Studying the role of EBV in cancer pathogenesis and the host cell–virus interaction, in both humans and animals, has provided principle insights into the mechanisms that drive the oncogenic process. This understanding has also raised the possibility for therapeutic and prophylactic intervention targeting EBV latent/lytic proteins, EBV miRNAs, and the tumor microenvironment, which includes the local cytokine milieu.
There are still many questions that cannot be answered at the moment. Although EBV remains the most common persistent, asymptomatic virus infection in humans, it is unclear why only a small fraction of people develop the viral-associated diseases/malignancies. There are also more studies needed to elucidate the oncogenic contributions to specific EBV lytic proteins in the context of latent infections in certain cancers. The precise role of EBV in the pathogenesis of epithelial cancers, as well as the complex interplay between EBV and the immune system, require additional investigations. However, the challenge would be to exploit these new mechanistic insights to gain a more comprehensive understanding of the biology of EBV infection in vivo and to develop novel therapies for treating virus-associated diseases.
Author Contributions
Conceptualization: H.Y. and E.S.R.; writing—original draft preparation: H.Y.; writing—review and editing: H.Y. and E.S.R.; funding acquisition: E.S.R. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the National Cancer Institute (NCI) at the National Institutes of Health (NIH) public health service grants R01-CA171979, and R01-CA244074 (E.S.R.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to thank our colleagues in the departments of Otorhinolaryngology-Head and Neck Surgery and Microbiology for their support and NCI for their funding assistance.
Conflicts of Interest
The authors declare no conflict of interest. The funder had no role in the design of the study, in the writing of the manuscript, or in the decision to publish the results.
References
- Cohen, J.I. Epstein-barr virus infection. N. Engl. J. Med. 2000, 343, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Luzuriaga, K.; Sullivan, J.L. Infectious mononucleosis. N. Engl. J. Med. 2010, 362, 1993–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the epstein-barr virus and the origins of associated lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Rickinson, A.B. Epstein-barr virus: 40 years on. Nat. Rev. Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. EBV Persistence—Introducing the virus. Curr. Top Microbiol. Immunol. 2015, 390 Pt 1, 151–209. [Google Scholar]
- Tonoyan, L.; Vincent-Bugnas, S.; Olivieri, C.V.; Doglio, A. New viral facets in oral diseases: The EBV paradox. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Robinson, W.H.; Steinman, L. Epstein-barr virus and multiple sclerosis. Science 2022, 375, 264–265. [Google Scholar] [CrossRef]
- Houen, G.; Trier, N.H. Epstein-barr virus and systemic autoimmune diseases. Front. Immunol. 2020, 11, 587380. [Google Scholar] [CrossRef]
- Farrell, P.J. Epstein-barr virus and cancer. Annu. Rev. Pathol. 2019, 14, 29–53. [Google Scholar] [CrossRef]
- de Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Old, L.J.; Boyse, E.A.; Oettgen, H.F.; Harven, E.D.; Geering, G.; Williamson, B.; Clifford, P. Precipitating antibody in human serum to an antigen present in cultured burkitt’s lymphoma cells. Proc. Natl. Acad. Sci. USA 1966, 56, 1699–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.A.; Henle, G. Indirect immunofluorescence tests with sera from African children and cultured Burkitt lymphoma cells. J. Bacteriol. 1966, 92, 275–276. [Google Scholar] [CrossRef] [Green Version]
- Henle, G.; Henle, W.; Diehl, V. Relation of burkitt’s tumor-associated herpes-ytpe virus to infectious mononucleosis. Proc. Natl. Acad. Sci. USA 1968, 59, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- zur Hausen, H.; Schulte-Holthausen, H.; Klein, G.; Henle, W.; Henle, G.; Clifford, P.; Santesson, L. EBV DNA in biopsies of burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature 1970, 228, 1056–1058. [Google Scholar] [CrossRef] [PubMed]
- Wolf, H.; zur Hausen, H.; Becker, V. EB viral genomes in epithelial nasopharyngeal carcinoma cells. Nat. New Biol. 1973, 244, 245–247. [Google Scholar] [PubMed] [Green Version]
- Ziegler, J.L.; Drew, W.L.; Miner, R.C.; Mintz, L.; Rosenbaum, E.; Gershow, J.; Lennette, E.T.; Greenspan, J.; Shillitoe, E.; Beckstead, J.; et al. Outbreak of burkitt’s-like lymphoma in homosexual men. Lancet 1982, 2, 631–633. [Google Scholar] [CrossRef]
- Greenspan, J.S.; Greenspan, D.; Lennette, E.T.; Abrams, D.I.; Conant, M.A.; Petersen, V.; Freese, U.K. Replication of epstein-barr virus within the epithelial cells of oral “hairy” leukoplakia, an AIDS-associated lesion. N. Engl. J. Med. 1985, 313, 1564–1571. [Google Scholar] [CrossRef]
- Nagington, J.; Gray, J. Cyclosporin A immunosuppression, epstein-barr antibody, and lymphoma. Lancet 1980, 1, 536–537. [Google Scholar] [CrossRef]
- Weiss, L.M.; Movahed, L.A.; Warnke, R.A.; Sklar, J. Detection of epstein-barr viral genomes in reed-sternberg cells of hodgkin’s disease. N. Engl. J. Med. 1989, 320, 502–506. [Google Scholar] [CrossRef]
- Jones, J.F.; Shurin, S.; Abramowsky, C.; Tubbs, R.R.; Sciotto, C.G.; Wahl, R.; Sands, J.; Gottman, D.; Katz, B.Z.; Sklar, J. T-cell lymphomas containing Epstein-Barr viral DNA in patients with chronic epstein-barr virus infections. N. Engl. J. Med. 1988, 318, 733–741. [Google Scholar] [CrossRef]
- Iwatsuki, K.; Miyake, T.; Hirai, Y.; Yamamoto, T. Hydroa vacciniforme: A distinctive form of epstein-barr virus-associated T-cell lymphoproliferative disorders. Eur. J. Dermatol. 2019, 29, 21–28. [Google Scholar] [PubMed]
- Hue, S.S.; Oon, M.L.; Wang, S.; Tan, S.Y.; Ng, S.B. Epstein-barr virus-associated T- and NK-cell lymphoproliferative diseases: An update and diagnostic approach. Pathology 2020, 52, 111–127. [Google Scholar] [CrossRef]
- Shibata, D.; Weiss, L.M. Epstein-barr virus-associated gastric adenocarcinoma. Am. J. Pathol. 1992, 140, 769–774. [Google Scholar] [PubMed]
- Bonnet, M.; Guinebretiere, J.M.; Kremmer, E.; Grunewald, V.; Benhamou, E.; Contesso, G.; Joab, I. Detection of epstein-barr virus in invasive breast cancers. J. Natl. Cancer Inst. 1999, 91, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, Y.; Mizugaki, Y.; Uchida, T.; Torii, T.; Imai, S.; Makuuchi, M.; Takada, K. Detection of epstein-barr virus (EBV) in hepatocellular carcinoma tissue: A novel EBV latency characterized by the absence of EBV-encoded small RNA expression. Virology 1999, 256, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, D.H. Biology and disease associations of epstein-barr virus. Philos. Trans. R Soc. Lond B Biol. Sci. 2001, 356, 461–473. [Google Scholar] [CrossRef] [Green Version]
- Thorley-Lawson, D.A. Epstein-barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef]
- Dunmire, S.K.; Grimm, J.M.; Schmeling, D.O.; Balfour, H.H.J.; Hogquist, K.A. The incubation period of primary epstein-barr virus infection: Viral dynamics and immunologic events. PLoS Pathog. 2015, 11, e1005286. [Google Scholar] [CrossRef]
- Tanner, J.; Weis, J.; Fearon, D.; Whang, Y.; Kieff, E. Epstein-barr virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorption, capping, and endocytosis. Cell 1987, 50, 203–213. [Google Scholar] [CrossRef]
- Savard, M.; Belanger, C.; Tardif, M.; Gourde, P.; Flamand, L.; Gosselin, J. Infection of primary human monocytes by epstein-barr virus. J. Virol. 2000, 74, 2612–2619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrei, G.; Trompet, E.; Snoeck, R. Novel therapeutics for epstein(-)barr virus. Molecules 2019, 24, 997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, H.C.; Mehta, D.; Lu, J.; El-Naccache, D.; Shukla, S.K.; Kovacsics, C.; Kolson, D.; Robertson, E.S. Gammaherpesvirus infection of human neuronal cells. mBio 2015, 6, e01844-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutt-Fletcher, L.M. Epstein-Barr virus entry. J. Virol. 2007, 81, 7825–7832. [Google Scholar] [CrossRef] [Green Version]
- Sixbey, J.W.; Vesterinen, E.H.; Nedrud, J.G.; Raab-Traub, N.; Walton, L.A.; Pagano, J.S. Replication of epstein-barr virus in human epithelial cells infected in vitro. Nature 1983, 306, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.X.; Young, L.S.; Niedobitek, G.; Dawson, C.W.; Birkenbach, M.; Wang, F.; Rickinson, A.B. Epstein-barr virus infection and replication in a human epithelial cell system. Nature 1992, 356, 347–350. [Google Scholar] [CrossRef]
- Fox, C.P.; Shannon-Lowe, C.; Rowe, M. Deciphering the role of epstein-barr virus in the pathogenesis of T and NK cell lymphoproliferations. Herpesviridae 2011, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Henle, W.; Diehl, V.; Kohn, G.; Hausen, H.; Henle, G. Herpes-type virus and chromosome marker in normal leukocytes after growth with irradiated Burkitt cells. Science 1967, 157, 1064–1065. [Google Scholar] [CrossRef]
- Diehl, V.; Henle, G.; Henle, W.; Kohn, G. Demonstration of a herpes group virus in cultures of peripheral leukocytes from patients with infectious mononucleosis. J. Virol. 1968, 2, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Baer, R.; Bankier, A.T.; Biggin, M.D.; Deininger, P.L.; Farrell, P.J.; Gibson, T.J.; Hatfull, G.; Hudson, G.S.; Satchwell, S.C.; Seguin, C.; et al. DNA sequence and expression of the B95-8 epstein-barr virus genome. Nature 1984, 310, 207–211. [Google Scholar] [CrossRef]
- Dambaugh, T.; Hennessy, K.; Chamnankit, L.; Kieff, E. U2 region of epstein-barr virus DNA may encode epstein-barr nuclear antigen 2. Proc. Natl. Acad. Sci. USA 1984, 81, 7632–7636. [Google Scholar] [CrossRef] [Green Version]
- Sample, J.; Young, L.; Martin, B.; Chatman, T.; Kieff, E.; Rickinson, A.; Kieff, E. Epstein-barr virus types 1 and 2 differ in their EBNA-3A, EBNA-3B, and EBNA-3C genes. J. Virol. 1990, 64, 4084–4092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrand, J.R.; Young, L.S.; Tugwood, J.D. Two families of sequences in the small RNA-encoding region of epstein-barr virus (EBV) correlate with EBV types A and B. J. Virol. 1989, 63, 983–986. [Google Scholar] [CrossRef] [Green Version]
- Zimber, U.; Adldinger, H.K.; Lenoir, G.M.; Vuillaume, M.; Knebel-Doeberitz, M.V.; Laux, G.; Desgranges, C.; Wittmann, P.; Freese, U.K.; Schneider, U.; et al. Geographical prevalence of two types of epstein-barr virus. Virology 1986, 154, 56–66. [Google Scholar] [CrossRef]
- Kuppers, R. B cells under influence: Transformation of B cells by epstein-barr virus. Nat. Rev. Immunol. 2003, 3, 801–812. [Google Scholar] [CrossRef]
- Hochberg, D.; Middeldorp, J.M.; Catalina, M.; Sullivan, J.L.; Luzuriaga, K.; Thorley-Lawson, D.A. Demonstration of the burkitt’s lymphoma epstein-barr virus phenotype in dividing latently infected memory cells in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 239–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, G. Viral latency and transformation: The strategy of epstein-barr virus. Cell 1989, 58, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Brooks, L.A.; Lear, A.L.; Young, L.S.; Rickinson, A.B. Transcripts from the epstein-barr virus bamHI A fragment are detectable in all three forms of virus latency. J. Virol. 1993, 67, 3182–3190. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.; Robertson, E.S. Mechanisms of B-cell oncogenesis induced by epstein-barr virus. J. Virol. 2019, 93, e00238-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munz, C. Latency and lytic replication in epstein-barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Hong, G.K.; Gulley, M.L.; Feng, W.H.; Delecluse, H.J.; Holley-Guthrie, E.; Kenney, S.C. Epstein-barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J. Virol. 2005, 79, 13993–14003. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A new model of epstein-barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frappier, L. Epstein-barr virus: Current questions and challenges. Tumour. Virus Res. 2021, 12, 200218. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.L.; Warren, N.; Sugden, B. Stable replication of plasmids derived from epstein-barr virus in various mammalian cells. Nature 1985, 313, 812–815. [Google Scholar] [CrossRef]
- Westhoff Smith, D.; Sugden, B. Potential cellular functions of epstein-barr nuclear antigen 1 (EBNA1) of epstein-barr virus. Viruses 2013, 5, 226–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, G.; Komano, J.; Sugden, B. Epstein-barr virus provides a survival factor to burkitt’s lymphomas. Proc. Natl. Acad. Sci. USA 2003, 100, 14269–14274. [Google Scholar] [CrossRef] [Green Version]
- Gruhne, B.; Sompallae, R.; Marescotti, D.; Kamranvar, S.A.; Gastaldello, S.; Masucci, M.G. The epstein-barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2009, 106, 2313–2318. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.B.; Bell, J.L.; Levine, A.J. Expression of epstein-barr virus nuclear antigen-1 induces B cell neoplasia in transgenic mice. EMBO J. 1996, 15, 3117–3126. [Google Scholar] [CrossRef]
- Levitskaya, J.; Coram, M.; Levitsky, V.; Imreh, S.; Steigerwald-Mullen, P.M.; Klein, G.; Kurilla, M.G.; Masucci, M.G. Inhibition of antigen processing by the internal repeat region of the epstein-barr virus nuclear antigen-1. Nature 1995, 375, 685–688. [Google Scholar] [CrossRef]
- Levitskaya, J.; Sharipo, A.; Leonchiks, A.; Ciechanover, A.; Masucci, M.G. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. USA 1997, 94, 12616–12621. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Y.; Saridakis, V.; Sarkari, F.; Duan, S.; Wu, T.; Arrowsmith, C.H.; Frappier, L. Molecular recognition of p53 and MDM2 by USP7/HAUSP. Nat. Struct. Mol. Biol. 2006, 13, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Lanz, T.V.; Brewer, R.C.; Ho, P.P.; Moon, J.S.; Jude, K.M.; Fernandez, D.; Fernandes, R.A.; Gomez, A.M.; Nadj, G.S.; Bartley, C.M.; et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022, 603, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.R.; Johannsen, E.; Tong, X.; Yalamanchili, R.; Kieff, E. The epstein-barr virus nuclear antigen 2 transactivator is directed to response elements by the j kappa recombination signal binding protein. Proc. Natl. Acad. Sci. USA 1994, 91, 7568–7572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, J.J.; Hayward, S.D. Masking of the CBF1/RBPJ kappa transcriptional repression domain by epstein-barr virus EBNA2. Science 1995, 268, 560–563. [Google Scholar] [CrossRef]
- Zhao, B.; Maruo, S.; Cooper, A.; R. Chase, M.; Johannsen, E.; Kieff, E.; Cahir-McFarland, E. RNAs induced by epstein-barr virus nuclear antigen 2 in lymphoblastoid cell lines. Proc. Natl. Acad. Sci. USA 2006, 103, 1900–1905. [Google Scholar] [CrossRef] [Green Version]
- Harley, J.B.; Chen, X.; Pujato, M.; Miller, D.; Maddox, A.; Forney, C.; Magnusen, A.F.; Lynch, A.; Chetal, K.; Yukawa, M.; et al. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat. Genet. 2018, 50, 699–707. [Google Scholar] [CrossRef]
- Hong, T.; Parameswaran, S.; Donmez, O.A.; Miller, D.; Forney, C.; Lape, M.; Saint Just Ribeiro, M.; Liang, J.; Edsall, L.E.; Magnusen, A.F.; et al. Epstein-barr virus nuclear antigen 2 extensively rewires the human chromatin landscape at autoimmune risk loci. Genome. Res. 2021, 31, 2185–2198. [Google Scholar] [CrossRef]
- Artavanis-Tsakonas, S.; Matsuno, K.; Fortini, M.E. Notch signaling. Science 1995, 268, 225–232. [Google Scholar] [CrossRef]
- Sakai, T.; Taniguchi, Y.; Tamura, K.; Minoguchi, S.; Fukuhara, T.; Strobl, L.J.; Zimber-Strobl, U.; Bornkamm, G.W.; Honjo, T. Functional replacement of the intracellular region of the Notch1 receptor by Epstein-Barr virus nuclear antigen 2. J. Virol. 1998, 72, 6034–6039. [Google Scholar] [CrossRef] [Green Version]
- Robertson, E.S.; Lin, J.; Kieff, E. The amino-terminal domains of epstein-barr virus nuclear proteins 3A, 3B, and 3C interact with RBPJ(kappa). J. Virol. 1996, 70, 3068–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radkov, S.A.; Touitou, R.; Brehm, A.; Rowe, M.; West, M.; Kouzarides, T.; Allday, M.J. Epstein-barr virus nuclear antigen 3C interacts with histone deacetylase to repress transcription. J. Virol. 1999, 73, 5688–5697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymula, A.; Palermo, R.D.; Bayoumy, A.; Groves, I.J.; Ba Abdullah, M.; Holder, B.; White, R.E. Epstein-barr virus nuclear antigen EBNA-LP is essential for transforming naive B cells, and facilitates recruitment of transcription factors to the viral genome. PLoS Pathog. 2018, 14, e1006890. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.I.; Wang, F.; Mannick, J.; Kieff, E. Epstein-barr virus nuclear protein 2 is a key determinant of lymphocyte transformation. Proc. Natl. Acad. Sci. USA 1989, 86, 9558–9562. [Google Scholar] [CrossRef] [Green Version]
- Tomkinson, B.; Robertson, E.; Kieff, E. Epstein-barr virus nuclear proteins EBNA-3A and EBNA-3C are essential for B-lymphocyte growth transformation. J. Virol. 1993, 67, 2014–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tursiella, M.L.; Bowman, E.R.; Wanzeck, K.C.; Throm, R.E.; Liao, J.; Zhu, J.; Sample, C.E. Epstein-barr virus nuclear antigen 3A promotes cellular proliferation by repression of the cyclin-dependent kinase inhibitor p21WAF1/CIP1. PLoS Pathog. 2014, 10, e1004415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, J.S.; Robertson, E.S. Epstein-barr virus nuclear antigen 3C regulates cyclin A/p27 complexes and enhances cyclin A-dependent kinase activity. J. Virol. 2004, 78, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, S.; Ghosh Roy, S.; Bose, P.; Saha, A. Role of EBNA-3 family proteins in EBV associated B-cell lymphomagenesis. Front. Microbiol. 2016, 7, 457. [Google Scholar] [CrossRef] [Green Version]
- Price, A.M.; Dai, J.; Bazot, Q.; Patel, L.; Nikitin, P.A.; Djavadian, R.; Winter, P.S.; Salinas, C.A.; Barry, A.P.; Wood, K.C.; et al. Epstein-barr virus ensures B cell survival by uniquely modulating apoptosis at early and late times after infection. Elife 2017, 6, e22509. [Google Scholar] [CrossRef]
- Paschos, K.; Smith, P.; Anderton, E.; Middeldorp, J.M.; White, R.E.; Allday, M.J. Epstein-barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene bim. PLoS Pathog. 2009, 5, e1000492. [Google Scholar] [CrossRef] [Green Version]
- Styles, C.T.; Bazot, Q.; Parker, G.A.; White, R.E.; Paschos, K.; Allday, M.J. EBV epigenetically suppresses the B cell-to-plasma cell differentiation pathway while establishing long-term latency. PLoS Biol. 2017, 15, e2001992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Styles, C.T.; Paschos, K.; White, R.E.; Farrell, P.J. The cooperative functions of the EBNA3 proteins are central to EBV persistence and latency. Pathogens 2018, 7, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gain, C.; Malik, S.; Bhattacharjee, S.; Ghosh, A.; Robertson, E.S.; Das, B.B.; Saha, A. Proteasomal inhibition triggers viral oncoprotein degradation via autophagy-lysosomal pathway. PLoS Pathog. 2020, 16, e1008105. [Google Scholar] [CrossRef] [Green Version]
- Kieser, A.; Sterz, K.R. The latent membrane protein 1 (LMP1). Curr. Top Microbiol. Immunol. 2015, 391, 119–149. [Google Scholar]
- Eliopoulos, A.G.; Stack, M.; Dawson, C.W.; Kaye, K.M.; Hodgkin, L.; Sihota, S.; Rowe, M.; Young, L.S. Epstein-barr virus-encoded LMP1 and CD40 mediate IL-6 production in epithelial cells via an NF-kappaB pathway involving TNF receptor-associated factors. Oncogene 1997, 14, 2899–2916. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, K.; Das, P.; Chattopadhyay, N.R.; Mal, S.; Choudhuri, T. The interplay between epstein-bar virus (EBV) with the p53 and its homologs during EBV associated malignancies. Heliyon 2019, 5, e02624. [Google Scholar] [CrossRef]
- Wang, L.W.; Jiang, S.; Gewurz, B.E. Epstein-barr virus LMP1-mediated oncogenicity. J. Virol. 2017, 91, e01718-16. [Google Scholar] [CrossRef] [Green Version]
- Chabay, P. Advances in the pathogenesis of EBV-associated diffuse large B cell lymphoma. Cancers 2021, 13, 2717. [Google Scholar] [CrossRef]
- Miller, W.E.; Mosialos, G.; Kieff, E.; Raab-Traub, N. Epstein-barr virus LMP1 induction of the epidermal growth factor receptor is mediated through a TRAF signaling pathway distinct from NF-kappaB activation. J. Virol. 1997, 71, 586–594. [Google Scholar] [CrossRef] [Green Version]
- Eliopoulos, A.G.; Young, L.S. LMP1 structure and signal transduction. Semin. Cancer Biol. 2001, 11, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Yoshizaki, T.; Sato, H.; Furukawa, M.; Pagano, J.S. The expression of matrix metalloproteinase 9 is enhanced by epstein-barr virus latent membrane protein 1. Proc. Natl. Acad. Sci. USA 1998, 95, 3621–3626. [Google Scholar] [CrossRef] [Green Version]
- Aravinth, S.P.; Rajendran, S.; Li, Y.; Wu, M.; Yi Wong, A.H.; Schwarz, H. Epstein-barr virus-encoded LMP1 induces ectopic CD137 expression on hodgkin and reed-sternberg cells via the PI3K-AKT-mTOR pathway. Leuk Lymphoma 2019, 60, 2697–2704. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.W.; Wang, H.; Zhang, W.W.; Wang, J.H.; Liu, W.J.; Xia, Z.J.; Huang, H.Q.; Jiang, W.Q.; Zhang, Y.J.; Wang, L. PD-L1 is upregulated by EBV-driven LMP1 through NF-kappaB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J. Hematol. Oncol. 2016, 9, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longnecker, R.; Kieff, E. A second epstein-barr virus membrane protein (LMP2) is expressed in latent infection and colocalizes with LMP1. J. Virol. 1990, 64, 2319–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longnecker, R. Epstein-barr virus latency: LMP2, a regulator or means for epstein-barr virus persistence? Adv. Cancer Res. 2000, 79, 175–200. [Google Scholar] [PubMed]
- Caldwell, R.G.; Wilson, J.B.; Anderson, S.J.; Longnecker, R. Epstein-barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity 1998, 9, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Casola, S.; Otipoby, K.L.; Alimzhanov, M.; Humme, S.; Uyttersprot, N.; Kutok, J.L.; Carroll, M.C.; Rajewsky, K. B cell receptor signal strength determines B cell fate. Nat. Immunol. 2004, 5, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Fish, K.; Comoglio, F.; Shaffer, A.L., III; Ji, Y.; Pan, K.T.; Scheich, S.; Oellerich, A.; Doebele, C.; Ikeda, M.; Schaller, S.J.; et al. Rewiring of B cell receptor signaling by epstein-barr virus LMP2A. Proc. Natl. Acad. Sci. USA 2020, 117, 26318–26327. [Google Scholar] [CrossRef]
- Scholle, F.; Bendt, K.M.; Raab-Traub, N. Epstein-barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt. J. Virol. 2000, 74, 10681–10689. [Google Scholar] [CrossRef] [Green Version]
- Shah, K.M.; Stewart, S.E.; Wei, W.; Woodman, C.B.; O’Neil, J.D.; Dawson, C.W.; Young, L.S. The EBV-encoded latent membrane proteins, LMP2A and LMP2B, limit the actions of interferon by targeting interferon receptors for degradation. Oncogene 2009, 28, 3903–3914. [Google Scholar] [CrossRef] [Green Version]
- Rovedo, M.; Longnecker, R. Epstein-barr virus latent membrane protein 2B (LMP2B) modulates LMP2A activity. J. Virol. 2007, 81, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Rechsteiner, M.P.; Berger, C.; Zauner, L.; Sigrist, J.A.; Weber, M.; Longnecker, R.; Bernasconi, M.; Nadal, D. Latent membrane protein 2B regulates susceptibility to induction of lytic epstein-barr virus infection. J. Virol. 2008, 82, 1739–1747. [Google Scholar] [CrossRef] [Green Version]
- Takada, K.; Nanbo, A. The role of EBERs in oncogenesis. Semin. Cancer Biol. 2001, 11, 461–467. [Google Scholar] [CrossRef]
- Niller, H.H.; Salamon, D.; Ilg, K.; Koroknai, A.; Banati, F.; Bauml, G.; Rucker, O.; Schwarzmann, F.; Wolf, H.; Minarovits, J. The in vivo binding site for oncoprotein c-Myc in the promoter for epstein-barr virus (EBV) encoding RNA (EBER) 1 suggests a specific role for EBV in lymphomagenesis. Med. Sci. Monit. 2003, 9, HY1-9. [Google Scholar]
- Komano, J.; Maruo, S.; Kurozumi, K.; Oda, T.; Takada, K. Oncogenic role of epstein-barr virus-encoded RNAs in burkitt’s lymphoma cell line Akata. J. Virol. 1999, 73, 9827–9831. [Google Scholar] [CrossRef] [Green Version]
- Ruf, I.K.; Rhyne, P.W.; Yang, C.; Cleveland, J.L.; Sample, J.T. Epstein-barr virus small RNAs potentiate tumorigenicity of burkitt lymphoma cells independently of an effect on apoptosis. J. Virol. 2000, 74, 10223–10228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, M.X.; de Turenne-Tessier, M.; Decaussin, G.; Benet, G.; Ooka, T. Establishment of a monkey kidney epithelial cell line with the BARF1 open reading frame from epstein-barr virus. Oncogene 1997, 14, 3073–3081. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; Decaussin, G.; Sumner, S.; Ooka, T. N-terminal domain of BARF1 gene encoded by epstein-barr virus is essential for malignant transformation of rodent fibroblasts and activation of BCL-2. Oncogene 2001, 20, 1176–1185. [Google Scholar] [CrossRef] [Green Version]
- Karran, L.; Gao, Y.; Smith, P.R.; Griffin, B.E. Expression of a family of complementary-strand transcripts in epstein-barr virus-infected cells. Proc. Natl. Acad. Sci. USA 1992, 89, 8058–8062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- zur Hausen, A.; Brink, A.A.; Craanen, M.E.; Middeldorp, J.M.; Meijer, C.J.; van den Brule, A.J. Unique transcription pattern of epstein-barr virus (EBV) in EBV-carrying gastric adenocarcinomas: Expression of the transforming BARF1 gene. Cancer Res. 2000, 60, 2745–2748. [Google Scholar] [PubMed]
- Stevens, S.J.; Verkuijlen, S.A.; Hariwiyanto, B.; Harijadi; Paramita, D.K.; Fachiroh, J.; Adham, M.; Tan, I.B.; Haryana, S.M.; Middeldorp, J.M. Noninvasive diagnosis of nasopharyngeal carcinoma: Nasopharyngeal brushings reveal high epstein-barr virus DNA load and carcinoma-specific viral BARF1 mRNA. Int. J. Cancer 2006, 119, 608–614. [Google Scholar] [CrossRef]
- Rosemarie, Q.; Sugden, B. Epstein-barr virus: How its lytic phase contributes to oncogenesis. Microorganisms 2020, 8, 1824. [Google Scholar] [CrossRef] [PubMed]
- Price, A.M.; Luftig, M.A. Dynamic epstein-barr virus gene expression on the path to B-cell transformation. Adv. Virus Res. 2014, 88, 279–313. [Google Scholar]
- Hu, L.; Lin, Z.; Wu, Y.; Dong, J.; Zhao, B.; Cheng, Y.; Huang, P.; Xu, L.; Xia, T.; Xiong, D.; et al. Comprehensive profiling of EBV gene expression in nasopharyngeal carcinoma through paired-end transcriptome sequencing. Front. Med. 2016, 10, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Matho, M.H.; Schlossman, A.; Meng, X.; Benhnia, M.R.; Kaever, T.; Buller, M.; Doronin, K.; Parker, S.; Peters, B.; Crotty, S.; et al. Structural and functional characterization of anti-A33 antibodies reveal a potent cross-species orthopoxviruses neutralizer. PLoS Pathog. 2015, 11, e1005148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strong, M.J.; Xu, G.; Coco, J.; Baribault, C.; Vinay, D.S.; Lacey, M.R.; Strong, A.L.; Lehman, T.A.; Seddon, M.B.; Lin, Z.; et al. Differences in gastric carcinoma microenvironment stratify according to EBV infection intensity: Implications for possible immune adjuvant therapy. PLoS Pathog. 2013, 9, e1003341. [Google Scholar] [CrossRef]
- Feederle, R.; Kost, M.; Baumann, M.; Janz, A.; Drouet, E.; Hammerschmidt, W.; Delecluse, H.J. The epstein-barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 2000, 19, 3080–3089. [Google Scholar] [CrossRef] [Green Version]
- Xue, S.A.; Labrecque, L.G.; Lu, Q.L.; Ong, S.K.; Lampert, I.A.; Kazembe, P.; Molyneux, E.; Broadhead, R.L.; Borgstein, E.; Griffin, B.E. Promiscuous expression of epstein-barr virus genes in burkitt’s lymphoma from the central African country Malawi. Int. J. Cancer 2002, 99, 635–643. [Google Scholar] [CrossRef]
- Cochet, C.; Martel-Renoir, D.; Grunewald, V.; Bosq, J.; Cochet, G.; Schwaab, G.; Bernaudin, J.F.; Joab, I. Expression of the epstein-barr virus immediate early gene, BZLF1, in nasopharyngeal carcinoma tumor cells. Virology 1993, 197, 358–365. [Google Scholar] [CrossRef]
- Borozan, I.; Zapatka, M.; Frappier, L.; Ferretti, V. Analysis of epstein-barr virus genomes and expression profiles in gastric adenocarcinoma. J. Virol. 2018, 92, e1003341. [Google Scholar] [CrossRef] [Green Version]
- Germini, D.; Sall, F.B.; Shmakova, A.; Wiels, J.; Dokudovskaya, S.; Drouet, E.; Vassetzky, Y. Oncogenic properties of the EBV ZEBRA protein. Cancers 2020, 12, 1479. [Google Scholar] [CrossRef]
- Marshall, W.L.; Yim, C.; Gustafson, E.; Graf, T.; Sage, D.R.; Hanify, K.; Williams, L.; Fingeroth, J.; Finberg, R.W. Epstein-barr virus encodes a novel homolog of the bcl-2 oncogene that inhibits apoptosis and associates with bax and bak. J. Virol. 1999, 73, 5181–5185. [Google Scholar] [CrossRef] [Green Version]
- Kelly, G.L.; Long, H.M.; Stylianou, J.; Thomas, W.A.; Leese, A.; Bell, A.I.; Bornkamm, G.W.; Mautner, J.; Rickinson, A.B.; Rowe, M. An epstein-barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: The Wp/BHRF1 link. PLoS Pathog. 2009, 5, e1000341. [Google Scholar] [CrossRef]
- Altmann, M.; Hammerschmidt, W. Epstein-barr virus provides a new paradigm: A requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005, 3, e404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beisser, P.S.; Verzijl, D.; Gruijthuijsen, Y.K.; Beuken, E.; Smit, M.J.; Leurs, R.; Bruggeman, C.A.; Vink, C. The epstein-barr virus BILF1 gene encodes a G protein-coupled receptor that inhibits phosphorylation of RNA-dependent protein kinase. J. Virol. 2005, 79, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Deng, W.; Hau, P.M.; Liu, J.; Lau, V.M.; Cheung, A.L.; Huen, M.S.; Tsao, S.W. Epstein-barr virus BZLF1 protein impairs accumulation of host DNA damage proteins at damage sites in response to DNA damage. Lab Investig. 2015, 95, 937–950. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Zhu, J.; Xie, Z.; Liao, G.; Liu, J.; Chen, M.R.; Hu, S.; Woodard, C.; Lin, J.; Taverna, S.D.; et al. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe. 2011, 10, 390–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.C.; Liu, M.T.; Chang, Y.T.; Fang, C.Y.; Chou, S.P.; Liao, H.W.; Kuo, K.L.; Hsu, S.L.; Chen, Y.R.; Wang, P.W.; et al. Epstein-barr virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic Acids Res. 2010, 38, 1932–1949. [Google Scholar] [CrossRef]
- Shumilov, A.; Tsai, M.H.; Schlosser, Y.T.; Kratz, A.S.; Bernhardt, K.; Fink, S.; Mizani, T.; Lin, X.; Jauch, A.; Mautner, J.; et al. Epstein-barr virus particles induce centrosome amplification and chromosomal instability. Nat. Commun. 2017, 8, 14257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, O.F.; Pagano, J.S.; Whitehurst, C.B. The translesion polymerase pol eta is required for efficient epstein-barr virus infectivity and is regulated by the viral deubiquitinating enzyme BPLF1. J. Virol. 2017, 91, e00600-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, T.H.; Sitz, J.; Shen, Q.; Leblanc-Lacroix, A.; Campos, E.I.; Borozan, I.; Marcon, E.; Greenblatt, J.; Fradet-Turcotte, A.; Jin, D.Y.; et al. A screen for epstein-barr virus proteins that inhibit the DNA damage response reveals a novel histone binding protein. J. Virol. 2018, 92, e00262-18. [Google Scholar] [CrossRef] [Green Version]
- van Gent, M.; Braem, S.G.; de Jong, A.; Delagic, N.; Peeters, J.G.; Boer, I.G.; Moynagh, P.N.; Kremmer, E.; Wiertz, E.J.; Ovaa, H.; et al. Epstein-barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with toll-like receptor signaling. PLoS Pathog. 2014, 10, e1003960. [Google Scholar] [CrossRef] [PubMed]
- Hislop, A.D.; Ressing, M.E.; van Leeuwen, D.; Pudney, V.A.; Horst, D.; Koppers-Lalic, D.; Croft, N.P.; Neefjes, J.J.; Rickinson, A.B.; Wiertz, E.J. A CD8+ T cell immune evasion protein specific to epstein-barr virus and its close relatives in old world primates. J. Exp. Med. 2007, 204, 1863–1873. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Currin, A.; Griffin, B.D.; Shannon-Lowe, C.; Thomas, W.A.; Ressing, M.E.; Wiertz, E.J.; Rowe, M. The epstein-barr virus G-protein-coupled receptor contributes to immune evasion by targeting MHC class I molecules for degradation. PLoS Pathog. 2009, 5, e1000255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, L.L.; Zuo, J.; Abbott, R.J.; Shannon-Lowe, C.; Tierney, R.J.; Hislop, A.D.; Rowe, M. Cooperation between epstein-barr virus immune evasion proteins spreads protection from CD8+ T cell recognition across all three phases of the lytic cycle. PLoS Pathog. 2014, 10, e1004322. [Google Scholar] [CrossRef]
- Rowe, M.; Glaunsinger, B.; van Leeuwen, D.; Zuo, J.; Sweetman, D.; Ganem, D.; Middeldorp, J.; Wiertz, E.J.; Ressing, M.E. Host shutoff during productive epstein-barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. USA 2007, 104, 3366–3371. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Fossum, E.; Joo, C.H.; Inn, K.S.; Shin, Y.C.; Johannsen, E.; Hutt-Fletcher, L.M.; Hass, J.; Jung, J.U. Epstein-barr virus LF2: An antagonist to type I interferon. J. Virol. 2009, 83, 1140–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorley-Lawson, D.A.; Allday, M.J. The curious case of the tumour virus: 50 years of burkitt’s lymphoma. Nat. Rev. Microbiol. 2008, 6, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.D.; Veenstra, H.; Khasnis, S.; Gunnell, A.; Webb, H.M.; Shannon-Lowe, C.; Andrews, S.; Osborne, C.S.; West, M.J. MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. Elife 2016, 5, e18270. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the world health organization classification of haematolymphoid tumours: Lymphoid neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; IARC: Lyon, France, 2017. [Google Scholar]
- Wu, T.C.; Mann, R.B.; Charache, P.; Hayward, S.D.; Staal, S.; Lambe, B.C.; Ambinder, R.F. Detection of EBV gene expression in reed-sternberg cells of hodgkin’s disease. Int. J. Cancer 1990, 46, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Coates, P.J.; Slavin, G.; D’Ardenne, A.J. Persistence of epstein-barr virus in reed-sternberg cells throughout the course of hodgkin’s disease. J. Pathol. 1991, 164, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.G.; Young, L.S. An etiological role for the epstein-barr virus in the pathogenesis of classical hodgkin lymphoma. Blood 2019, 134, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Kuppers, R.; Rajewsky, K.; Zhao, M.; Simons, G.; Laumann, R.; Fischer, R.; Hansmann, M.L. Hodgkin disease: Hodgkin and reed-sternberg cells picked from histological sections show clonal immunoglobulin gene rearrangements and appear to be derived from B cells at various stages of development. Proc. Natl. Acad. Sci. USA 1994, 91, 10962–10966. [Google Scholar] [CrossRef] [Green Version]
- Ok, C.Y.; Papathomas, T.G.; Medeiros, L.J.; Young, K.H. EBV-positive diffuse large B-cell lymphoma of the elderly. Blood 2013, 122, 328–340. [Google Scholar] [CrossRef] [Green Version]
- Malpica, L.; Marques-Piubelli, M.L.; Beltran, B.E.; Chavez, J.C.; Miranda, R.N.; Castillo, J.J. EBV-positive diffuse large B-cell lymphoma, not otherwise specified: 2022 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2022, 97, 951–965. [Google Scholar] [CrossRef]
- Kato, H.; Karube, K.; Yamamoto, K.; Takizawa, J.; Tsuzuki, S.; Yatabe, Y.; Kanda, T.; Katayama, M.; Ozawa, Y.; Ishitsuka, K.; et al. Gene expression profiling of epstein-barr virus-positive diffuse large B-cell lymphoma of the elderly reveals alterations of characteristic oncogenetic pathways. Cancer Sci. 2014, 105, 537–544. [Google Scholar] [CrossRef]
- Yoon, H.; Park, S.; Ju, H.; Ha, S.Y.; Sohn, I.; Jo, J.; Do, I.G.; Min, S.; Kim, S.J.; Kim, W.S.; et al. Integrated copy number and gene expression profiling analysis of epstein-barr virus-positive diffuse large B-cell lymphoma. Genes Chromosomes Cancer 2015, 54, 383–396. [Google Scholar] [CrossRef]
- White, R.E.; Ramer, P.C.; Naresh, K.N.; Meixlsperger, S.; Pinaud, L.; Rooney, C.; Savoldo, B.; Coutinho, R.; Bodor, C.; Gribben, J.; et al. EBNA3B-deficient EBV promotes B cell lymphomagenesis in humanized mice and is found in human tumors. J. Clin. Invest. 2012, 122, 1487–1502. [Google Scholar] [CrossRef]
- Montes-Mojarro, I.A.; Fend, F.; Quintanilla-Martinez, L. EBV and the pathogenesis of NK/T cell lymphoma. Cancers 2021, 13, 1414. [Google Scholar] [CrossRef]
- Kanavaros, P.; Briere, J.; Emile, J.F.; Gaulard, P. Epstein-barr virus in T and natural killer (NK) cell non-hodgkin’s lymphomas. Leukemia 1996, 10 (Suppl. 2), s84–s87. [Google Scholar]
- Wang, H.; Fu, B.B.; Gale, R.P.; Liang, Y. NK-/T-cell lymphomas. Leukemia 2021, 35, 2460–2468. [Google Scholar] [CrossRef]
- Kingma, D.W.; Weiss, W.B.; Jaffe, E.S.; Kumar, S.; Frekko, K.; Raffeld, M. Epstein-barr virus latent membrane protein-1 oncogene deletions: Correlations with malignancy in epstein-barr virus—Associated lymphoproliferative disorders and malignant lymphomas. Blood 1996, 88, 242–251. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, R.; Donahue, H.; Garcia, D.; Tan, J.; Shimizu, N.; Rice, A.P.; Ling, P.D. Epstein-barr virus BART9 miRNA modulates LMP1 levels and affects growth rate of nasal NK T cell lymphomas. PLoS ONE 2011, 6, e27271. [Google Scholar] [CrossRef]
- Huang, Y.; de Leval, L.; Gaulard, P. Molecular underpinning of extranodal NK/T-cell lymphoma. Best Pract. Res. Clin. Haematol. 2013, 26, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Bahri, R.; Boyer, F.; Halabi, M.A.; Chaunavel, A.; Feuillard, J.; Jaccard, A.; Ranger-Rogez, S. Epstein-barr virus (EBV) is mostly latent and clonal in angioimmunoblastic T cell lymphoma (AITL). Cancers 2022, 14, 2899. [Google Scholar] [CrossRef] [PubMed]
- Dunleavy, K.; Wilson, W.H. Angioimmunoblastic T-cell lymphoma: Immune modulation as a therapeutic strategy. Leuk Lymphoma 2007, 48, 449–451. [Google Scholar] [CrossRef]
- Bayda, N.; Tilloy, V.; Chaunavel, A.; Bahri, R.; Halabi, M.A.; Feuillard, J.; Jaccard, A.; Ranger-Rogez, S. Comprehensive epstein-barr virus transcriptome by RNA-sequencing in angioimmunoblastic T cell lymphoma (AITL) and other lymphomas. Cancers 2021, 13, 610. [Google Scholar] [CrossRef]
- Longnecker, R.M.; Kieff, E.; Cohen, J.I. Epstein-barr virus. In Fileds Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Wlliams, and Wilkins: Philadelphia, PA, USA, 2013; pp. 1898–1959. [Google Scholar]
- Rickinson, A.B.; Lo, K.W. Nasopharyngeal carcinoma: A history. In Nasopharyngeal Carcinoma; Lee, A.W.M., Lung, M.L., Ng, W.T., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 1–16. [Google Scholar]
- Pathmanathan, R.; Prasad, U.; Sadler, R.; Flynn, K.; Raab-Traub, N. Clonal proliferations of cells infected with epstein-barr virus in preinvasive lesions related to nasopharyngeal carcinoma. N. Engl. J. Med. 1995, 333, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Lu, J.; Pei, Y.; Robertson, E.S. Transcriptome reprogramming of epstein-barr virus infected epithelial and B cells reveals distinct host-virus interaction profiles. Cell Death Dis. 2022, 13, 894. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Yip, Y.L.; Jia, L.; Deng, W.; Zheng, H.; Dai, W.; Ko, J.M.Y.; Lo, K.W.; Chung, G.T.Y.; Yip, K.Y.; et al. Establishment and characterization of new tumor xenografts and cancer cell lines from EBV-positive nasopharyngeal carcinoma. Nat. Commun. 2018, 9, 4663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, L.S.; Dawson, C.W.; Clark, D.; Rupani, H.; Busson, P.; Tursz, T.; Johnson, A.; Rickinson, A.B. Epstein-barr virus gene expression in nasopharyngeal carcinoma. J. Gen. Virol. 1988, 69 Pt 5, 1051–1065. [Google Scholar] [CrossRef] [PubMed]
- Brooks, L.; Yao, Q.Y.; Rickinson, A.B.; Young, L.S. Epstein-barr virus latent gene transcription in nasopharyngeal carcinoma cells: Coexpression of EBNA1, LMP1, and LMP2 transcripts. J. Virol. 1992, 66, 2689–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, S.; Dawson, C.W.; Takada, K.; Curnow, J.; Moody, C.A.; Sixbey, J.W.; Young, L.S. Epstein-barr virus-encoded LMP2A regulates viral and cellular gene expression by modulation of the NF-kappaB transcription factor pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 15730–15735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.C.; Meng, X.; Hazawa, M.; Nagata, Y.; Varela, A.M.; Xu, L.; Sato, Y.; Liu, L.Z.; Ding, L.W.; Sharma, A.; et al. The genomic landscape of nasopharyngeal carcinoma. Nat. Genet. 2014, 46, 866–871. [Google Scholar] [CrossRef]
- Sun, K.; Jia, K.; Lv, H.; Wang, S.Q.; Wu, Y.; Lei, H.; Chen, X. EBV-positive gastric cancer: Current knowledge and future perspectives. Front. Oncol. 2020, 10, 583463. [Google Scholar] [CrossRef]
- Murphy, G.; Pfeiffer, R.; Camargo, M.C.; Rabkin, C.S. Meta-analysis shows that prevalence of epstein-barr virus-positive gastric cancer differs based on sex and anatomic location. Gastroenterology 2009, 137, 824–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Interaction between Epstein–Barr virus and the human host, and virus latent infection in B lymphocytes. Upper panel: Primary infection. Epstein–Barr virus transmitted via saliva establishes a primary lytic replication in the oropharyngeal mucosal epithelium, then the virus spreads throughout the lymphoid tissues in Waldeyer’s ring. There are two putative theories to explain how the virus enters memory B cells. One speculation proposes that EBV infects tonsillar naive B cells, leading to a latency 3 program; in this scenario, a full spectrum of latent proteins is expressed. The majority of these proliferating cells are eliminated by natural killer cells and the emerging latent-antigen-specific primary-T-cell response. However, some infected cells escape from immune surveillance by downregulating antigen expression and undergo germinal center (GC) reaction, where a more limited set of viral genes are expressed (the default program or latency 2). A stable reservoir of resting viral-genome positive memory B cells is established when these EBV-infected GC B cells migrate to peripheral blood, where viral antigen expression is silenced (latency 0). When EBNA1 is expressed intermittently during the division of these memory B cells, the viral genome is distributed to the daughter memory B cells (latency 1). Another view envisages that EBV directly infects pre-existing memory B cells in the memory B-cell reservoir. Memory B cells can terminally differentiate into plasma cells (solid arrow), possibly moving to the oropharyngeal mucosal basolateral side and, in the process, triggering the viral lytic replication. Virions produced at these sites are efficiently shed into the saliva and transmitted both to other hosts and to previously uninfected naive B cells within the same host. EBV-infected GC B cells might also differentiate directly into plasma cells (dashed arrow). It is also reported that EBV can also infect T cells, NK cells to form T-cell leukemia/lymphoma, NK-cell leukemia, and NK/T-cell lymphoma [37,38]. Lower panel: Persistent infection. The reservoir of EBV-infected memory B cells are normally in a dormant status. Under certain circumstances, these cells might be recruited into GC reactions, after which they might either re-enter the reservoir as silent memory B cells or return to the lymphoid tissue and undergo plasma cell differentiation, shedding EBV virions. This may initiate the growth-transforming latency III program on infection of naive and/or memory B cells. These new infections are more likely to be efficiently removed by the well-established memory-T-cell response. This figure was created with BioRender.com (accessed on 5 December 2022).
Figure 1.
Interaction between Epstein–Barr virus and the human host, and virus latent infection in B lymphocytes. Upper panel: Primary infection. Epstein–Barr virus transmitted via saliva establishes a primary lytic replication in the oropharyngeal mucosal epithelium, then the virus spreads throughout the lymphoid tissues in Waldeyer’s ring. There are two putative theories to explain how the virus enters memory B cells. One speculation proposes that EBV infects tonsillar naive B cells, leading to a latency 3 program; in this scenario, a full spectrum of latent proteins is expressed. The majority of these proliferating cells are eliminated by natural killer cells and the emerging latent-antigen-specific primary-T-cell response. However, some infected cells escape from immune surveillance by downregulating antigen expression and undergo germinal center (GC) reaction, where a more limited set of viral genes are expressed (the default program or latency 2). A stable reservoir of resting viral-genome positive memory B cells is established when these EBV-infected GC B cells migrate to peripheral blood, where viral antigen expression is silenced (latency 0). When EBNA1 is expressed intermittently during the division of these memory B cells, the viral genome is distributed to the daughter memory B cells (latency 1). Another view envisages that EBV directly infects pre-existing memory B cells in the memory B-cell reservoir. Memory B cells can terminally differentiate into plasma cells (solid arrow), possibly moving to the oropharyngeal mucosal basolateral side and, in the process, triggering the viral lytic replication. Virions produced at these sites are efficiently shed into the saliva and transmitted both to other hosts and to previously uninfected naive B cells within the same host. EBV-infected GC B cells might also differentiate directly into plasma cells (dashed arrow). It is also reported that EBV can also infect T cells, NK cells to form T-cell leukemia/lymphoma, NK-cell leukemia, and NK/T-cell lymphoma [37,38]. Lower panel: Persistent infection. The reservoir of EBV-infected memory B cells are normally in a dormant status. Under certain circumstances, these cells might be recruited into GC reactions, after which they might either re-enter the reservoir as silent memory B cells or return to the lymphoid tissue and undergo plasma cell differentiation, shedding EBV virions. This may initiate the growth-transforming latency III program on infection of naive and/or memory B cells. These new infections are more likely to be efficiently removed by the well-established memory-T-cell response. This figure was created with BioRender.com (accessed on 5 December 2022).
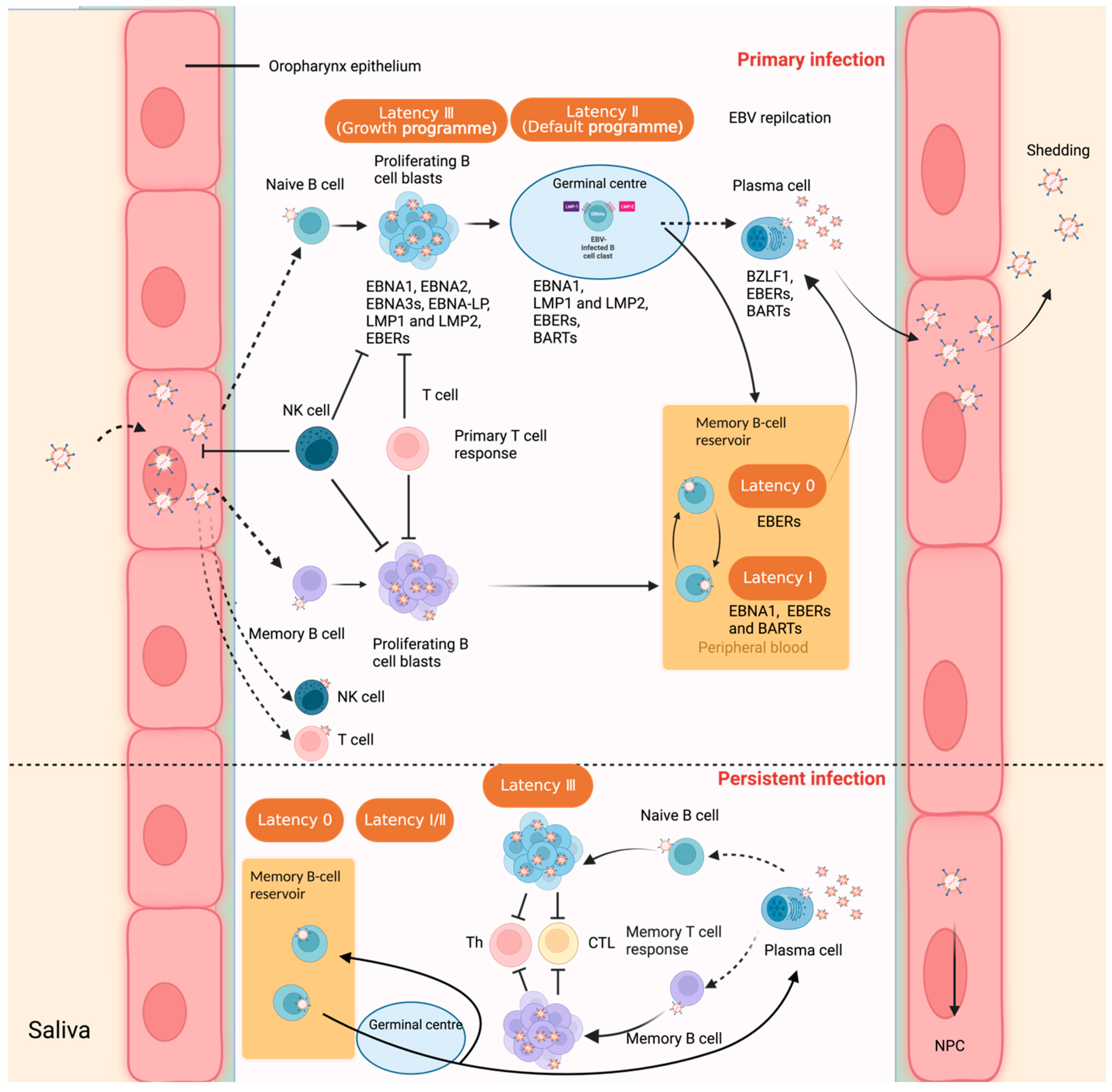
Figure 2.
Pathogenetic model of AITL. In AITL, a complex network of interactions take place between the tumor cells and profound surrounding inflammatory tumor microenvironment. FDC, follicular dendritic cell; HEV, high endothelial venule; TFH, follicular helper T cell; Treg, regulatory T cell.
Figure 2.
Pathogenetic model of AITL. In AITL, a complex network of interactions take place between the tumor cells and profound surrounding inflammatory tumor microenvironment. FDC, follicular dendritic cell; HEV, high endothelial venule; TFH, follicular helper T cell; Treg, regulatory T cell.


{kind=link}
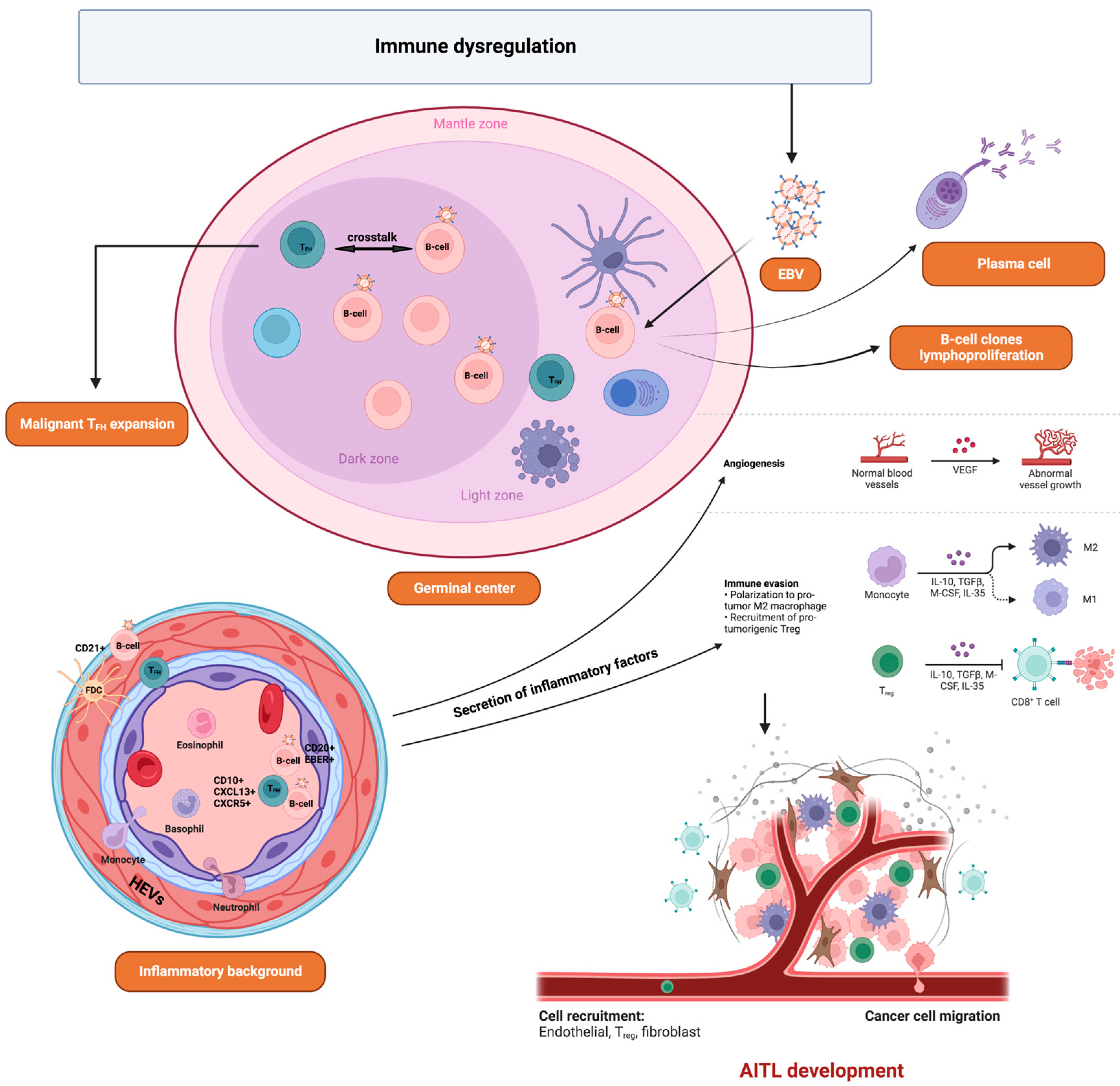{kind=link}
Table 1.
EBV viral gene expression patterns during different types of latent infection.
| Latency Pattern | Latency III | Latency II | Latency I | Latency 0 | |
|---|---|---|---|---|---|
| Genes | |||||
| EBNA1 | + | + | + | ND | |
| EBNA2 | + | ND | ND | ND | |
| EBNA3s | + | ND | ND | ND | |
| EBNA-LP | + | ND | ND | ND | |
| LMP1 | + | + | ND | ND | |
| LMP2 | + | + | ND | ND | |
| EBERs | + | + | + | + | |
| BHRF1 miRNAs | + | ND | ND | ND | |
| BARTs miRNAs | + | + | + | + | |
ND: not detected. +: positive.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yu, H.; Robertson, E.S. Epstein–Barr Virus History and Pathogenesis. Viruses 2023, 15, 714. https://doi.org/10.3390/v15030714
AMA Style
Yu H, Robertson ES. Epstein–Barr Virus History and Pathogenesis. Viruses. 2023; 15(3):714. https://doi.org/10.3390/v15030714
Chicago/Turabian StyleYu, Hui, and Erle S. Robertson. 2023. "Epstein–Barr Virus History and Pathogenesis" Viruses 15, no. 3: 714. https://doi.org/10.3390/v15030714
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.