

Epstein–Barr Virus B Cell Growth Transformation: The Nuclear Events

{kind=link}
Abstract
:1. Introduction
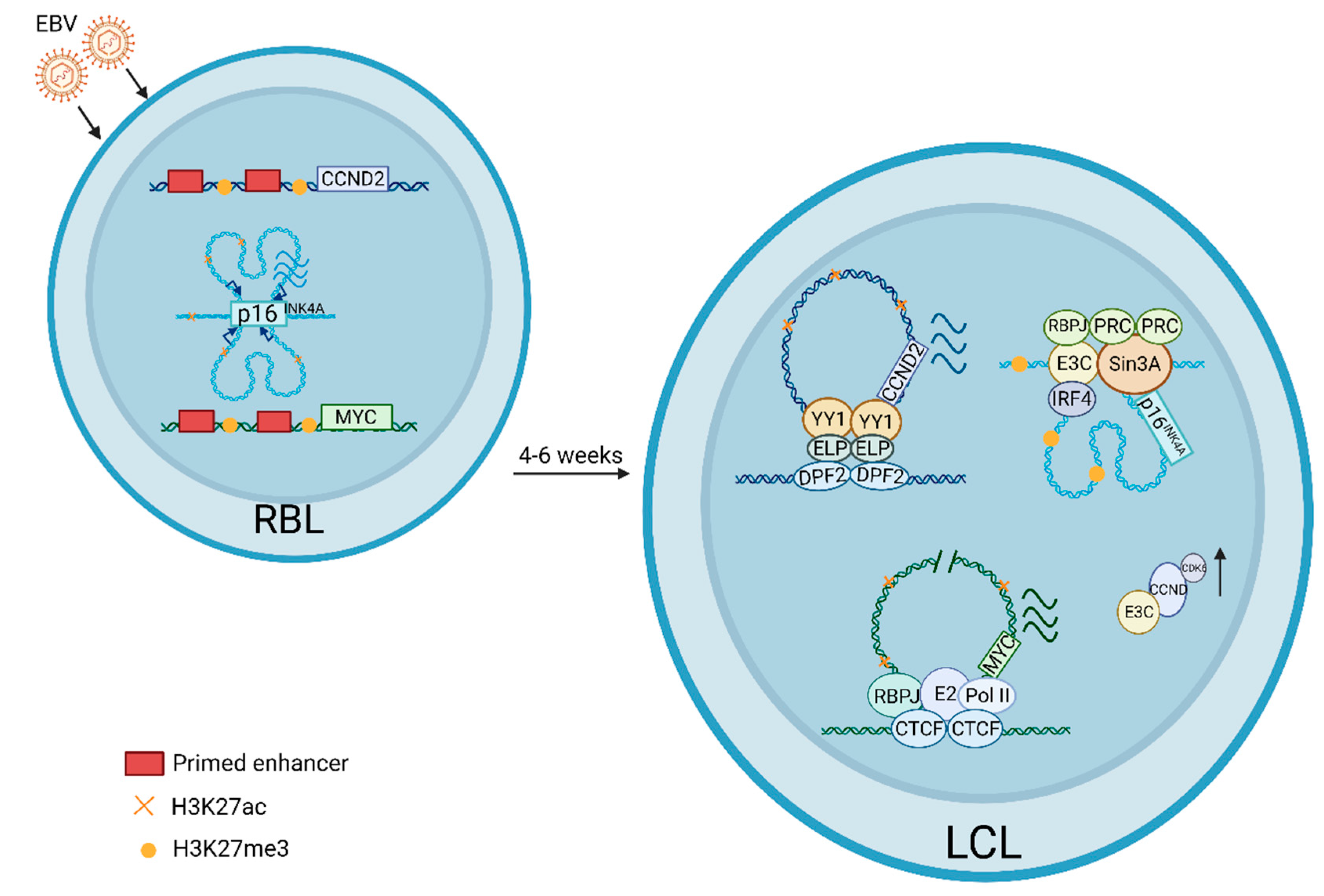
2. EBV Transformation from RBL to LCL Timeline
3. EBNA2 Is the Major EBV Transcription Activator
4. EBNALP in Transcription Regulation
5. EBNA3A and EBNA3C
6. EBNA1
7. LMP1
8. Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I.; Fauci, A.S.; Varmus, H.; Nabel, G.J. Epstein-Barr virus: An important vaccine target for cancer prevention. Sci. Transl. Med. 2011, 3, 107fs7. [Google Scholar] [CrossRef] [Green Version]
- Longnecker, R.; Kieff, E.; Cohen, J.I. Epstein-Barr Virus, 8th ed.; Lippincott Williams & Wilkins, a Wolters Kluwer Business: Philadelphia, PA, USA, 2013; Volume 2, pp. 1898–1959. [Google Scholar]
- Pich, D.; Mrozek-Gorska, P.; Bouvet, M.; Sugimoto, A.; Akidil, E.; Grundhoff, A.; Hamperl, S.; Ling, P.D.; Hammerschmidt, W. First Days in the Life of Naive Human B Lymphocytes Infected with Epstein-Barr Virus. mBio 2019, 10, e01723-19. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.W.; Wang, Z.; Ersing, I.; Nobre, L.; Guo, R.; Jiang, S.; Trudeau, S.; Zhao, B.; Weekes, M.P.; Gewurz, B.E. Epstein-Barr virus subverts mevalonate and fatty acid pathways to promote infected B-cell proliferation and survival. PLoS Pathog. 2019, 15, e1008030. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Li, D.; Zhang, L.; Jiang, S.; Liang, J.; Narita, Y.; Hou, I.; Zhong, Q.; Zheng, Z.; Xiao, H.; et al. RNA Sequencing Analyses of Gene Expression during Epstein-Barr Virus Infection of Primary B Lymphocytes. J. Virol. 2019, 93, e00226-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrozek-Gorska, P.; Buschle, A.; Pich, D.; Schwarzmayr, T.; Fechtner, R.; Scialdone, A.; Hammerschmidt, W. Epstein-Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc. Natl. Acad. Sci. USA 2019, 116, 16046–16055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, A.M.; Tourigny, J.P.; Forte, E.; Salinas, R.E.; Dave, S.S.; Luftig, M.A. Analysis of Epstein-Barr virus-regulated host gene expression changes through primary B-cell outgrowth reveals delayed kinetics of latent membrane protein 1-mediated NF-kappaB activation. J. Virol. 2012, 86, 11096–11106. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.W.; Shen, H.; Nobre, L.; Ersing, I.; Paulo, J.A.; Trudeau, S.; Wang, Z.; Smith, N.A.; Ma, Y.; Reinstadler, B.; et al. Epstein-Barr-Virus-Induced One-Carbon Metabolism Drives B Cell Transformation. Cell Metab. 2019, 30, 539–555.e11. [Google Scholar] [CrossRef] [Green Version]
- Alfieri, C.; Birkenbach, M.; Kieff, E. Early events in Epstein-Barr virus infection of human B lymphocytes. Virology 1991, 181, 595–608. [Google Scholar] [CrossRef]
- Sample, J.; Hummel, M.; Braun, D.; Birkenbach, M.; Kieff, E. Nucleotide sequences of mRNAs encoding Epstein-Barr virus nuclear proteins: A probable transcriptional initiation site. Proc. Natl. Acad. Sci. USA 1986, 83, 5096–5100. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Petti, L.; Braun, D.; Seung, S.; Kieff, E. A bicistronic Epstein-Barr virus mRNA encodes two nuclear proteins in latently infected, growth-transformed lymphocytes. J. Virol. 1987, 61, 945–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammerschmidt, W.; Sugden, B. Genetic analysis of immortalizing functions of Epstein-Barr virus in human B lymphocytes. Nature 1989, 340, 393–397. [Google Scholar] [CrossRef]
- Cohen, J.I.; Wang, F.; Mannick, J.; Kieff, E. Epstein-Barr virus nuclear protein 2 is a key determinant of lymphocyte transformation. Proc. Natl. Acad. Sci. USA 1989, 86, 9558–9562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Gregory, C.D.; Rowe, M.; Rickinson, A.B.; Wang, D.; Birkenbach, M.; Kikutani, H.; Kishimoto, T.; Kieff, E. Epstein-Barr virus nuclear antigen 2 specifically induces expression of the B-cell activation antigen CD23. Proc. Natl. Acad. Sci. USA 1987, 84, 3452–3456. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Tsang, S.F.; Kurilla, M.G.; Cohen, J.I.; Kieff, E. Epstein-Barr virus nuclear antigen 2 transactivates latent membrane protein LMP1. J. Virol. 1990, 64, 3407–3416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooney, C.M.; Brimmell, M.; Buschle, M.; Allan, G.; Farrell, P.J.; Kolman, J.L. Host cell and EBNA-2 regulation of Epstein-Barr virus latent-cycle promoter activity in B lymphocytes. J. Virol. 1992, 66, 496–504. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, C.; Laux, G.; Eick, D.; Jochner, N.; Bornkamm, G.W.; Kempkes, B. The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2. J. Virol. 1999, 73, 4481–4484. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Zou, J.; Wang, H.; Johannsen, E.; Peng, C.W.; Quackenbush, J.; Mar, J.C.; Morton, C.C.; Freedman, M.L.; Blacklow, S.C.; et al. Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc. Natl. Acad. Sci. USA 2011, 108, 14902–14907. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Maruo, S.; Cooper, A.; Chase, M.R.; Johannsen, E.; Kieff, E.; Cahir-McFarland, E. RNAs induced by Epstein-Barr virus nuclear antigen 2 in lymphoblastoid cell lines. Proc. Natl. Acad. Sci. USA 2006, 103, 1900–1905. [Google Scholar] [CrossRef] [Green Version]
- Schlee, M.; Krug, T.; Gires, O.; Zeidler, R.; Hammerschmidt, W.; Mailhammer, R.; Laux, G.; Sauer, G.; Lovric, J.; Bornkamm, G.W. Identification of Epstein-Barr virus (EBV) nuclear antigen 2 (EBNA2) target proteins by proteome analysis: Activation of EBNA2 in conditionally immortalized B cells reflects early events after infection of primary B cells by EBV. J. Virol. 2004, 78, 3941–3952. [Google Scholar] [CrossRef] [Green Version]
- Kempkes, B.; Spitkovsky, D.; Jansen-Durr, P.; Ellwart, J.W.; Kremmer, E.; Delecluse, H.J.; Rottenberger, C.; Bornkamm, G.W.; Hammerschmidt, W. B-cell proliferation and induction of early G1-regulating proteins by Epstein-Barr virus mutants conditional for EBNA2. EMBO J. 1995, 14, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Spender, L.C.; Lucchesi, W.; Bodelon, G.; Bilancio, A.; Karstegl, C.E.; Asano, T.; Dittrich-Breiholz, O.; Kracht, M.; Vanhaesebroeck, B.; Farrell, P.J. Cell target genes of Epstein-Barr virus transcription factor EBNA-2: Induction of the p55alpha regulatory subunit of PI3-kinase and its role in survival of EREB2.5 cells. J. Gen. Virol. 2006, 87, 2859–2867. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.; Staffler, G.; Hartmann, A.; Hock, J.; Henning, K.; Grabusic, K.; Mailhammer, R.; Hoffmann, R.; Wilmanns, M.; Lang, R.; et al. Cellular target genes of Epstein-Barr virus nuclear antigen 2. J. Virol. 2006, 80, 9761–9771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dambaugh, T.; Hennessy, K.; Chamnankit, L.; Kieff, E. U2 region of Epstein-Barr virus DNA may encode Epstein-Barr nuclear antigen 2. Proc. Natl. Acad. Sci. USA 1984, 81, 7632–7636. [Google Scholar] [CrossRef] [Green Version]
- Harada, S.; Yalamanchili, R.; Kieff, E. Epstein-Barr virus nuclear protein 2 has at least two N-terminal domains that mediate self-association. J. Virol. 2001, 75, 2482–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.W.; Zhao, B.; Kieff, E. Four EBNA2 domains are important for EBNALP coactivation. J. Virol. 2004, 78, 11439–11442. [Google Scholar] [CrossRef] [Green Version]
- Friberg, A.; Thumann, S.; Hennig, J.; Zou, P.; Nossner, E.; Ling, P.D.; Sattler, M.; Kempkes, B. The EBNA-2 N-Terminal Transactivation Domain Folds into a Dimeric Structure Required for Target Gene Activation. PLoS Pathog. 2015, 11, e1004910. [Google Scholar] [CrossRef] [Green Version]
- Ling, P.D.; Rawlins, D.R.; Hayward, S.D. The Epstein-Barr virus immortalizing protein EBNA-2 is targeted to DNA by a cellular enhancer-binding protein. Proc. Natl. Acad. Sci. USA 1993, 90, 9237–9241. [Google Scholar] [CrossRef] [Green Version]
- Grossman, S.R.; Johannsen, E.; Tong, X.; Yalamanchili, R.; Kieff, E. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the J kappa recombination signal binding protein. Proc. Natl. Acad. Sci. USA 1994, 91, 7568–7572. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.I.; Kieff, E. An Epstein-Barr virus nuclear protein 2 domain essential for transformation is a direct transcriptional activator. J. Virol. 1991, 65, 5880–5885. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.I.; Wang, F.; Kieff, E. Epstein-Barr virus nuclear protein 2 mutations define essential domains for transformation and transactivation. J. Virol. 1991, 65, 2545–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzellos, S.; Correia, P.B.; Karstegl, C.E.; Cancian, L.; Cano-Flanagan, J.; McClellan, M.J.; West, M.J.; Farrell, P.J. A single amino acid in EBNA-2 determines superior B lymphoblastoid cell line growth maintenance by Epstein-Barr virus type 1 EBNA-2. J. Virol. 2014, 88, 8743–8753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnusamy, R.; Khatri, R.; Correia, P.B.; Wood, C.D.; Mancini, E.J.; Farrell, P.J.; West, M.J. Increased association between Epstein-Barr virus EBNA2 from type 2 strains and the transcriptional repressor BS69 restricts EBNA2 activity. PLoS Pathog. 2019, 15, e1007458. [Google Scholar] [CrossRef]
- Harter, M.R.; Liu, C.D.; Shen, C.L.; Gonzalez-Hurtado, E.; Zhang, Z.M.; Xu, M.; Martinez, E.; Peng, C.W.; Song, J. BS69/ZMYND11 C-Terminal Domains Bind and Inhibit EBNA2. PLoS Pathog. 2016, 12, e1005414. [Google Scholar] [CrossRef] [Green Version]
- Henkel, T.; Ling, P.D.; Hayward, S.D.; Peterson, M.G. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa. Science 1994, 265, 92–95. [Google Scholar] [CrossRef]
- Wang, H.; Zou, J.; Zhao, B.; Johannsen, E.; Ashworth, T.; Wong, H.; Pear, W.S.; Schug, J.; Blacklow, S.C.; Arnett, K.L.; et al. Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc. Natl. Acad. Sci. USA 2011, 108, 14908–14913. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Hayward, S.D. Nuclear localization of CBF1 is regulated by interactions with the SMRT corepressor complex. Mol. Cell. Biol. 2001, 21, 6222–6232. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, J.J.; Hayward, S.D. Masking of the CBF1/RBPJ kappa transcriptional repression domain by Epstein-Barr virus EBNA2. Science 1995, 268, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Portal, D.; Zhao, B.; Calderwood, M.A.; Sommermann, T.; Johannsen, E.; Kieff, E. EBV nuclear antigen EBNALP dismisses transcription repressors NCoR and RBPJ from enhancers and EBNA2 increases NCoR-deficient RBPJ DNA binding. Proc. Natl. Acad. Sci. USA 2011, 108, 7808–7813. [Google Scholar] [CrossRef] [Green Version]
- Johannsen, E.; Koh, E.; Mosialos, G.; Tong, X.; Kieff, E.; Grossman, S.R. Epstein-Barr virus nuclear protein 2 transactivation of the latent membrane protein 1 promoter is mediated by J kappa and PU.1. J. Virol. 1995, 69, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Yue, W.; Davenport, M.G.; Shackelford, J.; Pagano, J.S. Mitosis-specific hyperphosphorylation of Epstein-Barr virus nuclear antigen 2 suppresses its function. J. Virol. 2004, 78, 3542–3552. [Google Scholar] [CrossRef] [Green Version]
- Beer, S.; Wange, L.E.; Zhang, X.; Kuklik-Roos, C.; Enard, W.; Hammerschmidt, W.; Scialdone, A.; Kempkes, B. EBNA2-EBF1 complexes promote MYC expression and metabolic processes driving S-phase progression of Epstein-Barr virus-infected B cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2200512119. [Google Scholar] [CrossRef]
- Tong, X.; Drapkin, R.; Reinberg, D.; Kieff, E. The 62- and 80-kDa subunits of transcription factor IIH mediate the interaction with Epstein-Barr virus nuclear protein 2. Proc. Natl. Acad. Sci. USA 1995, 92, 3259–3263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, X.; Drapkin, R.; Yalamanchili, R.; Mosialos, G.; Kieff, E. The Epstein-Barr virus nuclear protein 2 acidic domain forms a complex with a novel cellular coactivator that can interact with TFIIE. Mol. Cell. Biol. 1995, 15, 4735–4744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, X.; Wang, F.; Thut, C.J.; Kieff, E. The Epstein-Barr virus nuclear protein 2 acidic domain can interact with TFIIB, TAF40, and RPA70 but not with TATA-binding protein. J. Virol. 1995, 69, 585–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Grossman, S.R.; Kieff, E. Epstein-Barr virus nuclear protein 2 interacts with p300, CBP, and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc. Natl. Acad. Sci. USA 2000, 97, 430–435. [Google Scholar]
- Wu, D.Y.; Kalpana, G.V.; Goff, S.P.; Schubach, W.H. Epstein-Barr virus nuclear protein 2 (EBNA2) binds to a component of the human SNF-SWI complex, hSNF5/Ini1. J. Virol. 1996, 70, 6020–6028. [Google Scholar] [CrossRef] [Green Version]
- Chabot, P.R.; Raiola, L.; Lussier-Price, M.; Morse, T.; Arseneault, G.; Archambault, J.; Omichinski, J.G. Structural and functional characterization of a complex between the acidic transactivation domain of EBNA2 and the Tfb1/p62 subunit of TFIIH. PLoS Pathog. 2014, 10, e1004042. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.D.; Chen, Y.L.; Min, Y.L.; Zhao, B.; Cheng, C.P.; Kang, M.S.; Chiu, S.J.; Kieff, E.; Peng, C.W. The nuclear chaperone nucleophosmin escorts an Epstein-Barr Virus nuclear antigen to establish transcriptional cascades for latent infection in human B cells. PLoS Pathog. 2012, 8, e1003084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.D.; Cheng, C.P.; Fang, J.S.; Chen, L.C.; Zhao, B.; Kieff, E.; Peng, C.W. Modulation of Epstein-Barr virus nuclear antigen 2-dependent transcription by protein arginine methyltransferase 5. Biochem. Biophys. Res. Commun. 2013, 430, 1097–1102. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.; Chen, H.S.; Kossenkov, A.V.; DeWispeleare, K.; Won, K.J.; Lieberman, P.M. EBNA2 Drives Formation of New Chromosome Binding Sites and Target Genes for B-Cell Master Regulatory Transcription Factors RBP-jkappa and EBF1. PLoS Pathog. 2016, 12, e1005339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClellan, M.J.; Wood, C.D.; Ojeniyi, O.; Cooper, T.J.; Kanhere, A.; Arvey, A.; Webb, H.M.; Palermo, R.D.; Harth-Hertle, M.L.; Kempkes, B.; et al. Modulation of enhancer looping and differential gene targeting by Epstein-Barr virus transcription factors directs cellular reprogramming. PLoS Pathog. 2013, 9, e1003636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Schmidt, S.C.; Jiang, S.; Willox, B.; Bernhardt, K.; Liang, J.; Johannsen, E.C.; Kharchenko, P.; Gewurz, B.E.; Kieff, E.; et al. Epstein-Barr virus oncoprotein super-enhancers control B cell growth. Cell Host Microbe 2015, 17, 205–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, C.D.; Veenstra, H.; Khasnis, S.; Gunnell, A.; Webb, H.M.; Shannon-Lowe, C.; Andrews, S.; Osborne, C.S.; West, M.J. MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. eLIFE 2016, 5, e18270. [Google Scholar] [CrossRef]
- Jiang, S.; Zhou, H.; Liang, J.; Gerdt, C.; Wang, C.; Ke, L.; Schmidt, S.C.S.; Narita, Y.; Ma, Y.; Wang, S.; et al. The Epstein-Barr Virus Regulome in Lymphoblastoid Cells. Cell Host Microbe 2017, 22, 561–573.e4. [Google Scholar] [CrossRef] [Green Version]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Gunnell, A.; Webb, H.M.; Wood, C.D.; McClellan, M.J.; Wichaidit, B.; Kempkes, B.; Jenner, R.G.; Osborne, C.; Farrell, P.J.; West, M.J. RUNX super-enhancer control through the Notch pathway by Epstein-Barr virus transcription factors regulates B cell growth. Nucleic Acids Res. 2016, 44, 4636–4650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boija, A.; Klein, I.A.; Sabari, B.R.; Dall’Agnese, A.; Coffey, E.L.; Zamudio, A.V.; Li, C.H.; Shrinivas, K.; Manteiga, J.C.; Hannett, N.M.; et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 2018, 175, 1842–1855.e16. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Wang, L.; Qin, Z.; Wang, J.; Zheng, X.; Wei, L.; Zhang, X.; Zhang, X.; Liu, C.; Li, Z.; et al. Phase Separation of Epstein-Barr Virus EBNA2 and Its Coactivator EBNALP Controls Gene Expression. J. Virol. 2020, 94, e01771-19. [Google Scholar] [CrossRef] [PubMed]
- McCann, E.M.; Kelly, G.L.; Rickinson, A.B.; Bell, A.I. Genetic analysis of the Epstein-Barr virus-coded leader protein EBNA-LP as a co-activator of EBNA2 function. J. Gen. Virol. 2001, 82, 3067–3079. [Google Scholar] [CrossRef]
- Harada, S.; Kieff, E. Epstein-Barr virus nuclear protein LP stimulates EBNA-2 acidic domain-mediated transcriptional activation. J. Virol. 1997, 71, 6611–6618. [Google Scholar] [CrossRef] [Green Version]
- Mannick, J.B.; Cohen, J.I.; Birkenbach, M.; Marchini, A.; Kieff, E. The Epstein-Barr virus nuclear protein encoded by the leader of the EBNA RNAs is important in B-lymphocyte transformation. J. Virol. 1991, 65, 6826–6837. [Google Scholar] [CrossRef] [Green Version]
- Szymula, A.; Palermo, R.D.; Bayoumy, A.; Groves, I.J.; Ba Abdullah, M.; Holder, B.; White, R.E. Epstein-Barr virus nuclear antigen EBNA-LP is essential for transforming naive B cells, and facilitates recruitment of transcription factors to the viral genome. PLoS Pathog. 2018, 14, e1006890. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, A.J.; Palmero, I.; Peters, G.; Farrell, P.J. EBNA-2 and EBNA-LP cooperate to cause G0 to G1 transition during immortalization of resting human B lymphocytes by Epstein-Barr virus. EMBO J. 1994, 13, 3321–3328. [Google Scholar] [CrossRef]
- Sinclair, A.J.; Palmero, I.; Holder, A.; Peters, G.; Farrell, P.J. Expression of cyclin D2 in Epstein-Barr virus-positive Burkitt’s lymphoma cell lines is related to methylation status of the gene. J. Virol. 1995, 69, 1292–1295. [Google Scholar] [CrossRef] [Green Version]
- Nitsche, F.; Bell, A.; Rickinson, A. Epstein-Barr virus leader protein enhances EBNA-2-mediated transactivation of latent membrane protein 1 expression: A role for the W1W2 repeat domain. J. Virol. 1997, 71, 6619–6628. [Google Scholar] [CrossRef] [Green Version]
- Portal, D.; Rosendorff, A.; Kieff, E. Epstein-Barr nuclear antigen leader protein coactivates transcription through interaction with histone deacetylase 4. Proc. Natl. Acad. Sci. USA 2006, 103, 19278–19283. [Google Scholar] [CrossRef] [Green Version]
- Ling, P.D.; Peng, R.S.; Nakajima, A.; Yu, J.H.; Tan, J.; Moses, S.M.; Yang, W.H.; Zhao, B.; Kieff, E.; Bloch, K.D.; et al. Mediation of Epstein-Barr virus EBNA-LP transcriptional coactivation by Sp100. EMBO J. 2005, 24, 3565–3575. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhou, H.; Xue, Y.; Liang, J.; Narita, Y.; Gerdt, C.; Zheng, A.Y.; Jiang, R.; Trudeau, S.; Peng, C.W.; et al. Epstein-Barr Virus Nuclear Antigen Leader Protein Coactivates EP300. J. Virol. 2018, 92, e02155-17. [Google Scholar] [CrossRef] [Green Version]
- Peng, R.; Tan, J.; Ling, P.D. Conserved regions in the Epstein-Barr virus leader protein define distinct domains required for nuclear localization and transcriptional cooperation with EBNA2. J. Virol. 2000, 74, 9953–9963. [Google Scholar] [CrossRef] [Green Version]
- Han, I.; Xue, Y.; Harada, S.; Orstavik, S.; Skalhegg, B.; Kieff, E. Protein kinase A associates with HA95 and affects transcriptional coactivation by Epstein-Barr virus nuclear proteins. Mol. Cell. Biol. 2002, 22, 2136–2146. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, Y.; Nakajima, K.; Igarashi, M.; Morita, T.; Tanaka, M.; Suzuki, M.; Yokoyama, A.; Matsuda, G.; Kato, K.; Kanamori, M.; et al. Interaction of Epstein-Barr virus nuclear antigen leader protein (EBNA-LP) with HS1-associated protein X-1: Implication of cytoplasmic function of EBNA-LP. J. Virol. 2000, 74, 10104–10111. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.W.; Zhao, B.; Chen, H.C.; Chou, M.L.; Lai, C.Y.; Lin, S.Z.; Hsu, H.Y.; Kieff, E. Hsp72 up-regulates Epstein-Barr virus EBNALP coactivation with EBNA2. Blood 2007, 109, 5447–5454. [Google Scholar] [CrossRef]
- Portal, D.; Zhou, H.; Zhao, B.; Kharchenko, P.V.; Lowry, E.; Wong, L.; Quackenbush, J.; Holloway, D.; Jiang, S.; Lu, Y.; et al. Epstein-Barr virus nuclear antigen leader protein localizes to promoters and enhancers with cell transcription factors and EBNA2. Proc. Natl. Acad. Sci. USA 2013, 110, 18537–18542. [Google Scholar] [CrossRef] [Green Version]
- Huber, F.M.; Greenblatt, S.M.; Davenport, A.M.; Martinez, C.; Xu, Y.; Vu, L.P.; Nimer, S.D.; Hoelz, A. Histone-binding of DPF2 mediates its repressive role in myeloid differentiation. Proc. Natl. Acad. Sci. USA 2017, 114, 6016–6021. [Google Scholar] [CrossRef] [Green Version]
- Weintraub, A.S.; Li, C.H.; Zamudio, A.V.; Sigova, A.A.; Hannett, N.M.; Day, D.S.; Abraham, B.J.; Cohen, M.A.; Nabet, B.; Buckley, D.L.; et al. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 2017, 171, 1573–1588.e28. [Google Scholar] [CrossRef] [Green Version]
- Dalbies-Tran, R.; Stigger-Rosser, E.; Dotson, T.; Sample, C.E. Amino acids of Epstein-Barr virus nuclear antigen 3A essential for repression of Jkappa-mediated transcription and their evolutionary conservation. J. Virol. 2001, 75, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Marshall, D.R.; Sample, C.E. A conserved domain of the Epstein-Barr virus nuclear antigens 3A and 3C binds to a discrete domain of Jkappa. J. Virol. 1996, 70, 4228–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waltzer, L.; Logeat, F.; Brou, C.; Israel, A.; Sergeant, A.; Manet, E. The human J kappa recombination signal sequence binding protein (RBP-J kappa) targets the Epstein-Barr virus EBNA2 protein to its DNA responsive elements. EMBO J. 1994, 13, 5633–5638. [Google Scholar]
- Tomkinson, B.; Kieff, E. Second-site homologous recombination in Epstein-Barr virus: Insertion of type 1 EBNA 3 genes in place of type 2 has no effect on in vitro infection. J. Virol. 1992, 66, 780–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomkinson, B.; Robertson, E.; Kieff, E. Epstein-Barr virus nuclear proteins EBNA-3A and EBNA-3C are essential for B-lymphocyte growth transformation. J. Virol. 1993, 67, 2014–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruo, S.; Johannsen, E.; Illanes, D.; Cooper, A.; Kieff, E. Epstein-Barr Virus nuclear protein EBNA3A is critical for maintaining lymphoblastoid cell line growth. J. Virol. 2003, 77, 10437–10447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruo, S.; Wu, Y.; Ishikawa, S.; Kanda, T.; Iwakiri, D.; Takada, K. Epstein-Barr virus nuclear protein EBNA3C is required for cell cycle progression and growth maintenance of lymphoblastoid cells. Proc. Natl. Acad. Sci. USA 2006, 103, 19500–19505. [Google Scholar] [CrossRef] [Green Version]
- Maruo, S.; Johannsen, E.; Illanes, D.; Cooper, A.; Zhao, B.; Kieff, E. Epstein-Barr virus nuclear protein 3A domains essential for growth of lymphoblasts: Transcriptional regulation through RBP-Jkappa/CBF1 is critical. J. Virol. 2005, 79, 10171–10179. [Google Scholar] [CrossRef] [Green Version]
- Maruo, S.; Wu, Y.; Ito, T.; Kanda, T.; Kieff, E.D.; Takada, K. Epstein-Barr virus nuclear protein EBNA3C residues critical for maintaining lymphoblastoid cell growth. Proc. Natl. Acad. Sci. USA 2009, 106, 4419–4424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Sakakibara, S.; Maruo, S.; Zhao, B.; Calderwood, M.A.; Holthaus, A.M.; Lai, C.Y.; Takada, K.; Kieff, E.; Johannsen, E. Epstein-Barr virus nuclear protein 3C domains necessary for lymphoblastoid cell growth: Interaction with RBP-Jkappa regulates TCL1. J. Virol. 2009, 83, 12368–12377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, E.S.; Lin, J.; Kieff, E. The amino-terminal domains of Epstein-Barr virus nuclear proteins 3A, 3B, and 3C interact with RBPJ(kappa). J. Virol. 1996, 70, 3068–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waltzer, L.; Perricaudet, M.; Sergeant, A.; Manet, E. Epstein-Barr virus EBNA3A and EBNA3C proteins both repress RBP-J kappa-EBNA2-activated transcription by inhibiting the binding of RBP-J kappa to DNA. J. Virol. 1996, 70, 5909–5915. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.; Johannsen, E.; Maruo, S.; Cahir-McFarland, E.; Illanes, D.; Davidson, D.; Kieff, E. EBNA3A association with RBP-Jkappa down-regulates c-myc and Epstein-Barr virus-transformed lymphoblast growth. J. Virol. 2003, 77, 999–1010. [Google Scholar] [CrossRef] [Green Version]
- Cludts, I.; Farrell, P.J. Multiple functions within the Epstein-Barr virus EBNA-3A protein. J. Virol. 1998, 72, 1862–1869. [Google Scholar] [CrossRef] [Green Version]
- Hickabottom, M.; Parker, G.A.; Freemont, P.; Crook, T.; Allday, M.J. Two nonconsensus sites in the Epstein-Barr virus oncoprotein EBNA3A cooperate to bind the co-repressor carboxyl-terminal-binding protein (CtBP). J. Biol. Chem. 2002, 277, 47197–47204. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, M.; Holthaus, A.M.; Calderwood, M.A.; Lai, C.Y.; Krastins, B.; Sarracino, D.; Johannsen, E. The EBNA3 family of Epstein-Barr virus nuclear proteins associates with the USP46/USP12 deubiquitination complexes to regulate lymphoblastoid cell line growth. PLoS Pathog. 2015, 11, e1004822. [Google Scholar] [CrossRef] [PubMed]
- Hertle, M.L.; Popp, C.; Petermann, S.; Maier, S.; Kremmer, E.; Lang, R.; Mages, J.; Kempkes, B. Differential gene expression patterns of EBV infected EBNA-3A positive and negative human B lymphocytes. PLoS Pathog. 2009, 5, e1000506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderton, E.; Yee, J.; Smith, P.; Crook, T.; White, R.E.; Allday, M.J. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: Clues to the pathogenesis of Burkitt’s lymphoma. Oncogene 2008, 27, 421–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harth-Hertle, M.L.; Scholz, B.A.; Erhard, F.; Glaser, L.V.; Dolken, L.; Zimmer, R.; Kempkes, B. Inactivation of intergenic enhancers by EBNA3A initiates and maintains polycomb signatures across a chromatin domain encoding CXCL10 and CXCL9. PLoS Pathog. 2013, 9, e1003638. [Google Scholar] [CrossRef] [PubMed]
- Tursiella, M.L.; Bowman, E.R.; Wanzeck, K.C.; Throm, R.E.; Liao, J.; Zhu, J.; Sample, C.E. Epstein-Barr Virus Nuclear Antigen 3A Promotes Cellular Proliferation by Repression of the Cyclin-Dependent Kinase Inhibitor p21WAF1/CIP1. PLoS Pathog. 2014, 10, e1004415. [Google Scholar] [CrossRef] [Green Version]
- Bazot, Q.; Deschamps, T.; Tafforeau, L.; Siouda, M.; Leblanc, P.; Harth-Hertle, M.L.; Rabourdin-Combe, C.; Lotteau, V.; Kempkes, B.; Tommasino, M.; et al. Epstein-Barr virus nuclear antigen 3A protein regulates CDKN2B transcription via interaction with MIZ-1. Nucleic Acids Res. 2014, 42, 9700–9716. [Google Scholar] [CrossRef]
- Bazot, Q.; Paschos, K.; Skalska, L.; Kalchschmidt, J.S.; Parker, G.A.; Allday, M.J. Epstein-Barr Virus Proteins EBNA3A and EBNA3C Together Induce Expression of the Oncogenic MicroRNA Cluster miR-221/miR-222 and Ablate Expression of Its Target p57KIP2. PLoS Pathog. 2015, 11, e1005031. [Google Scholar] [CrossRef] [Green Version]
- Maruo, S.; Zhao, B.; Johannsen, E.; Kieff, E.; Zou, J.; Takada, K. Epstein-Barr virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing p16INK4A and p14ARF expression. Proc. Natl. Acad. Sci. USA 2011, 108, 1919–1924. [Google Scholar] [CrossRef] [Green Version]
- Styles, C.T.; Bazot, Q.; Parker, G.A.; White, R.E.; Paschos, K.; Allday, M.J. EBV epigenetically suppresses the B cell-to-plasma cell differentiation pathway while establishing long-term latency. PLoS Biol. 2017, 15, e2001992. [Google Scholar] [CrossRef] [Green Version]
- Paschos, K.; Bazot, Q.; Lees, J.; Farrell, P.J.; Allday, M.J. Requirement for PRC1 subunit BMI1 in host gene activation by Epstein-Barr virus protein EBNA3C. Nucleic Acids Res. 2019, 47, 2807–2821. [Google Scholar] [CrossRef]
- Paschos, K.; Smith, P.; Anderton, E.; Middeldorp, J.M.; White, R.E.; Allday, M.J. Epstein-barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene Bim. PLoS Pathog. 2009, 5, e1000492. [Google Scholar] [CrossRef] [Green Version]
- Skalska, L.; White, R.E.; Franz, M.; Ruhmann, M.; Allday, M.J. Epigenetic repression of p16(INK4A) by latent Epstein-Barr virus requires the interaction of EBNA3A and EBNA3C with CtBP. PLoS Pathog. 2010, 6, e1000951. [Google Scholar] [CrossRef] [Green Version]
- Price, A.M.; Dai, J.; Bazot, Q.; Patel, L.; Nikitin, P.A.; Djavadian, R.; Winter, P.S.; Salinas, C.A.; Barry, A.P.; Wood, K.C.; et al. Epstein-Barr virus ensures B cell survival by uniquely modulating apoptosis at early and late times after infection. Elife 2017, 6, e22509. [Google Scholar] [CrossRef]
- Sommermann, T.; Yasuda, T.; Ronen, J.; Wirtz, T.; Weber, T.; Sack, U.; Caeser, R.; Zhang, J.; Li, X.; Chu, V.T.; et al. Functional interplay of Epstein-Barr virus oncoproteins in a mouse model of B cell lymphomagenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 14421–14432. [Google Scholar] [CrossRef]
- Schmidt, S.C.; Jiang, S.; Zhou, H.; Willox, B.; Holthaus, A.M.; Kharchenko, P.V.; Johannsen, E.C.; Kieff, E.; Zhao, B. Epstein-Barr virus nuclear antigen 3A partially coincides with EBNA3C genome-wide and is tethered to DNA through BATF complexes. Proc. Natl. Acad. Sci. USA 2015, 112, 554–559. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Welch, R.; Zhao, B.; Ta, T.; Keles, S.; Johannsen, E. Epstein-Barr Virus Nuclear Antigen 3 (EBNA3) Proteins Regulate EBNA2 Binding to Distinct RBPJ Genomic Sites. J. Virol. 2015, 90, 2906–2919. [Google Scholar] [CrossRef] [Green Version]
- McClellan, M.J.; Khasnis, S.; Wood, C.D.; Palermo, R.D.; Schlick, S.N.; Kanhere, A.S.; Jenner, R.G.; West, M.J. Downregulation of integrin receptor-signaling genes by Epstein-Barr virus EBNA 3C via promoter-proximal and -distal binding elements. J. Virol. 2012, 86, 5165–5178. [Google Scholar] [CrossRef] [Green Version]
- Skalska, L.; White, R.E.; Parker, G.A.; Turro, E.; Sinclair, A.J.; Paschos, K.; Allday, M.J. Induction of p16(INK4a) is the major barrier to proliferation when Epstein-Barr virus (EBV) transforms primary B cells into lymphoblastoid cell lines. PLoS Pathog. 2013, 9, e1003187. [Google Scholar] [CrossRef]
- Ohashi, M.; Hayes, M.; McChesney, K.; Johannsen, E. Epstein-Barr virus nuclear antigen 3C (EBNA3C) interacts with the metabolism sensing C-terminal binding protein (CtBP) repressor to upregulate host genes. PLoS Pathog. 2021, 17, e1009419. [Google Scholar] [CrossRef]
- Jiang, S.; Willox, B.; Zhou, H.; Holthaus, A.M.; Wang, A.; Shi, T.T.; Maruo, S.; Kharchenko, P.V.; Johannsen, E.C.; Kieff, E.; et al. Epstein-Barr virus nuclear antigen 3C binds to BATF/IRF4 or SPI1/IRF4 composite sites and recruits Sin3A to repress CDKN2A. Proc. Natl. Acad. Sci. USA 2014, 111, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Allday, M.J.; Farrell, P.J. Epstein-Barr virus nuclear antigen EBNA3C/6 expression maintains the level of latent membrane protein 1 in G1-arrested cells. J. Virol. 1994, 68, 3491–3498. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Johannsen, E.; Robertson, E.; Kieff, E. Epstein-Barr virus nuclear antigen 3C putative repression domain mediates coactivation of the LMP1 promoter with EBNA-2. J. Virol. 2002, 76, 232–242. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Sample, C.E. Epstein-barr virus nuclear antigen 3C activates the latent membrane protein 1 promoter in the presence of Epstein-Barr virus nuclear antigen 2 through sequences encompassing an spi-1/Spi-B binding site. J. Virol. 2000, 74, 5151–5160. [Google Scholar] [CrossRef]
- Kalchschmidt, J.S.; Bashford-Rogers, R.; Paschos, K.; Gillman, A.C.; Styles, C.T.; Kellam, P.; Allday, M.J. Epstein-Barr virus nuclear protein EBNA3C directly induces expression of AID and somatic mutations in B cells. J. Exp. Med. 2016, 213, 921–928. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Mar, J.C.; Maruo, S.; Lee, S.; Gewurz, B.E.; Johannsen, E.; Holton, K.; Rubio, R.; Takada, K.; Quackenbush, J.; et al. Epstein-Barr virus nuclear antigen 3C regulated genes in lymphoblastoid cell lines. Proc. Natl. Acad. Sci. USA 2011, 108, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Paschos, K.; Parker, G.A.; Watanatanasup, E.; White, R.E.; Allday, M.J. BIM promoter directly targeted by EBNA3C in polycomb-mediated repression by EBV. Nucleic Acids Res. 2012, 40, 7233–7246. [Google Scholar] [CrossRef] [Green Version]
- Kalchschmidt, J.S.; Gillman, A.C.; Paschos, K.; Bazot, Q.; Kempkes, B.; Allday, M.J. EBNA3C Directs Recruitment of RBPJ (CBF1) to Chromatin during the Process of Gene Repression in EBV Infected B Cells. PLoS Pathog. 2016, 12, e1005383. [Google Scholar] [CrossRef] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR-Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Lu, J.; Cai, Q.; Saha, A.; Jha, H.C.; Dzeng, R.K.; Robertson, E.S. The EBV Latent Antigen 3C Inhibits Apoptosis through Targeted Regulation of Interferon Regulatory Factors 4 and 8. PLoS Pathog. 2013, 9, e1003314. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Walsh, M.J.; Bernhardt, K.; Ashbaugh, C.W.; Trudeau, S.J.; Ashbaugh, I.Y.; Jiang, S.; Jiang, C.; Zhao, B.; Root, D.E.; et al. CRISPR/Cas9 Screens Reveal Epstein-Barr Virus-Transformed B Cell Host Dependency Factors. Cell Host Microbe 2017, 21, 580–591.e7. [Google Scholar] [CrossRef] [Green Version]
- Knight, J.S.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C regulates cyclin A/p27 complexes and enhances cyclin A-dependent kinase activity. J. Virol. 2004, 78, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Knight, J.S.; Sharma, N.; Kalman, D.E.; Robertson, E.S. A cyclin-binding motif within the amino-terminal homology domain of EBNA3C binds cyclin A and modulates cyclin A-dependent kinase activity in Epstein-Barr virus-infected cells. J. Virol. 2004, 78, 12857–12867. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.K.; Jha, H.C.; El-Naccache, D.W.; Robertson, E.S. An EBV recombinant deleted for residues 130-159 in EBNA3C can deregulate p53/Mdm2 and Cyclin D1/CDK6 which results in apoptosis and reduced cell proliferation. Oncotarget 2016, 7, 18116–18134. [Google Scholar] [CrossRef] [Green Version]
- Pei, Y.; Singh, R.K.; Shukla, S.K.; Lang, F.; Zhang, S.; Robertson, E.S. Epstein-Barr Virus Nuclear Antigen 3C Facilitates Cell Proliferation by Regulating Cyclin D2. J. Virol. 2018, 92, e00663-18. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.; Halder, S.; Upadhyay, S.K.; Lu, J.; Kumar, P.; Murakami, M.; Cai, Q.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C facilitates G1-S transition by stabilizing and enhancing the function of cyclin D1. PLoS Pathog. 2011, 7, e1001275. [Google Scholar] [CrossRef]
- Knight, J.S.; Sharma, N.; Robertson, E.S. SCFSkp2 complex targeted by Epstein-Barr virus essential nuclear antigen. Mol. Cell. Biol. 2005, 25, 1749–1763. [Google Scholar] [CrossRef] [Green Version]
- Knight, J.S.; Sharma, N.; Robertson, E.S. Epstein-Barr virus latent antigen 3C can mediate the degradation of the retinoblastoma protein through an SCF cellular ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2005, 102, 18562–18566. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.; Bamidele, A.; Murakami, M.; Robertson, E.S. EBNA3C attenuates the function of p53 through interaction with inhibitor of growth family proteins 4 and 5. J. Virol. 2011, 85, 2079–2088. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Lu, J.; Cai, Q.; Sun, Z.; Jha, H.C.; Robertson, E.S. EBNA3C augments Pim-1 mediated phosphorylation and degradation of p21 to promote B-cell proliferation. PLoS Pathog. 2014, 10, e1004304. [Google Scholar] [CrossRef] [Green Version]
- Rosendorff, A.; Illanes, D.; David, G.; Lin, J.; Kieff, E.; Johannsen, E. EBNA3C coactivation with EBNA2 requires a SUMO homology domain. J. Virol. 2004, 78, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Knight, J.S.; Lan, K.; Subramanian, C.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C recruits histone deacetylase activity and associates with the corepressors mSin3A and NCoR in human B-cell lines. J. Virol. 2003, 77, 4261–4272. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.S.; Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef] [Green Version]
- Westhoff Smith, D.; Sugden, B. Potential cellular functions of Epstein-Barr Nuclear Antigen 1 (EBNA1) of Epstein-Barr Virus. Viruses 2013, 5, 226–240. [Google Scholar] [CrossRef] [Green Version]
- De Leo, A.; Calderon, A.; Lieberman, P.M. Control of Viral Latency by Episome Maintenance Proteins. Trends Microbiol. 2020, 28, 150–162. [Google Scholar] [CrossRef]
- Altmann, M.; Pich, D.; Ruiss, R.; Wang, J.; Sugden, B.; Hammerschmidt, W. Transcriptional activation by EBV nuclear antigen 1 is essential for the expression of EBV’s transforming genes. Proc. Natl. Acad. Sci. USA 2006, 103, 14188–14193. [Google Scholar] [CrossRef] [Green Version]
- Frappier, L. Ebna1. Curr. Top. Microbiol. Immunol. 2015, 391, 3–34. [Google Scholar] [CrossRef]
- Frappier, L. EBNA1 and host factors in Epstein-Barr virus latent DNA replication. Curr. Opin. Virol. 2012, 2, 733–739. [Google Scholar] [CrossRef]
- Dresang, L.R.; Vereide, D.T.; Sugden, B. Identifying sites bound by Epstein-Barr virus nuclear antigen 1 (EBNA1) in the human genome: Defining a position-weighted matrix to predict sites bound by EBNA1 in viral genomes. J. Virol. 2009, 83, 2930–2940. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.; Wikramasinghe, P.; Norseen, J.; Tsai, K.; Wang, P.; Showe, L.; Davuluri, R.V.; Lieberman, P.M. Genome-wide analysis of host-chromosome binding sites for Epstein-Barr Virus Nuclear Antigen 1 (EBNA1). Virol. J. 2010, 7, 262. [Google Scholar] [CrossRef] [Green Version]
- Tempera, I.; De Leo, A.; Kossenkov, A.V.; Cesaroni, M.; Song, H.; Dawany, N.; Showe, L.; Lu, F.; Wikramasinghe, P.; Lieberman, P.M. Identification of MEF2B, EBF1, and IL6R as Direct Gene Targets of Epstein-Barr Virus (EBV) Nuclear Antigen 1 Critical for EBV-Infected B-Lymphocyte Survival. J. Virol. 2016, 90, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Lamontagne, R.J.; Soldan, S.S.; Su, C.; Wiedmer, A.; Won, K.J.; Lu, F.; Goldman, A.R.; Wickramasinghe, J.; Tang, H.Y.; Speicher, D.W.; et al. A multi-omics approach to Epstein-Barr virus immortalization of B-cells reveals EBNA1 chromatin pioneering activities targeting nucleotide metabolism. PLoS Pathog. 2021, 17, e1009208. [Google Scholar] [CrossRef]
- Dheekollu, J.; Wiedmer, A.; Ayyanathan, K.; Deakyne, J.S.; Messick, T.E.; Lieberman, P.M. Cell-cycle-dependent EBNA1-DNA crosslinking promotes replication termination at oriP and viral episome maintenance. Cell 2021, 184, 643–654.e13. [Google Scholar] [CrossRef]
- Messick, T.E.; Smith, G.R.; Soldan, S.S.; McDonnell, M.E.; Deakyne, J.S.; Malecka, K.A.; Tolvinski, L.; van den Heuvel, A.P.J.; Gu, B.W.; Cassel, J.A.; et al. Structure-based design of small-molecule inhibitors of EBNA1 DNA binding blocks Epstein-Barr virus latent infection and tumor growth. Sci. Transl. Med. 2019, 11, eaau5612. [Google Scholar] [CrossRef]
- Soldan, S.S.; Anderson, E.M.; Frase, D.M.; Zhang, Y.; Caruso, L.B.; Wang, Y.; Deakyne, J.S.; Gewurz, B.E.; Tempera, I.; Lieberman, P.M.; et al. EBNA1 inhibitors have potent and selective antitumor activity in xenograft models of Epstein-Barr virus-associated gastric cancer. Gastric Cancer 2021, 24, 1076–1088. [Google Scholar] [CrossRef]
- Lee, E.K.; Kim, S.Y.; Noh, K.W.; Joo, E.H.; Zhao, B.; Kieff, E.; Kang, M.S. Small molecule inhibition of Epstein-Barr virus nuclear antigen-1 DNA binding activity interferes with replication and persistence of the viral genome. Antivir. Res. 2014, 104, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.W.; Jiang, S.; Gewurz, B.E. Epstein-Barr Virus LMP1-Mediated Oncogenicity. J. Virol. 2017, 91, e01718-16. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Barrera, L.A.; Ersing, I.; Willox, B.; Schmidt, S.C.; Greenfeld, H.; Zhou, H.; Mollo, S.B.; Shi, T.T.; Takasaki, K.; et al. The NF-kappaB genomic landscape in lymphoblastoid B cells. Cell Rep. 2014, 8, 1595–1606. [Google Scholar] [CrossRef] [Green Version]
- Henley, M.J.; Koehler, A.N. Advances in targeting ‘undruggable’ transcription factors with small molecules. Nat. Rev. Drug Discov. 2021, 20, 669–688. [Google Scholar] [CrossRef]
- Liu, J.; Chen, H.; Kaniskan, H.U.; Xie, L.; Chen, X.; Jin, J.; Wei, W. TF-PROTACs Enable Targeted Degradation of Transcription Factors. J. Am. Chem. Soc. 2021, 143, 8902–8910. [Google Scholar] [CrossRef]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondeson, D.P.; Mares, A.; Smith, I.E.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, B. Epstein–Barr Virus B Cell Growth Transformation: The Nuclear Events. Viruses 2023, 15, 832. https://doi.org/10.3390/v15040832
Zhao B. Epstein–Barr Virus B Cell Growth Transformation: The Nuclear Events. Viruses. 2023; 15(4):832. https://doi.org/10.3390/v15040832
Chicago/Turabian StyleZhao, Bo. 2023. "Epstein–Barr Virus B Cell Growth Transformation: The Nuclear Events" Viruses 15, no. 4: 832. https://doi.org/10.3390/v15040832
APA StyleZhao, B. (2023). Epstein–Barr Virus B Cell Growth Transformation: The Nuclear Events. Viruses, 15(4), 832. https://doi.org/10.3390/v15040832