
An Automated Bioinformatics Pipeline Informing Near-Real-Time Public Health Responses to New HIV Diagnoses in a Statewide HIV Epidemic

 , ,
, ,
Abstract
:1. Background
2. Methods
2.1. State-Wide HIV Databases
2.2. Sequence QC and Analyses
2.3. Data Integration
2.4. Report Generation
2.5. Case Management
3. Results
3.1. Database and Sequence QC
3.2. Bioinformatic Pipeline
3.3. Reports and Case Management
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fauci, A.S.; Redfield, R.R.; Sigounas, G. Ending the HIV epidemic: A plan for the United States. JAMA 2019, 321, 844–845. [Google Scholar] [CrossRef] [Green Version]
- Castro-Nallar, E.; Pérez-Losada, M.; Burton, G.F.; Crandall, K.A. The evolution of HIV: Inferences using phylogenetics. Mol. Phylogenet. Evol. 2012, 62, 777–792. [Google Scholar] [CrossRef] [Green Version]
- Adler, M.W.; Johnson, A.M. Contact tracing for HIV infection. Br. Med. J. 1988, 296, 1420–1421. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.M.; May, S.; Tweeten, S.; Drumright, L.; Pacold, M.E.; Pond, S.L.; Pesano, R.L.; Lie, Y.S.; Richman, D.D.; Frost, S.D.; et al. A public health model for the molecular surveillance of HIV transmission in San Diego, California. AIDS 2009, 23, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Kantor, R.; Fulton, J.P.; Steingrimsson, J.; Novitsky, V.; Howison, M.; Gillani, F.; Li, Y.; Manne, A.; Parillo, Z.; Spence, M.; et al. Challenges in evaluating the use of viral sequence data to identify HIV transmission networks for public health. Stat. Commun. Infect. Dis. 2020, 12, 20190019. [Google Scholar] [CrossRef]
- Hassan, A.S.; Pybus, O.G.; Sanders, E.J.; Albert, J.; Esbjörnsson, J. Defining HIV-1 transmission clusters based on sequence data. AIDS 2017, 31, 1211–1222. [Google Scholar] [CrossRef]
- Wertheim, J.O.; Chato, C.; Poon, A.F.Y. Comparative analysis of HIV sequences in real time for public health. Curr. Opin. HIV AIDS 2019, 14, 213–220. [Google Scholar] [CrossRef]
- Poon, A.F.; Gustafson, R.; Daly, P.; Zerr, L.; Demlow, S.E.; Wong, J.; Woods, C.K.; Hogg, R.S.; Krajden, M.; Moore, D.; et al. Near real-time monitoring of HIV transmission hotspots from routine HIV genotyping: An implementation case study. Lancet HIV 2016, 3, e231–e238. [Google Scholar] [CrossRef] [Green Version]
- Steingrimsson, J.A.; Fulton, J.; Howison, M.; Novitsky, V.; Gillani, F.S.; Bertrand, T.; Civitarese, A.; Howe, K.; Ronquillo, G.; Lafazia, B.; et al. Beyond HIV outbreaks: Protocol, rationale and implementation of a prospective study quantifying the benefit of incorporating viral sequence clustering analysis into routine public health interventions. BMJ Open 2022, 12, e060184. [Google Scholar] [CrossRef] [PubMed]
- DeLong, A.K.; Wu, M.; Bennett, D.; Parkin, N.; Wu, Z.; Hogan, J.W.; Kantor, R. Sequence quality analysis tool for HIV type 1 protease and reverse transcriptase. AIDS Res. Hum. Retrovir. 2012, 28, 894–901. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.F.; Shafer, R.W. Web Resources for HIV type 1 Genotypic-Resistance Test Interpretation. Clin. Infect. Dis. 2006, 42, 1608–1618. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novitsky, V.; Steingrimsson, J.A.; Howison, M.; Gillani, F.S.; Li, Y.; Manne, A.; Fulton, J.; Spence, M.; Parillo, Z.; Marak, T.; et al. Empirical comparison of analytical approaches for identifying molecular HIV-1 clusters. Sci. Rep. 2020, 10, 18547. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Ragonnet-Cronin, M.; Hodcroft, E.; Hué, S.; Fearnhill, E.; Delpech, V.; Brown, A.J.; Lycett, S. Automated analysis of phylogenetic clusters. BMC Bioinform. 2013, 14, 317. [Google Scholar] [CrossRef] [Green Version]
- Kosakovsky Pond, S.L.; Weaver, S.; Leigh Brown, A.J.; Wertheim, J.O. HIV-TRACE (TRAnsmission Cluster Engine): A Tool for Large Scale Molecular Epidemiology of HIV-1 and Other Rapidly Evolving Pathogens. Mol. Biol. Evol. 2018, 35, 1812–1819. [Google Scholar] [CrossRef] [Green Version]
- Oster, A.M.; France, A.M.; Panneer, N.; Bañez Ocfemia, M.C.; Campbell, E.; Dasgupta, S.; Switzer, W.M.; Wertheim, J.O.; Hernandez, A.L. Identifying Clusters of Recent and Rapid HIV Transmission Through Analysis of Molecular Surveillance Data. JAIDS 2018, 79, 543–550. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Detecting and Responding to HIV Transmission Clusters: A Guide for Health Departments. 2018. Available online: https://www.cdc.gov/hiv/pdf/funding/announcements/ps18-1802/CDC-HIV-PS18-1802-AttachmentE-Detecting-Investigating-and-Responding-to-HIV-Transmission-Clusters.pdf (accessed on 30 June 2022).
- Yu, G.; Smith, D.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Wang, L.G.; Lam, T.T.; Xu, S.; Dai, Z.; Zhou, L.; Feng, T.; Guo, P.; Dunn, C.W.; Jones, B.R.; Bradley, T.; et al. treeio: An R package for phylogenetic tree input and output with richly annotated and associated data. Mol. Biol. Evol. 2020, 37, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Bouamrane, M.M.; Tao, C.; Sarkar, I.N. Managing interoperability and complexity in health systems. Methods Inf. Med. 2015, 54, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Hamburg, M.A.; Cohen, M.; DeSalvo, K.; Gerberding, J.; Khaldun, J.; Lakey, D.; MacKenzie, E.; Palacio, H.; Shah, N.R. Building a National Public Health System in the United States. N. Engl. J. Med. 2022, 387, 385–388. [Google Scholar] [CrossRef]
- Peters, P.J.; Pontones, P.; Hoover, K.W.; Patel, M.R.; Galang, R.R.; Shields, J.; Blosser, S.J.; Spiller, M.W.; Combs, B.; Switzer, W.M.; et al. HIV Infection Linked to Injection Use of Oxymorphone in Indiana, 2014-2015. N. Engl. J. Med. 2016, 375, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Monterosso, A.; Minnerly, S.; Goings, S.; Morris, A.; France, A.M.; Dasgupta, S.; Oster, A.M.; Fanning, M. Identifying and investigating a rapidly growing HIV transmission cluster in Texas [CROI Abstract 845LB]. Top. Antivir. Med. 2017, 25, 1359s. [Google Scholar]
- Novitsky, V.; Steingrimsson, J.; Howison, M.; Dunn, C.W.; Gillani, F.S.; Fulton, J.; Bertrand, T.; Howe, K.; Bhattarai, L.; Ronquillo, G.; et al. Not all clusters are equal: Dynamics of molecular HIV-1 clusters in a statewide Rhode Island epidemic. AIDS 2023, 37, 389–399. [Google Scholar] [CrossRef]
- Smith, S.A.; Beaulieu, J.M.; Donoghue, M.J. Mega-phylogeny approach for comparative biology: An alternative to supertree and supermatrix approaches. BMC Evol. Biol. 2009, 9, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wertheim, J.O.; Murrell, B.; Mehta, S.R.; Forgione, L.A.; Kosakovsky Pond, S.L.; Smith, D.M.; Torian, L.V. Growth of HIV-1 Molecular Transmission Clusters in New York City. J. Infect. Dis. 2018, 218, 1943–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guang, A.; Howison, M.; Ledingham, L.; D’Antuono, M.; Chan, P.A.; Lawrence, C.; Dunn, C.W.; Kantor, R. Incorporating Within-Host Diversity in Phylogenetic Analyses for Detecting Clusters of New HIV Diagnoses. Front. Microbiol. 2022, 12, 803190. [Google Scholar] [CrossRef] [PubMed]


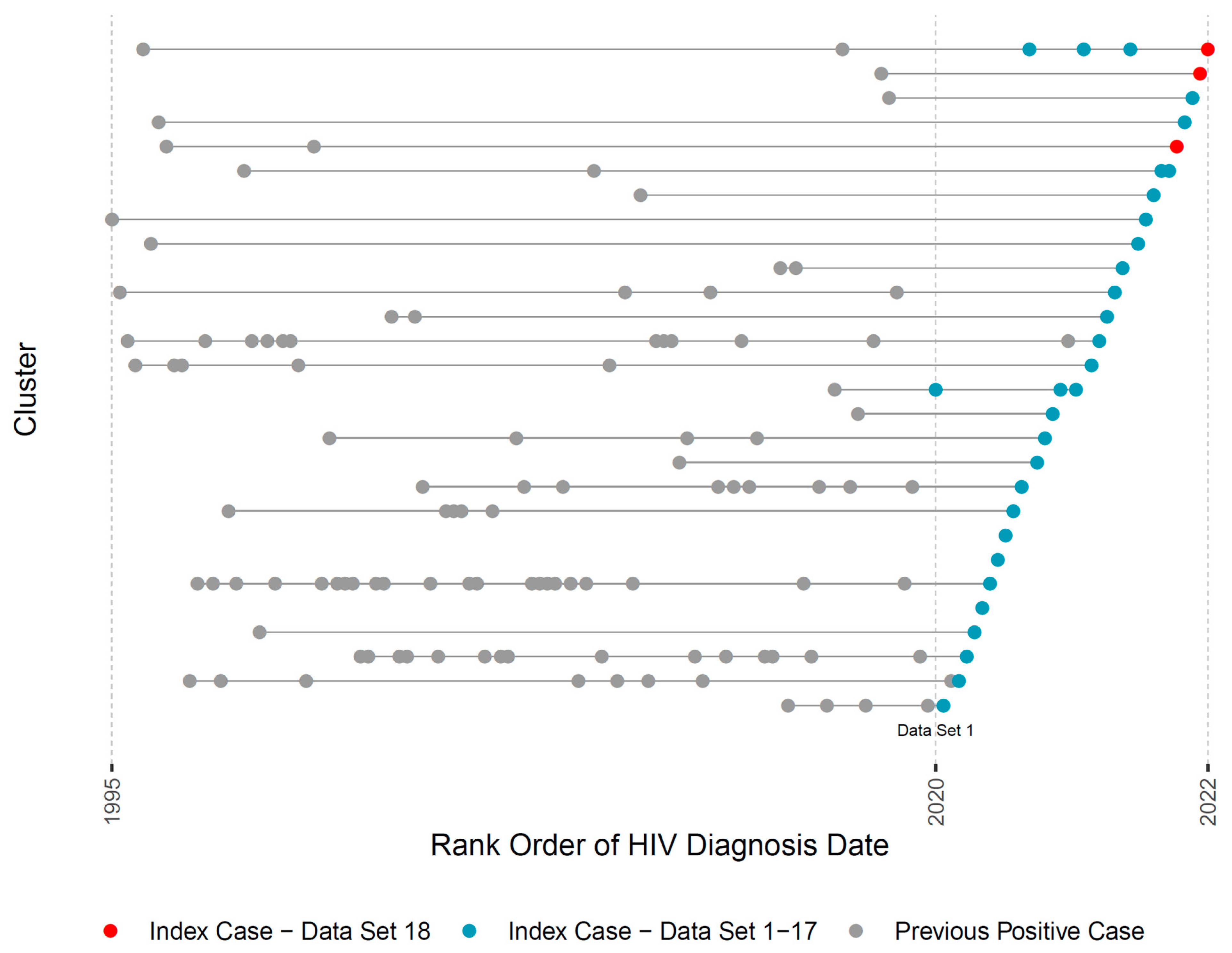

{kind=link}
{kind=link}
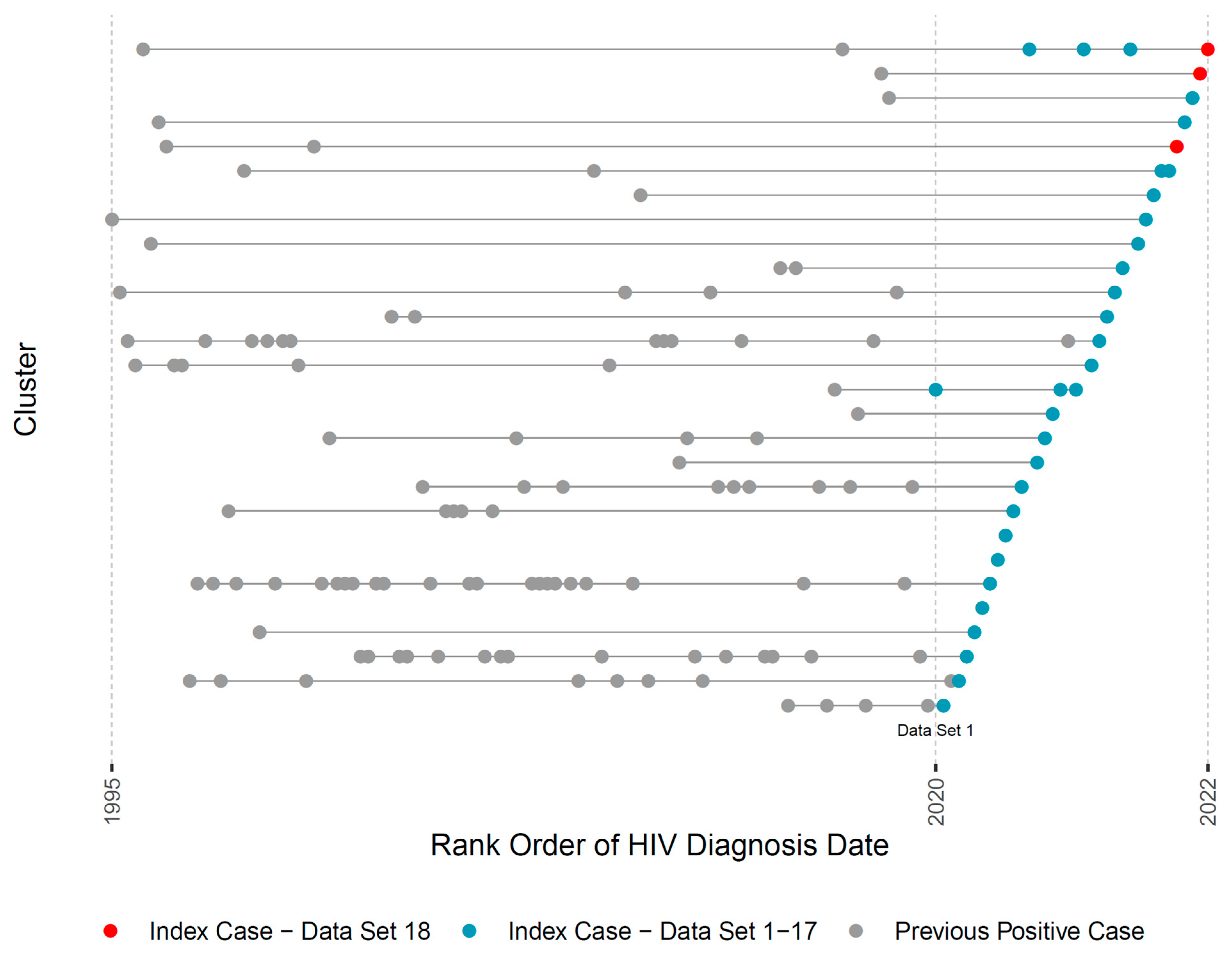{kind=link}
| Dataset Number | |||||||||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | Total | |
| Index Cases | 1 | 1 | 1 | 5 | 6 | 5 | 5 | 5 | 3 | 5 | 0 | 3 | 3 | 3 | 0 | 2 | 5 | 4 | 57 |
| Clustered in Statewide Dataset: | |||||||||||||||||||
| Any Phylogenetic Method | 1 | 1 | 1 | 2 | 5 | 2 | 4 | 1 | 2 | 2 | 0 | 3 | 2 | 3 | 0 | 2 | 3 | 3 | 37 |
| RAxML | 1 | 1 | 1 | 2 | 5 | 2 | 4 | 1 | 2 | 2 | 0 | 3 | 2 | 3 | 0 | 2 | 3 | 2 | 36 |
| IQ-Tree | 1 | 1 | 1 | 1 | 5 | 2 | 3 | 1 | 2 | 1 | 0 | 2 | 2 | 1 | 0 | 2 | 3 | 2 | 30 |
| FastTree | 1 | 1 | 1 | 1 | 4 | 2 | 4 | 1 | 2 | 1 | 0 | 3 | 2 | 2 | 0 | 2 | 3 | 3 | 33 |
| FastTree (ALRT) | 1 | 1 | 1 | 1 | 5 | 2 | 3 | 1 | 2 | 1 | 0 | 2 | 1 | 1 | 0 | 2 | 3 | 2 | 29 |
| MEGA | 1 | 1 | 1 | 2 | 5 | 2 | 4 | 1 | 2 | 1 | 0 | 3 | 2 | 2 | 0 | 2 | 3 | 1 | 33 |
| HIV-TRACE (1.5%) | 1 | 1 | 1 | 1 | 4 | 2 | 1 | 1 | 2 | 0 | 0 | 1 | 0 | 1 | 0 | 2 | 3 | 0 | 21 |
| CDC Cluster of Concern (0.5%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Only by Phylogenetic Methods | 0 | 0 | 0 | 1 | 1 | 0 | 3 | 0 | 0 | 2 | 0 | 2 | 2 | 2 | 0 | 0 | 0 | 3 | 16 |
| Clustered in Immunology Center Subset: | |||||||||||||||||||
| Any Phylogenetic Method | 1 | 1 | 1 | 2 | 5 | 2 | 4 | 1 | 2 | 2 | 0 | 3 | 2 | 3 | 0 | 2 | 3 | 3 | 37 |
| RAxML | 1 | 1 | 1 | 2 | 5 | 2 | 4 | 1 | 2 | 2 | 0 | 3 | 2 | 3 | 0 | 2 | 3 | 2 | 36 |
| IQ-Tree | 1 | 1 | 1 | 1 | 5 | 2 | 3 | 1 | 2 | 1 | 0 | 2 | 2 | 2 | 0 | 2 | 3 | 2 | 31 |
| FastTree | 1 | 1 | 1 | 1 | 4 | 2 | 4 | 1 | 2 | 0 | 0 | 3 | 1 | 2 | 0 | 2 | 3 | 3 | 31 |
| FastTree (ALRT) | 1 | 1 | 1 | 1 | 5 | 2 | 3 | 1 | 2 | 1 | 0 | 2 | 1 | 2 | 0 | 2 | 3 | 2 | 30 |
| MEGA | 1 | 1 | 1 | 2 | 5 | 2 | 4 | 1 | 2 | 1 | 0 | 3 | 1 | 2 | 0 | 2 | 3 | 1 | 32 |
| HIV-TRACE (1.5%) | 1 | 1 | 1 | 1 | 4 | 2 | 1 | 1 | 2 | 0 | 0 | 1 | 0 | 1 | 0 | 2 | 3 | 0 | 21 |
| CDC Cluster of Concern (0.5%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Only by Phylogenetic Methods | 0 | 0 | 0 | 1 | 1 | 0 | 3 | 0 | 0 | 2 | 0 | 2 | 2 | 2 | 0 | 0 | 0 | 3 | 16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Howison, M.; Gillani, F.S.; Novitsky, V.; Steingrimsson, J.A.; Fulton, J.; Bertrand, T.; Howe, K.; Civitarese, A.; Bhattarai, L.; MacAskill, M.; et al. An Automated Bioinformatics Pipeline Informing Near-Real-Time Public Health Responses to New HIV Diagnoses in a Statewide HIV Epidemic. Viruses 2023, 15, 737. https://doi.org/10.3390/v15030737
Howison M, Gillani FS, Novitsky V, Steingrimsson JA, Fulton J, Bertrand T, Howe K, Civitarese A, Bhattarai L, MacAskill M, et al. An Automated Bioinformatics Pipeline Informing Near-Real-Time Public Health Responses to New HIV Diagnoses in a Statewide HIV Epidemic. Viruses. 2023; 15(3):737. https://doi.org/10.3390/v15030737
Chicago/Turabian StyleHowison, Mark, Fizza S. Gillani, Vlad Novitsky, Jon A. Steingrimsson, John Fulton, Thomas Bertrand, Katharine Howe, Anna Civitarese, Lila Bhattarai, Meghan MacAskill, and et al. 2023. "An Automated Bioinformatics Pipeline Informing Near-Real-Time Public Health Responses to New HIV Diagnoses in a Statewide HIV Epidemic" Viruses 15, no. 3: 737. https://doi.org/10.3390/v15030737
APA StyleHowison, M., Gillani, F. S., Novitsky, V., Steingrimsson, J. A., Fulton, J., Bertrand, T., Howe, K., Civitarese, A., Bhattarai, L., MacAskill, M., Ronquillo, G., Hague, J., Dunn, C. W., Bandy, U., Hogan, J. W., & Kantor, R. (2023). An Automated Bioinformatics Pipeline Informing Near-Real-Time Public Health Responses to New HIV Diagnoses in a Statewide HIV Epidemic. Viruses, 15(3), 737. https://doi.org/10.3390/v15030737