


Membraneless Compartmentalization of Nuclear Assembly Sites during Murine Cytomegalovirus Infection

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Viruses and Infection Conditions
2.3. Antibodies and Reagents
2.4. Immunofluorescence and Microscopy
2.5. Semiquantitative Analysis of Viral DNA Synthesis
2.6. Immunofluorescence and “Click Chemistry”
2.7. Image Analysis
2.8. Western Blot
2.9. Detection of Liquid–Liquid Phase Separation (LLPS) Drivers in MCMV Protein Sequences
2.10. Prediction of LLPS Propensity in MCMV-Encoded Proteins
2.11. Data Presentation and Statistics
3. Results
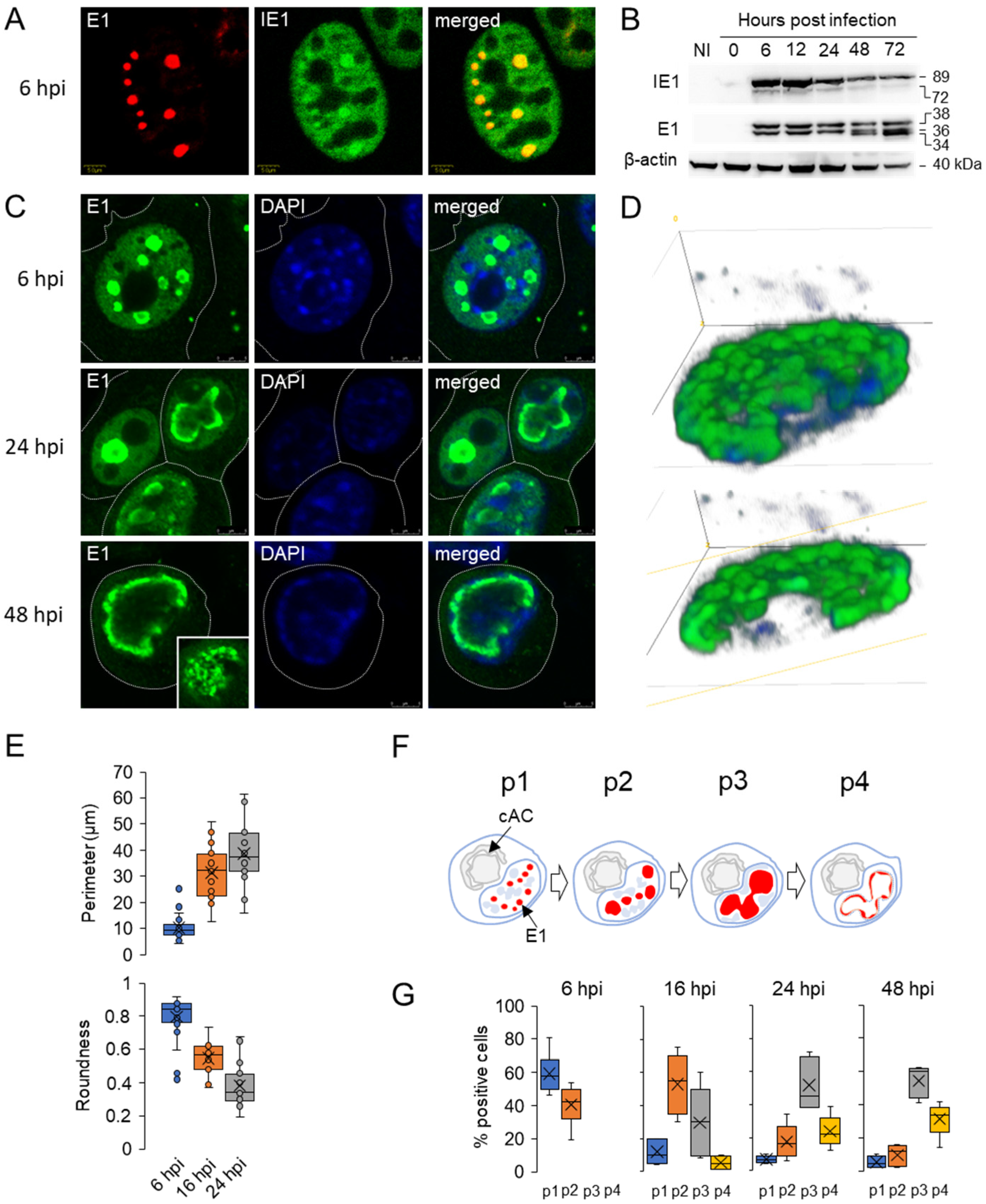
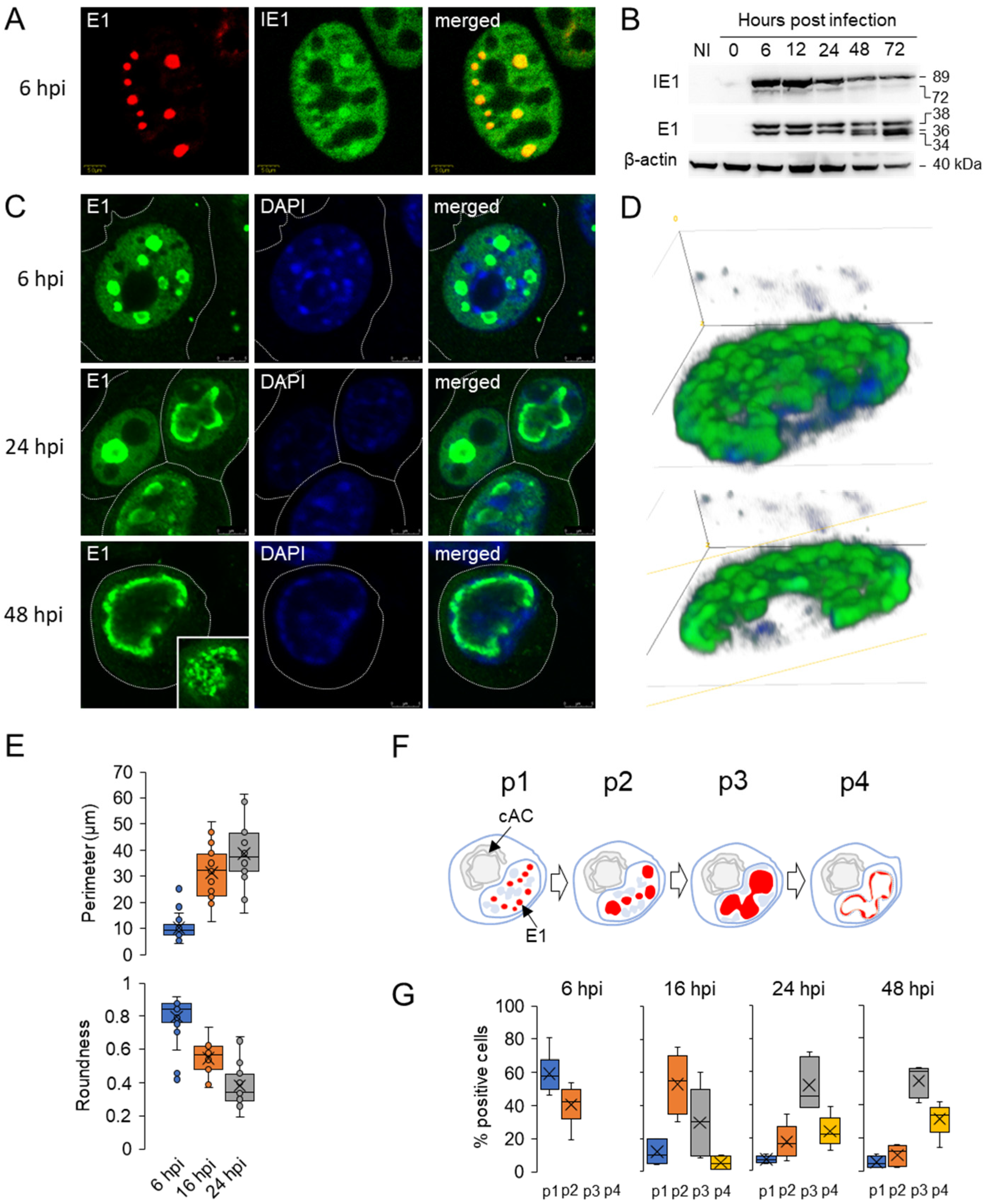
3.1. Diffuse Nuclear Distribution of Immediate-Early 1 (IE1) Protein and Nuclear Membraneless Assemblies of Early 1 (E1) Proteins
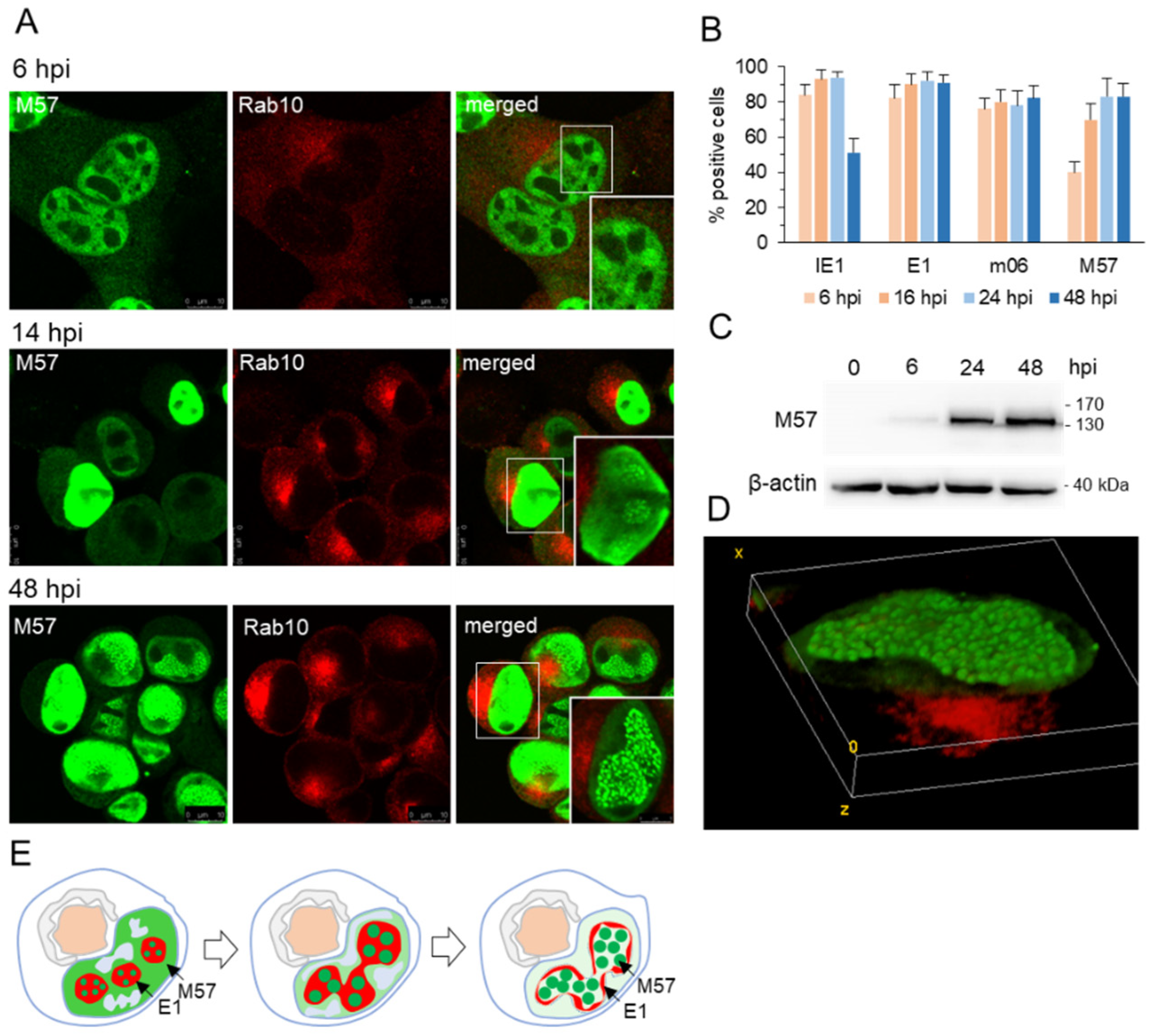
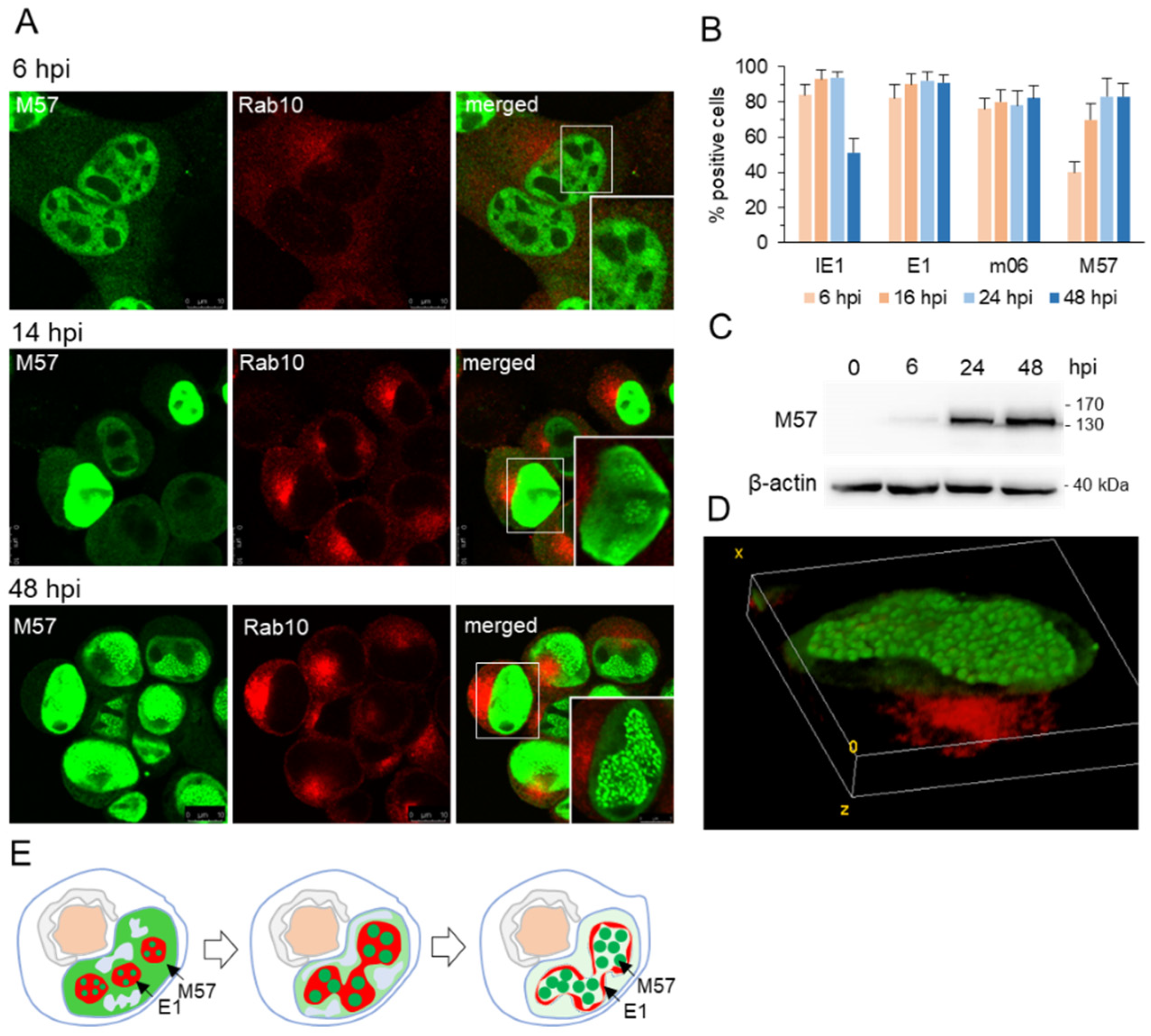
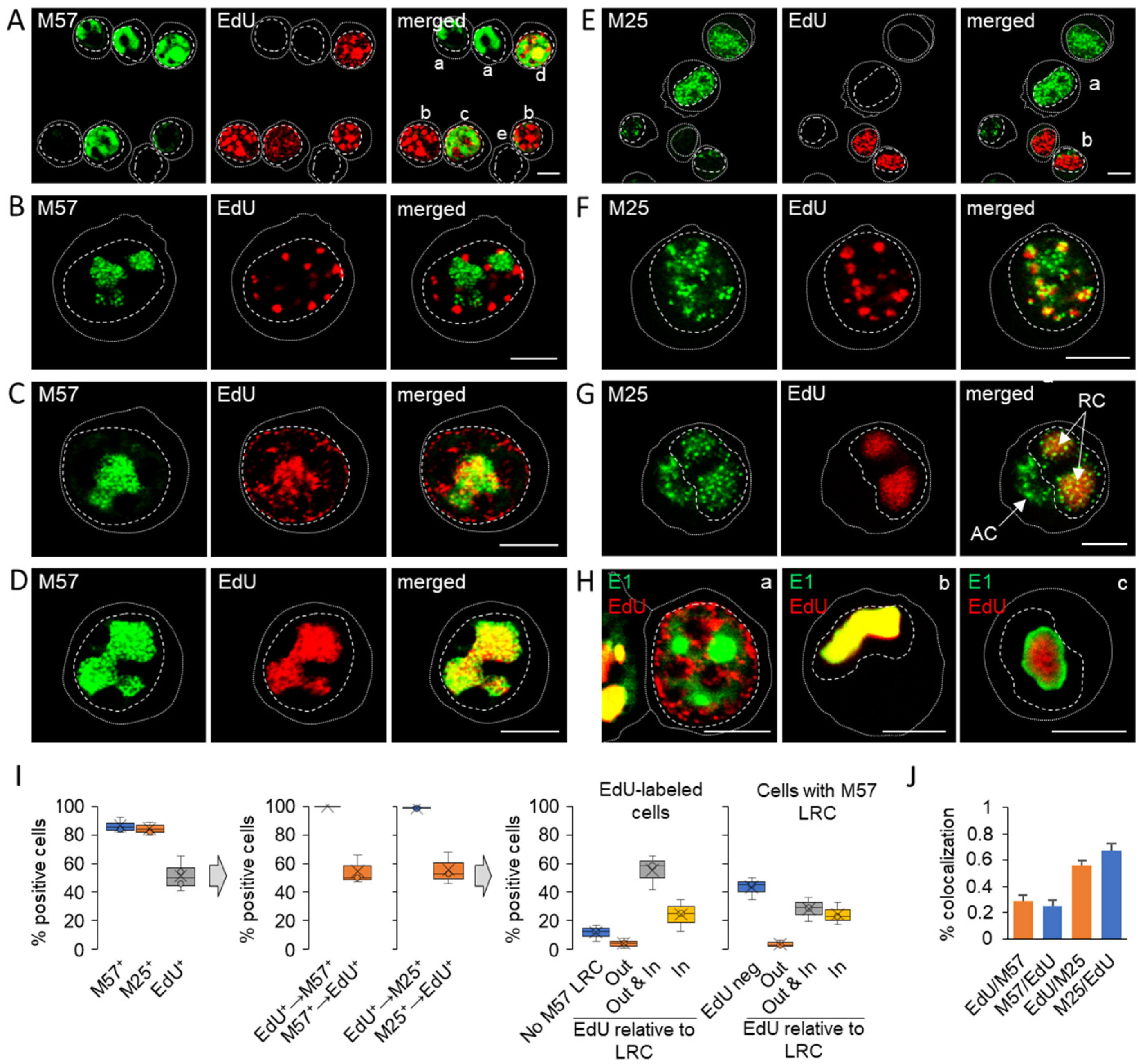
3.2. Nuclear Membraneless Assemblies of pM57
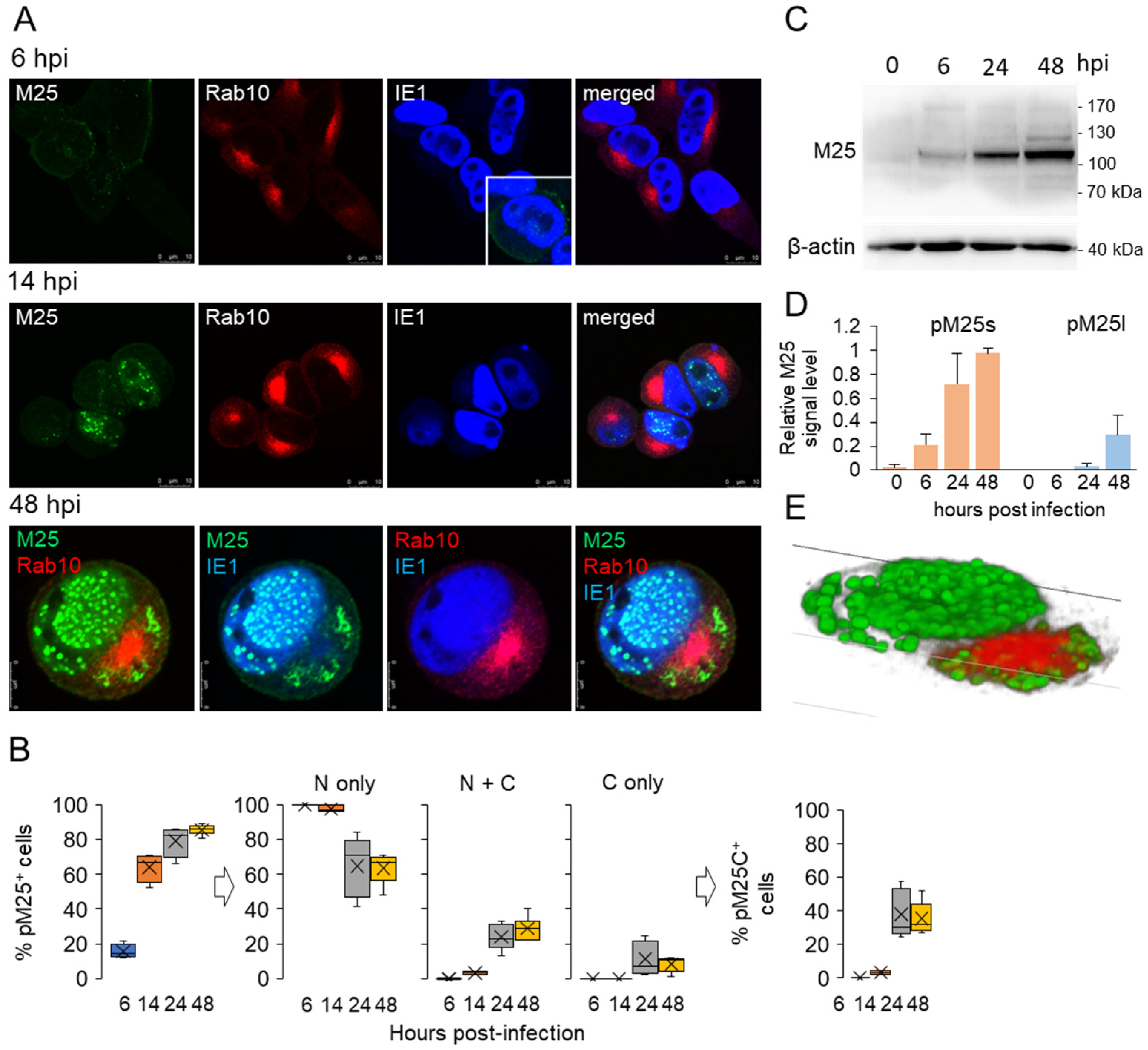
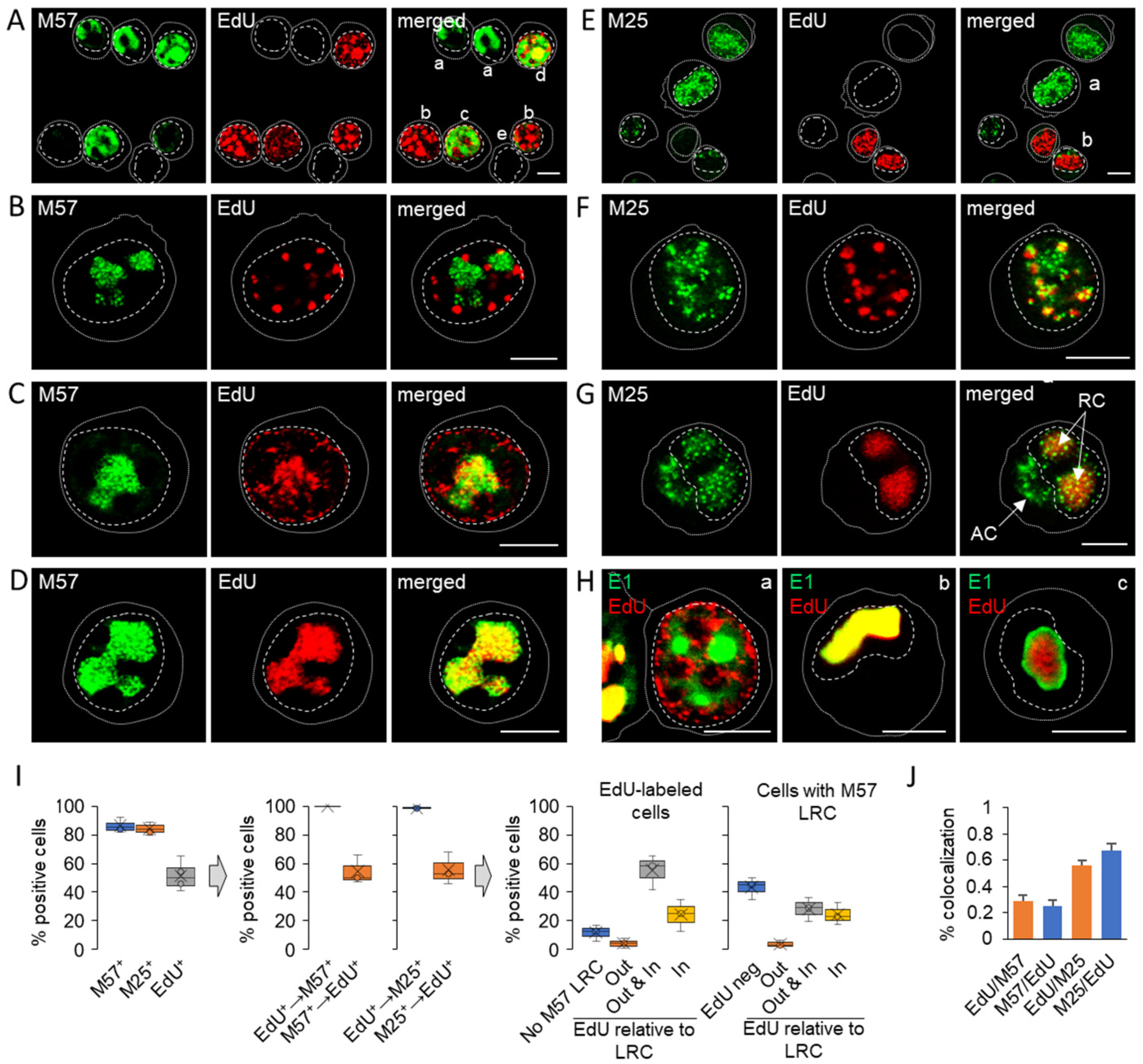
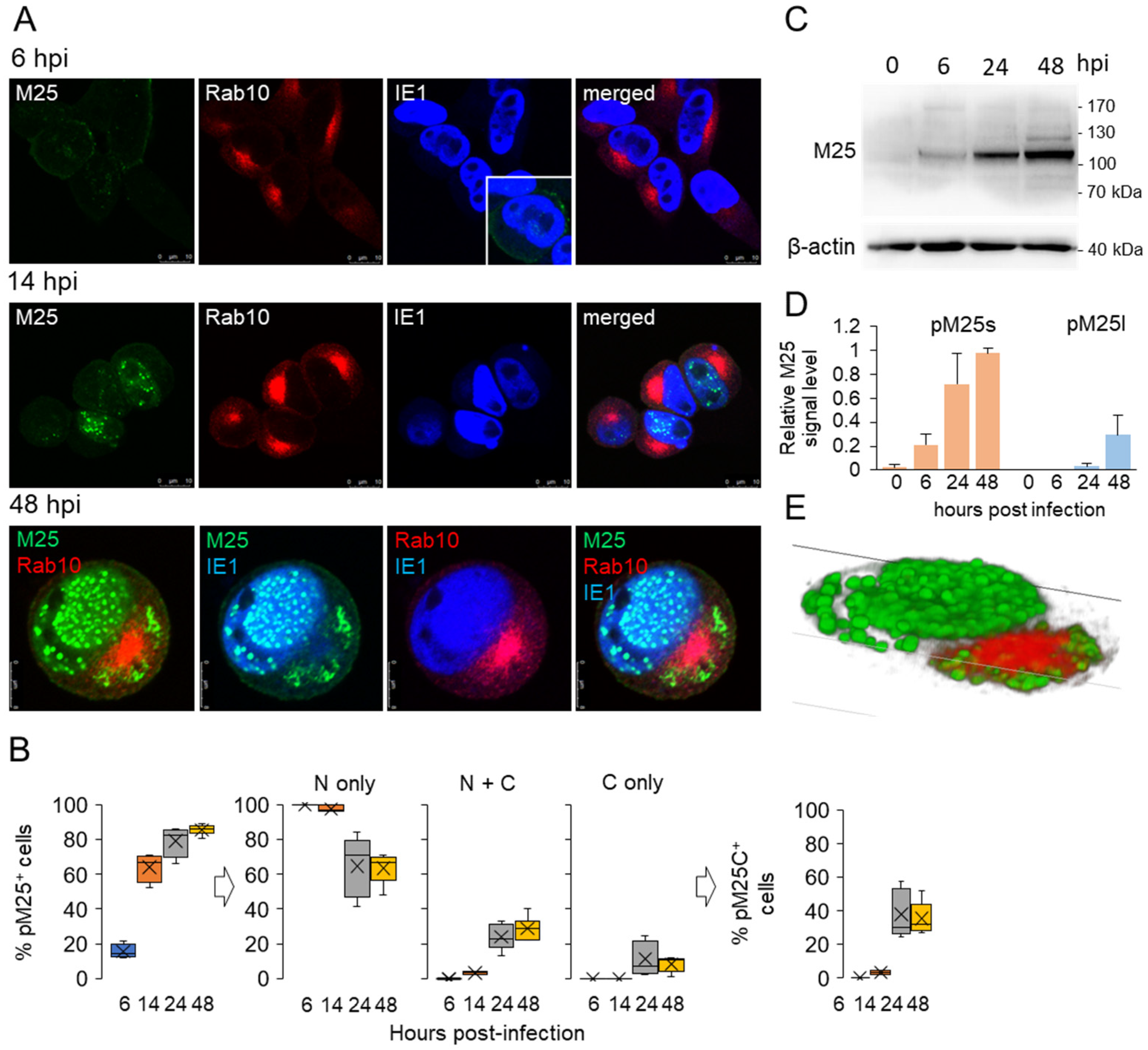
3.3. Nuclear and Cytoplasmic Membraneless Assemblies of pM25
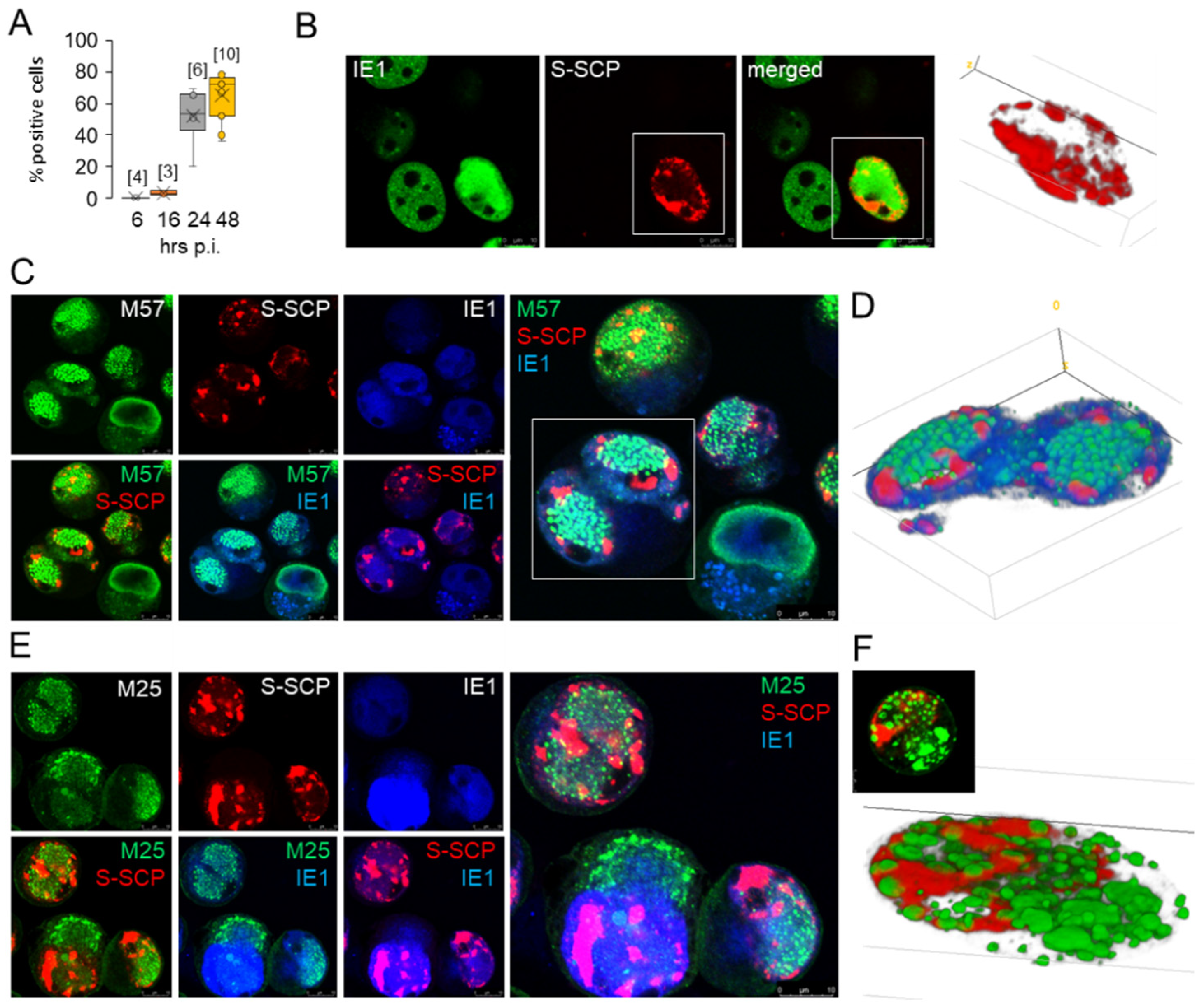
3.4. Nucleocapsid Assembly Sites and Nuclear Assemblies of the Small Capsid Protein
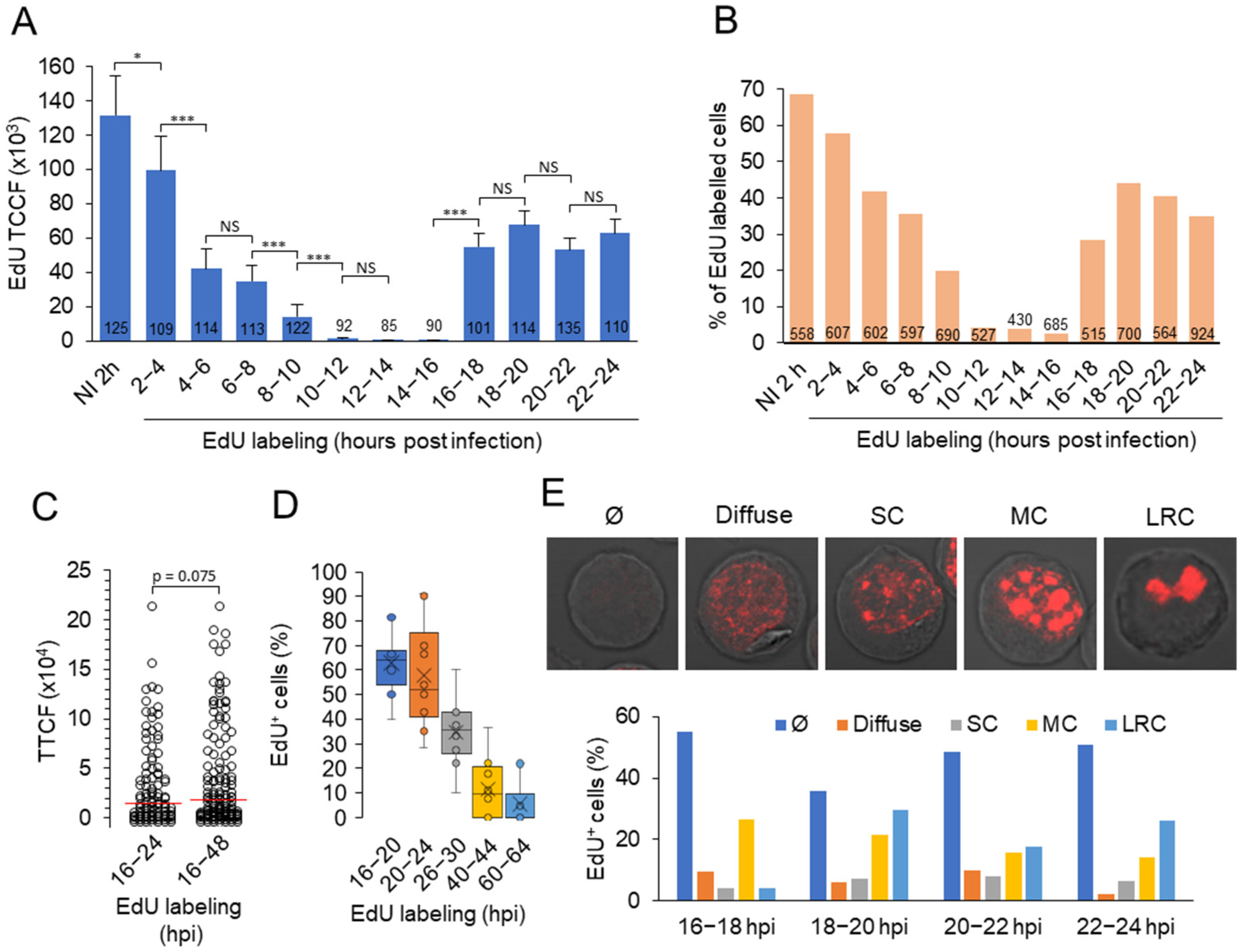
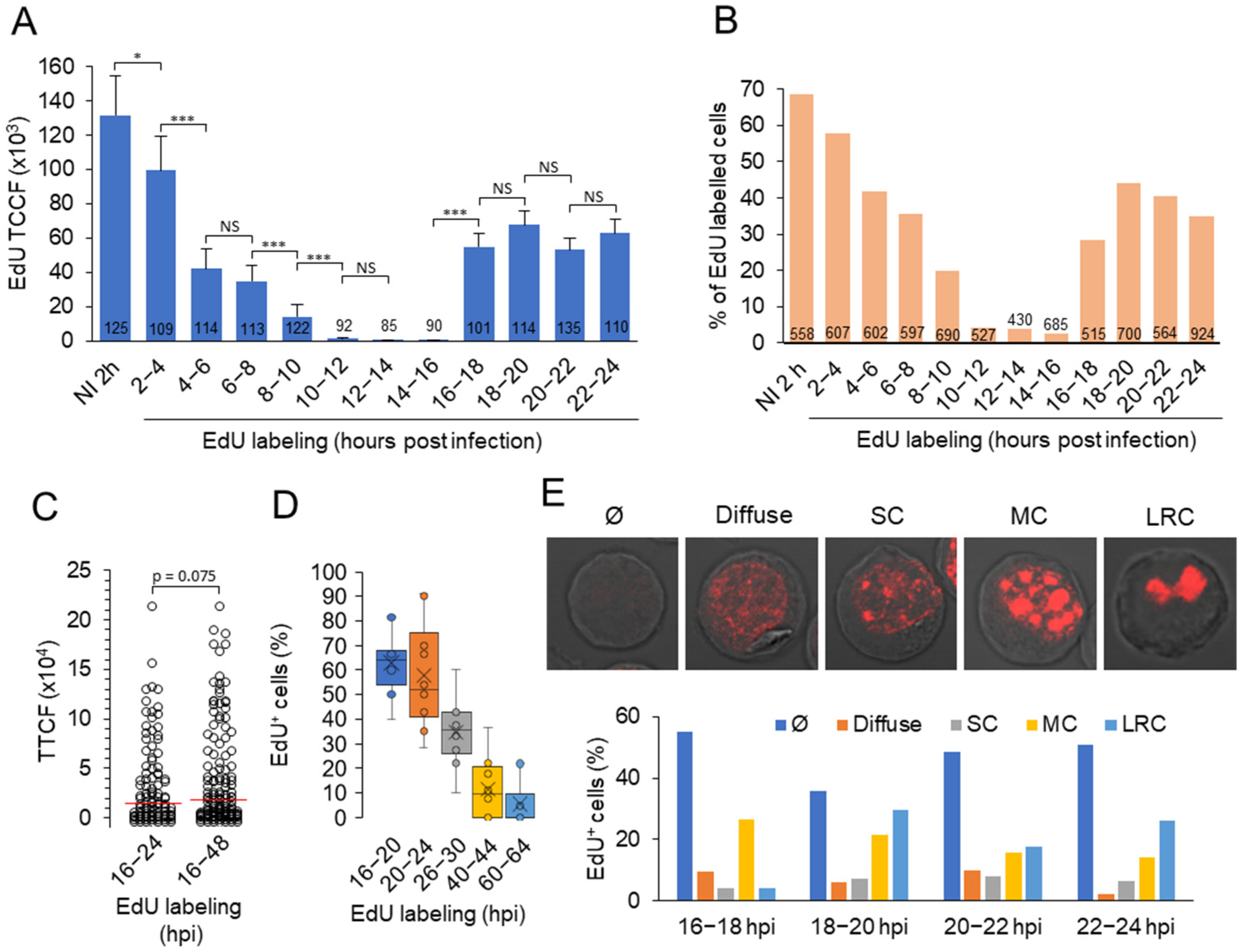
3.5. Viral DNA Synthesis and Visualization of Viral DNA within Nuclear Replication Compartments
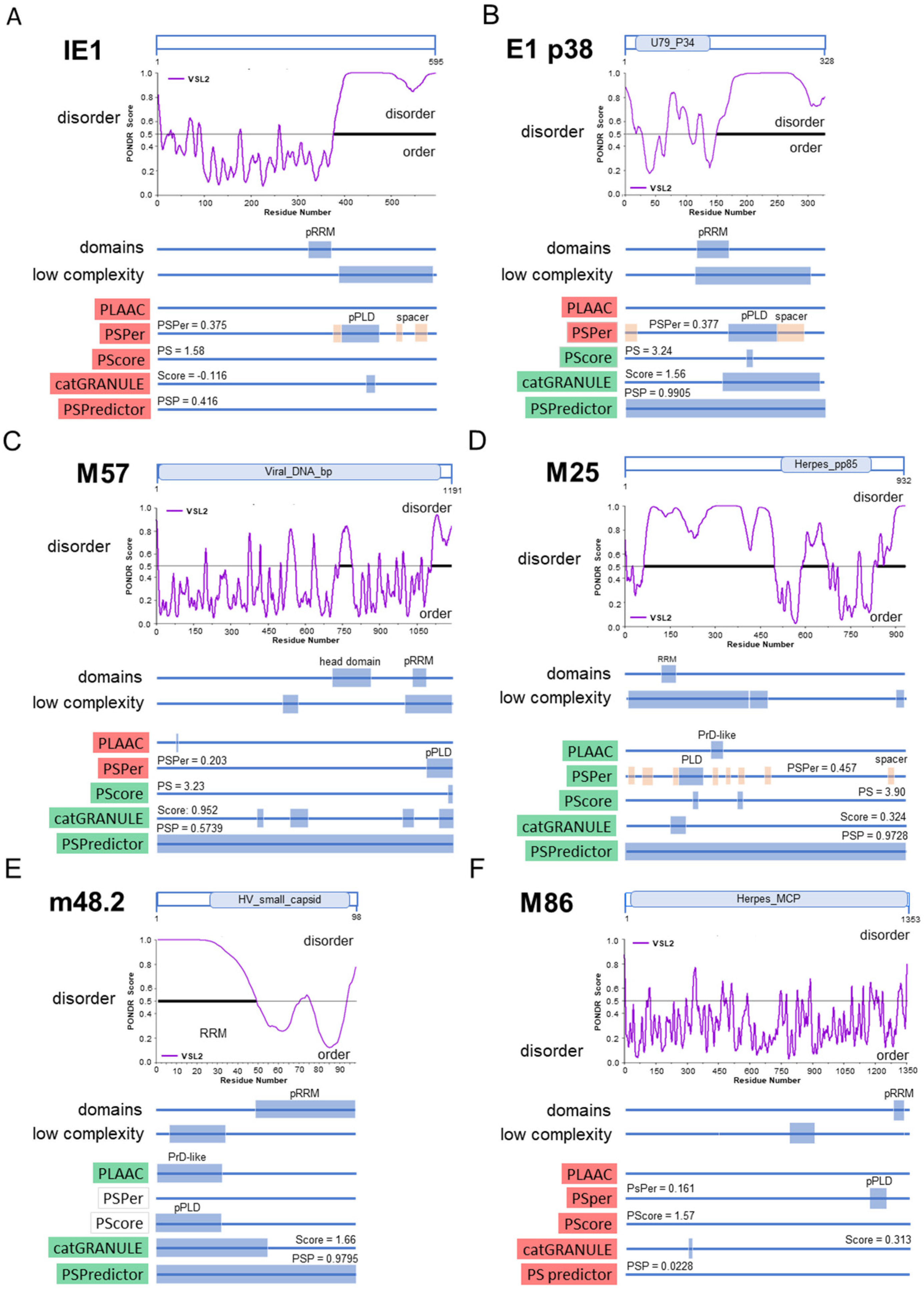
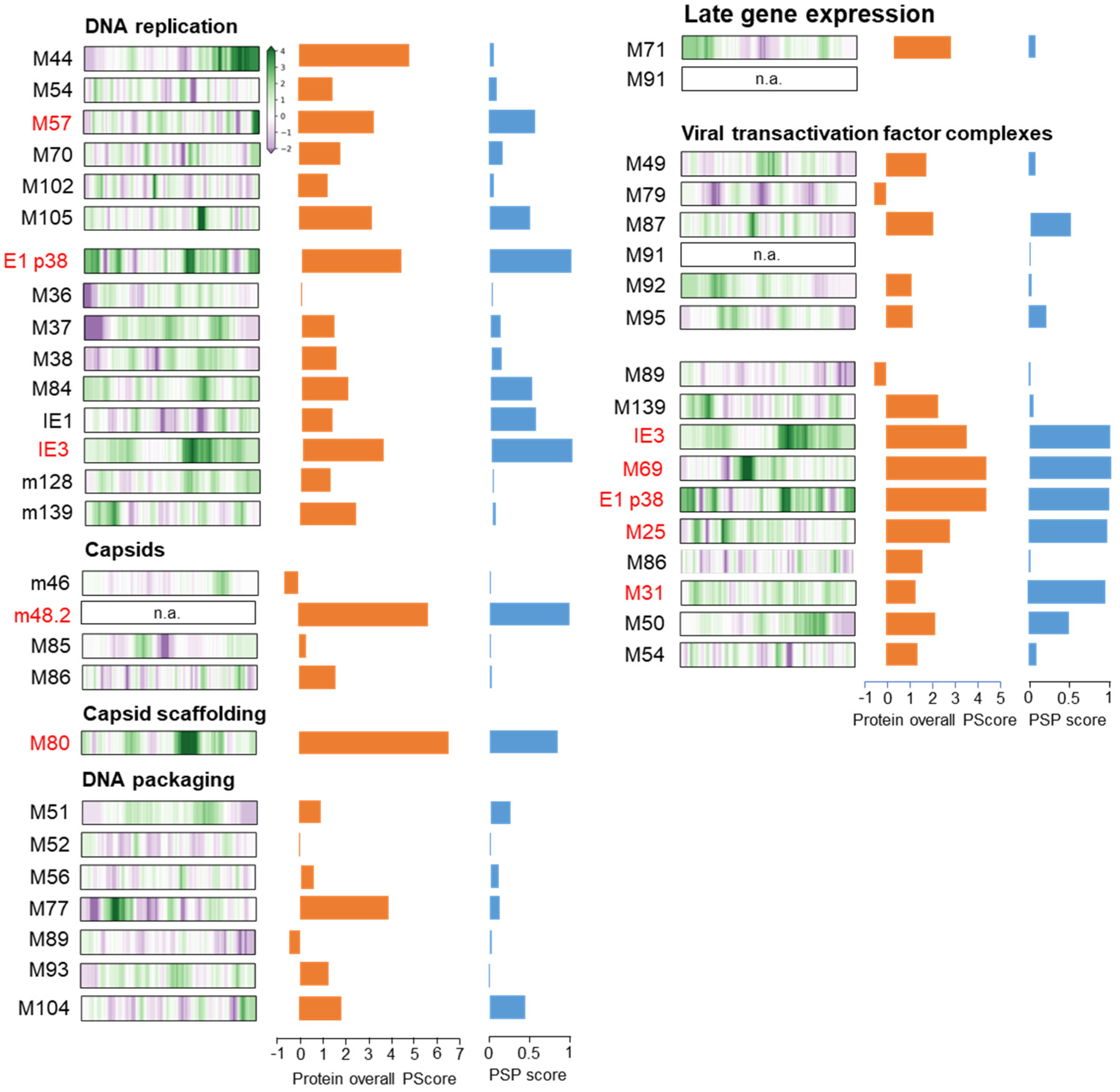
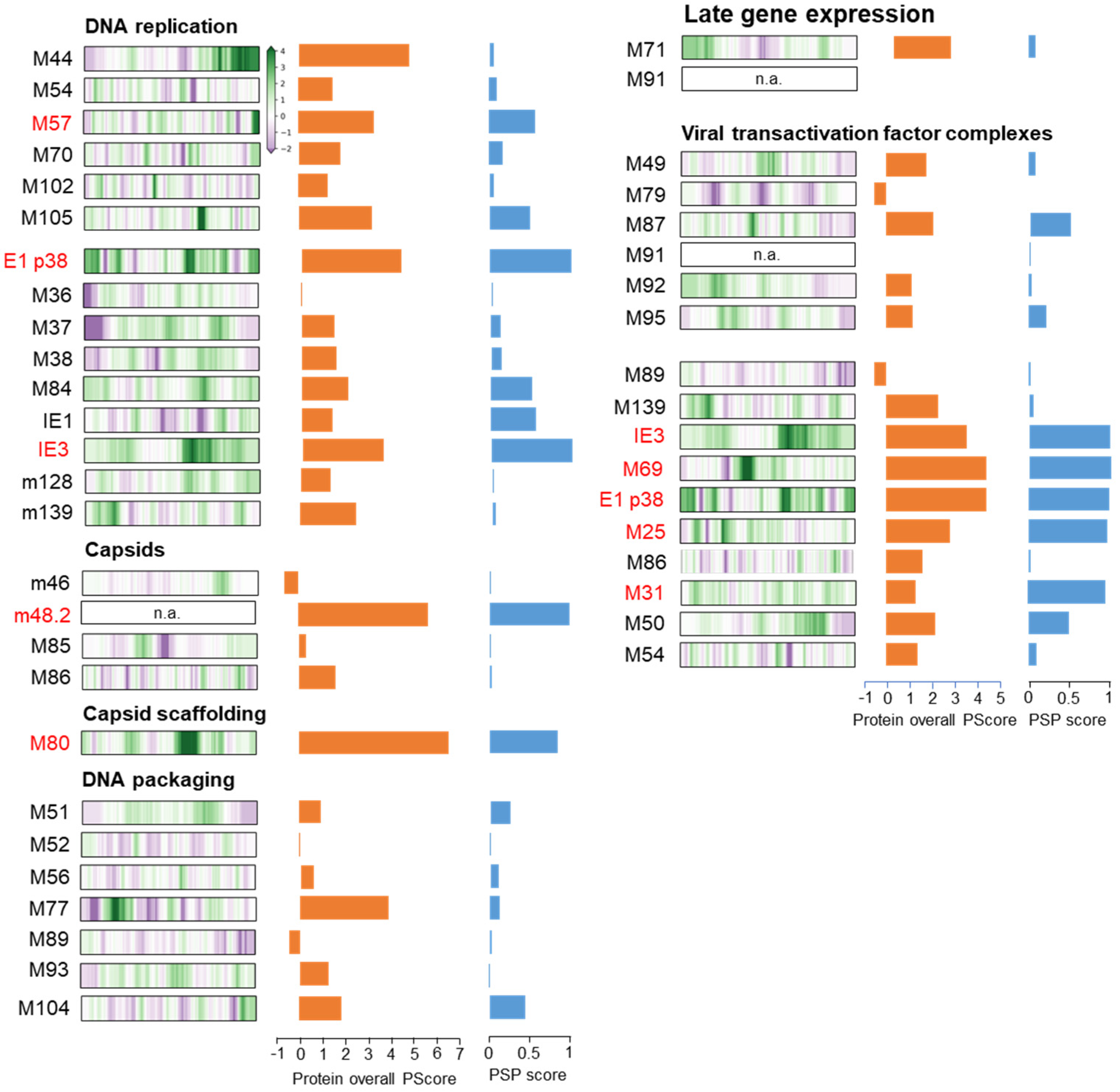
3.6. Membraneless Assemblies Correlate with LLPS Propensity of MCMV Proteins
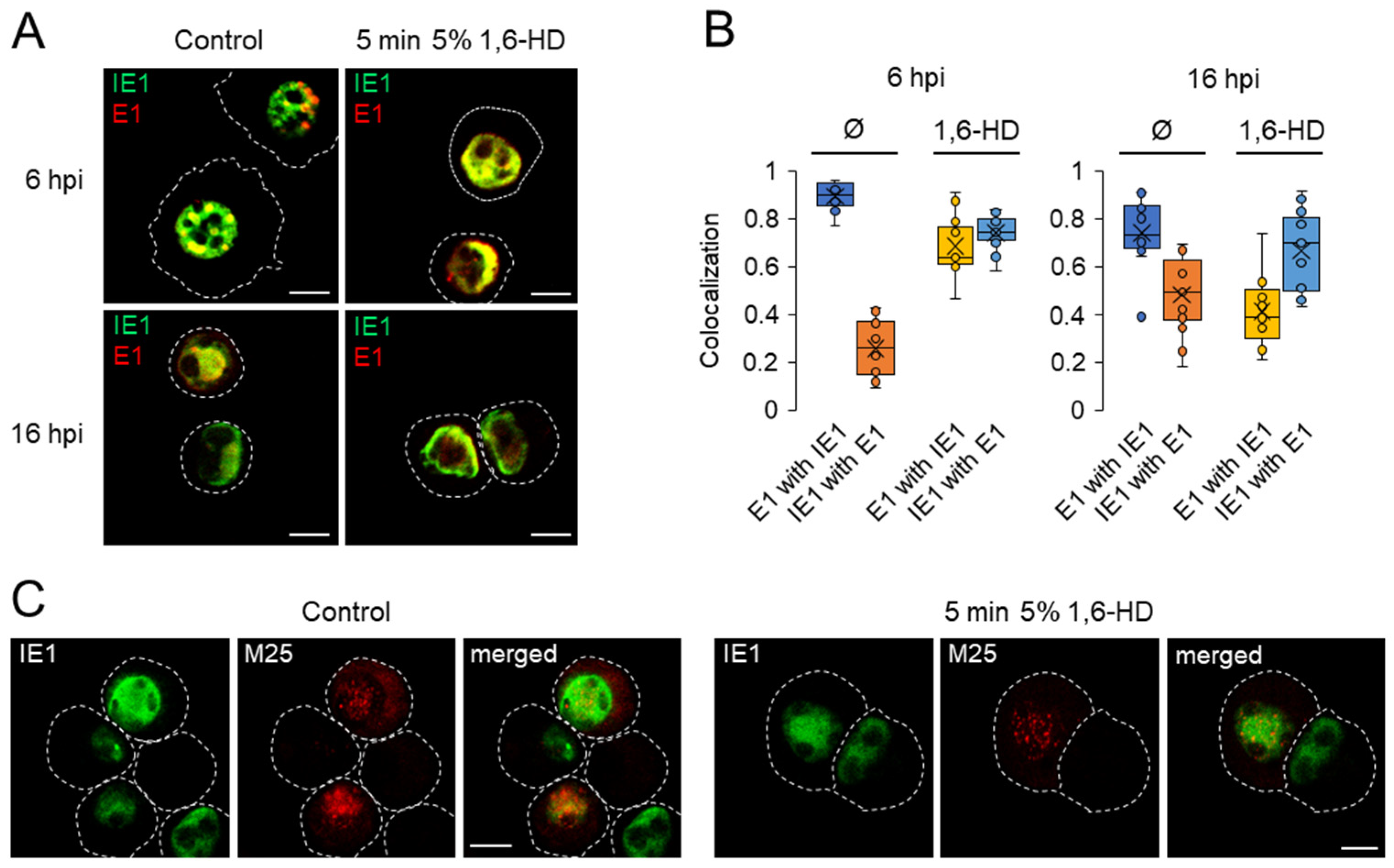
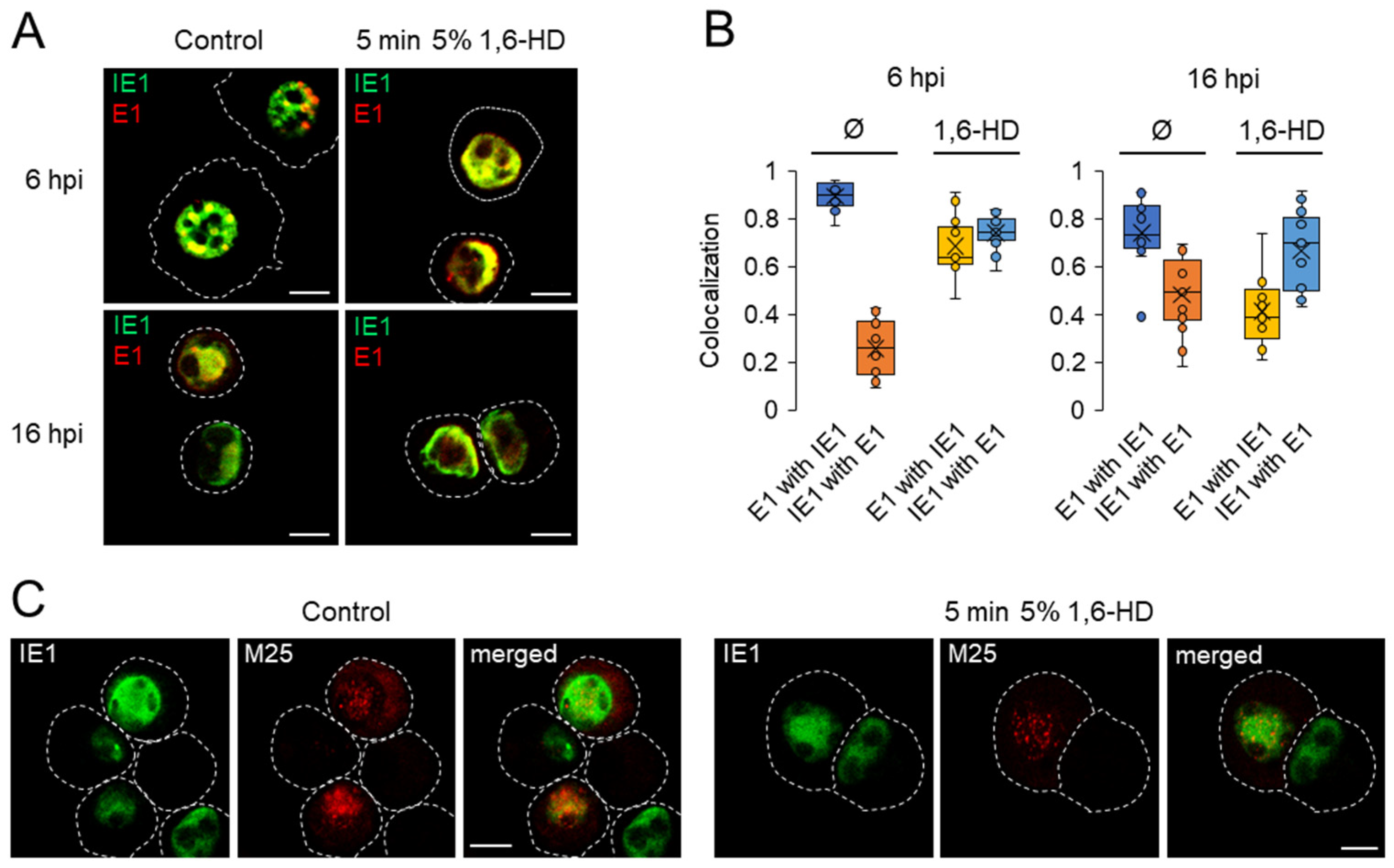
3.7. Investigation of the Physical Properties of Membraneless Nuclear Compartments MCMV-infected Cells with 1,6-Hexanediol
4. Discussion
4.1. Nuclear Compartmentalization by MLAs
4.2. Inefficient Compartmentalization of Nuclear and Cytoplasmic Virus Manufacturing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gugliesi, F.; Coscia, A.; Griffante, G.; Galitska, G.; Pasquero, S.; Albano, C.; Biolatti, M. Where Do We Stand after Decades of Studying Human Cytomegalovirus? Microorganisms 2020, 8, 685. [Google Scholar] [CrossRef]
- Griffiths, P.; Reeves, M. Pathogenesis of Human Cytomegalovirus in the Immunocompromised Host. Nat. Rev. Microbiol. 2021, 19, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Kabani, N.; Ross, S.A. Congenital Cytomegalovirus Infection. J. Infect. Dis. 2020, 221, S9–S14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wofford, A.S.; McCusker, I.; Green, J.C.; Vensko, T.A.; Pellett, P.E. Betaherpesvirus Assembly and Egress: Recent Advances Illuminate the Path, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; Volume 108, ISBN 9780128207611. [Google Scholar]
- Sanchez, V.; Britt, W. Human Cytomegalovirus Egress: Overcoming Barriers and Co-Opting Cellular Functions. Viruses 2022, 14, 15. [Google Scholar] [CrossRef]
- Charman, M.; Weitzman, M.D. Replication Compartments of DNA Viruses in the Nucleus: Location, Location, Location. Viruses 2020, 12, 151. [Google Scholar] [CrossRef] [Green Version]
- Caragliano, E.; Brune, W.; Bosse, J.B. Herpesvirus Replication Compartments: Dynamic Biomolecular Condensates? Viruses 2022, 14, 960. [Google Scholar] [CrossRef]
- Tandon, R.; Mocarski, E.S.; Conway, J.F. The A, B, Cs of Herpesvirus Capsids. Viruses 2015, 7, 899–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, A.S.; Peeples, W.B.; Rosen, M.K. A Framework for Understanding the Functions of Biomolecular Condensates across Scales. Nat. Rev. Mol. Cell Biol. 2021, 22, 215–235. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Erdos, G.; Szabó, B.; Schád, É.; Tantos, Á.; Abukhairan, R.; Horváth, T.; Murvai, N.; Kovács, O.P.; Kovács, M.; et al. PhaSePro: The Database of Proteins Driving Liquid–Liquid Phase Separation. Nucleic Acids Res. 2020, 48, D360–D367. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Etibor, T.A.; Yamauchi, Y.; Amorim, M.J. Liquid Biomolecular Condensates and Viral Lifecycles: Review and Perspectives. Viruses 2021, 13, 366. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.A.; Lloyd, M.L. A Review of Murine Cytomegalovirus as a Model for Human Cytomegalovirus Disease—Do Mice Lie? Int. J. Mol. Sci. 2021, 22, 214. [Google Scholar] [CrossRef] [PubMed]
- Lučin, P.; Jug Vučko, N.; Karleuša, L.; Mahmutefendić Lučin, H.; Blagojević Zagorac, G.; Lisnić, B.; Pavišić, V.; Marcelić, M.; Grabušić, K.; Brizić, I.; et al. Cytomegalovirus Generates Assembly Compartment in the Early Phase of Infection by Perturbation of Host-Cell Factors Recruitment at the Early Endosome/Endosomal Recycling Compartment/Trans-Golgi Interface. Front. Cell Dev. Biol. 2020, 8, 914. [Google Scholar] [CrossRef]
- Mahmutefendić Lučin, H.; Blagojević Zagorac, G.; Marcelić, M.; Lučin, P. Host Cell Signatures of the Envelopment Site within Beta-Herpes Virions. Int. J. Mol. Sci. 2022, 23, 9994. [Google Scholar] [CrossRef] [PubMed]
- Kutle, I.; Sengstake, S.; Templin, C.; Glaß, M.; Kubsch, T.; Keyser, K.A.; Binz, A.; Bauerfeind, R.; Sodeik, B.; Čičin-Šain, L.; et al. The M25 Gene Products Are Critical for the Cytopathic Effect of Mouse Cytomegalovirus. Sci. Rep. 2017, 7, 15588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutle, I.; Szymańska-de Wijs, K.M.; Bogdanow, B.; Cuvalo, B.; Steinbrück, L.; Jonjić, S.; Wagner, K.; Niedenthal, R.; Selbach, M.; Wiebusch, L.; et al. Murine Cytomegalovirus M25 Proteins Sequester the Tumor Suppressor Protein P53 in Nuclear Accumulations. J. Virol. 2020, 94, e00574-20. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Li, L.; Maul, G.G. Mouse Cytomegalovirus Early M112/113 Proteins Control the Repressive Effect of IE3 on the Major Immediate-Early Promoter. J. Virol. 2005, 79, 257–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciocco-Schmitt, G.M.; Karabekian, Z.; Godfrey, E.W.; Stenberg, R.M.; Campbell, A.E.; Kerry, J.A. Identification and Characterization of Novel Murine Cytomegalovirus M112–113 (E1) Gene Products. Virology 2002, 294, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Bosse, J.B.; Bauerfeind, R.; Popilka, L.; Marcinowski, L.; Taeglich, M.; Jung, C.; Striebinger, H.; von Einem, J.; Gaul, U.; Walther, P.; et al. A Beta-Herpesvirus with Fluorescent Capsids to Study Transport in Living Cells. PLoS ONE 2012, 7, e40585. [Google Scholar] [CrossRef] [Green Version]
- Martínez, F.P.; Cruz Cosme, R.S.; Tang, Q. Murine Cytomegalovirus Major Immediate-Early Protein 3 Interacts with Cellular and Viral Proteins in Viral DNA Replication Compartments and Is Important for Early Gene Activation. J. Gen. Virol. 2010, 91, 2664–2676. [Google Scholar] [CrossRef] [PubMed]
- Marcinowski, L.; Lidschreiber, M.; Windhager, L.; Rieder, M.; Bosse, J.B.; Rädle, B.; Bonfert, T.; Györy, I.; de Graaf, M.; da Costa, O.P.; et al. Real-Time Transcriptional Profiling of Cellular and Viral Gene Expression during Lytic Cytomegalovirus Infection. PLoS Pathog. 2012, 8, 6–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucin, P.; Jonjic, S. Cytomegalovirus Replication Cycle: An Overview. Period. Biol. 1995, 97, 13–22. [Google Scholar]
- Štimac, I.; Vučko, N.J.; Zagorac, G.B.; Marcelić, M.; Lučin, H.M.; Lučin, P. Dynamin Inhibitors Prevent the Establishment of the Cytomegalovirus Assembly Compartment in the Early Phase of Infection. Life 2021, 11, 876. [Google Scholar] [CrossRef]
- Marcelić, M.; Mahmutefendić Lučin, H.; Jurak Begonja, A.; Blagojević Zagorac, G.; Juranić Lisnić, V.; Lučin, P. Endosomal Phosphatidylinositol-3-Phosphate-Associated Functions Are Dispensable for Establishment of the Cytomegalovirus Pre-Assembly Compartment but Essential for the Virus Growth. Life 2021, 11, 859. [Google Scholar] [CrossRef] [PubMed]
- Karleuša, L.; Mahmutefendić, H.; Tomaš, M.I.; Zagorac, G.B.; Lučin, P. Landmarks of Endosomal Remodeling in the Early Phase of Cytomegalovirus Infection. Virology 2018, 515, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Bühler, B.; Keil, G.M.; Weiland, F.; Koszinowski, U.H. Characterization of the Murine Cytomegalovirus Early Transcription Unit E1 That Is Induced by Immediate-Early Proteins. J. Virol. 1990, 64, 1907–1919. [Google Scholar] [CrossRef] [Green Version]
- Lacaze, P.; Forster, T.; Ross, A.; Kerr, L.E.; Salvo-Chirnside, E.; Lisnic, V.J.; Lopez-Campos, G.H.; Garcia-Ramirez, J.J.; Messerle, M.; Trgovcich, J.; et al. Temporal Profiling of the Coding and Noncoding Murine Cytomegalovirus Transcriptomes. J. Virol. 2011, 85, 6065–6076. [Google Scholar] [CrossRef] [Green Version]
- Karleuša, L.; Lučin, H.M.; Zagorac, G.B.; Lučin, P. Cytoplasmic Virion Assembly Compartment of Betaherpesviruses. Period. Biol. 2020, 121–122, 97–106. [Google Scholar] [CrossRef]
- Lučin, P.; Kareluša, L.; Blagojević Zagorac, G.; Mahmutefendić Lučin, H.; Pavišić, V.; Jug Vučko, N.; Lukanović Jurić, S.; Marcelić, M.; Lisnić, B.; Jonjić, S. Cytomegaloviruses Exploit Recycling Rab Proteins in the Sequential Establishment of the Assembly Compartment. Front. Cell Dev. Biol. 2018, 6, 165. [Google Scholar] [CrossRef] [Green Version]
- Pancsa, R.; Vranken, W.; Mészáros, B. Computational Resources for Identifying and Describing Proteins Driving Liquid-Liquid Phase Separation. Brief. Bioinform. 2021, 22, bbaa408. [Google Scholar] [CrossRef] [PubMed]
- Brizić, I.; Lisnić, B.; Brune, W.; Hengel, H.; Jonjić, S. Cytomegalovirus Infection: Mouse Model. Curr. Protoc. Immunol. 2018, 122, e51. [Google Scholar] [CrossRef]
- Crnković-Mertens, I.; Messerle, M.; Milotić, I.; Szepan, U.; Kučić, N.; Krmpotić, A.; Jonjić, S.; Koszinowski, U.H. Virus Attenuation after Deletion of the Cytomegalovirus Fc Receptor Gene Is Not Due to Antibody Control. J. Virol. 1998, 72, 1377–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kučić, N.; Mahmutefendić, H.; Lučin, P. Inhibition of Protein Kinases C Prevents Murine Cytomegalovirus Replication. J. Gen. Virol. 2005, 86, 2153–2161. [Google Scholar] [CrossRef] [PubMed]
- Budt, M.; Niederstadt, L.; Valchanova, R.S.; Jonjić, S.; Brune, W. Specific Inhibition of the PKR-Mediated Antiviral Response by the Murine Cytomegalovirus Proteins M142 and M143. J. Virol. 2009, 83, 1260–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovis, T.L.; Brlic, P.K.; Kaynan, N.; Lisnic, V.J.; Brizic, I.; Jordan, S.; Tomic, A.; Kvestak, D.; Babic, M.; Tsukerman, P.; et al. Inflammatory Monocytes and NK Cells Play a Crucial Role in DNAM-1-Dependent Control of Cytomegalovirus Infection. J. Exp. Med. 2016, 213, 1835–1850. [Google Scholar] [CrossRef] [Green Version]
- Reusch, U.; Muranyi, W.; Lucin, P.; Burgert, H.-G.; Hengel, H.; Koszinowski, U.H. A Cytomegalovirus Glycoprotein Re-Routes MHC Class I Complexes to Lysosomes for Degradation. EMBO J. 1999, 18, 1081–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salic, A.; Mitchison, T.J. A Chemical Method for Fast and Sensitive Detection of DNA Synthesis in Vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 2415–2420. [Google Scholar] [CrossRef] [Green Version]
- McCloy, R.A.; Rogers, S.; Caldon, C.E.; Lorca, T.; Castro, A. Burgess, A. Partial Inhibition of Cdk1 in G2 Phase Overrides the SAC and Decouples Mitotic Events. Cell Cycle 2014, 13, 1400–1412. [Google Scholar] [CrossRef] [Green Version]
- Bolte, S. Cordelières, F.P. A Guided Tour into Subcellular Colocalization Analysis in Light Microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically Disordered Protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wootton, J.C.; Federhen, S. Statistics of Local Complexity in Amino Acid Sequences and Sequence Databases. Comput. Chem 1993, 17, 149–163. [Google Scholar] [CrossRef]
- Lancaster, A.K.; Nutter-Upham, A.; Lindquist, S.; King, O.D. PLAAC: A Web and Command-Line Application to Identify Proteins with Prion-like Amino Acid Composition. Bioinformatics 2014, 30, 2501–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlando, G.; Raimondi, D.; Tabaro, F.; Codicè, F.; Moreau, Y.; Vranken, W.F. Computational Identification of Prion-like RNA-Binding Proteins That Form Liquid Phase-Separated Condensates. Bioinformatics 2019, 35, 4617–4623. [Google Scholar] [CrossRef] [PubMed]
- Vernon, R.M.C.; Chong, P.A.; Tsang, B.; Kim, T.H.; Bah, A.; Farber, P.; Lin, H.; Forman-Kay, J.D. Pi-Pi Contacts Are an Overlooked Protein Feature Relevant to Phase Separation. Elife 2018, 7, e31486. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, B.; Gotor, N.L.; Dhar, R.; Cirillo, D.; Baldrighi, M.; Tartaglia, G.G.; Lehner, B. A Concentration-Dependent Liquid Phase Separation Can Cause Toxicity upon Increased Protein Expression. Cell Rep. 2016, 16, 222–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotor, N.L.; Armaos, A.; Calloni, G.; Burgas, M.T.; Martin Vabulas, R.; de Groot, N.S.; Tartaglia, G.G. RNA-Binding and Prion Domains: The Yin and Yang of Phase Separation. Nucleic Acids Res. 2020, 48, 9491–9504. [Google Scholar] [CrossRef]
- Chu, X.; Sun, T.; Li, Q.; Xu, Y.; Zhang, Z.; Lai, L.; Pei, J. Prediction of Liquid–Liquid Phase Separating Proteins Using Machine Learning. BMC Bioinform. 2022, 23, 72. [Google Scholar] [CrossRef]
- Busche, A.; Angulo, A.; Kay-Jackson, P.; Ghazal, P.; Messerle, M. Phenotypes of Major Immediate-Early Gene Mutants of Mouse Cytomegalovirus. Med. Microbiol. Immunol. 2008, 197, 233–240. [Google Scholar] [CrossRef]
- Wright, D.A.; Spector, D.H. Posttranscriptional Regulation of a Class of Human Cytomegalovirus Phosphoproteins Encoded by an Early Transcription Unit. J. Virol. 1989, 63, 3117–3127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Cosme, R.; Armstrong, N.; Tang, Q. One of the Triple Poly(A) Signals in the M112-113 Gene Is Important and Sufficient for Stabilizing the M112-113 MRNA and the Replication of Murine Cytomegalovirus. Viruses 2020, 12, 954. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-E.; Ahn, J.-H. Role of the Specific Interaction of UL112-113 P84 with UL44 DNA Polymerase Processivity Factor in Promoting DNA Replication of Human Cytomegalovirus. J. Virol. 2010, 84, 8409–8421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caragliano, E.; Bonazza, S.; Frascaroli, G.; Tang, J.; Soh, T.K.; Grünewald, K.; Bosse, J.B.; Brune, W. Human Cytomegalovirus Forms Phase-Separated Compartments at Viral Genomes to Facilitate Viral Replication. Cell Rep. 2022, 38, 110469. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Speiseder, T.; Dobner, T.; Gonzalez, R.A. DNA Virus Replication Compartments. J. Virol. 2014, 88, 1404–1420. [Google Scholar] [CrossRef] [Green Version]
- Penfold, M.E.T.; Mocarski, E.S. Formation of Cytomegalovirus DNA Replication Compartments Defined by Localization of Viral Proteins and DNA Synthesis. Virology 1997, 239, 46–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strang, B.L.; Boulant, S.; Chang, L.; Knipe, D.M.; Kirchhausen, T.; Coen, D.M. Human Cytomegalovirus UL44 Concentrates at the Periphery of Replication Compartments, the Site of Viral DNA Synthesis. J. Virol. 2012, 86, 2089–2095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.-H.; Jang, W.-J.; Hayward, G.S. The Human Cytomegalovirus IE2 and UL112-113 Proteins Accumulate in Viral DNA Replication Compartments That Initiate from the Periphery of Promyelocytic Leukemia Protein-Associated Nuclear Bodies (PODs or ND10). J. Virol. 1999, 73, 10458–10471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira, A.P.; Glauser, D.L.; Laimbacher, A.S.; Strasser, R.; Schraner, E.M.; Wild, P.; Ziegler, U.; Breakefield, X.O.; Ackermann, M.; Fraefel, C. Live Visualization of Herpes Simplex Virus Type 1 Compartment Dynamics. J. Virol. 2008, 82, 4974–4990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kattenhorn, L.M.; Mills, R.; Wagner, M.; Lomsadze, A.; Makeev, V.; Borodovsky, M.; Ploegh, H.L.; Kessler, B.M. Identification of Proteins Associated with Murine Cytomegalovirus Virions. J. Virol. 2004, 78, 11187–11197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.A.; Carlson, M.E.; Henry, S.C.; Shanley, J.D. The Murine Cytomegalovirus M25 Open Reading Frame Encodes a Component of the Tegument. Virology 1999, 262, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, T.J.; McNamee, E.E.; Day, C.; Knipe, D.M. Herpes Simplex Virus Replication Compartments Can Form by Coalescence of Smaller Compartments. Virology 2003, 309, 232–247. [Google Scholar] [CrossRef] [Green Version]
- Holehouse, A.S.; Das, R.K.; Ahad, J.N.; Richardson, M.O.G.; Pappu, R.V. CIDER: Resources to Analyze Sequence-Ensemble Relationships of Intrinsically Disordered Proteins. Biophys. J. 2017, 112, 16–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, L.; Britt, W.J. The Interaction between the Major Capsid Protein and the Smallest Capsid Protein of Human Cytomegalovirus Is Dependent on Two Linear Sequences in the Smallest Capsid Protein. J. Virol. 2003, 77, 2730–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroschwald, S.; Maharana, S.; Simon, A. Hexanediol: A Chemical Probe to Investigate the Material Properties of Membrane-Less Compartments. Matters 2017, e201702000010. [Google Scholar] [CrossRef] [Green Version]
- Düster, R.; Kaltheuner, I.H.; Schmitz, M.; Geyer, M. 1,6-Hexanediol, Commonly Used to Dissolve Liquid-Liquid Phase Separated Condensates, Directly Impairs Kinase and Phosphatase Activities. J. Biol. Chem. 2021, 296, 1009926. [Google Scholar] [CrossRef] [PubMed]
- Lopez, N.; Camporeale, G.; Salgueiro, M.; Borkosky, S.S.; Visentín, A.; Peralta-Martinez, R.; Loureiro, M.E.; de Prat-Gay, G. Deconstructing Virus Condensation. PLoS Pathog. 2021, 17, e1009926. [Google Scholar] [CrossRef]
- Tomer, E.; Cohen, E.M.; Drayman, N.; Afriat, A.; Weitzman, M.D.; Zaritsky, A.; Kobiler, O. Coalescing Replication Compartments Provide the Opportunity for Recombination between Coinfecting Herpesviruses. FASEB J. 2019, 33, 9388–9403. [Google Scholar] [CrossRef]
- McSwiggen, D.T.; Hansen, A.S.; Teves, S.S.; Marie-Nelly, H.; Hao, Y.; Heckert, A.B.; Umemoto, K.K.; Dugast-Darzacq, C.; Tjian, R.; Darzacq, X. Evidence for DNA-Mediated Nuclear Compartmentalization Distinct from Phase Separation. Elife 2019, 8, e47098. [Google Scholar] [CrossRef] [PubMed]
- Metrick, C.M.; Koenigsberg, A.L.; Heldwein, E.E. Conserved Outer Tegument Component Ul11 from Herpes Simplex Virus 1 Is an Intrinsically Disordered, Rna-Binding Protein. mBio 2020, 11, e00810-20. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Godinez, W.J.; Kim, I.H.; Tektonidis, M.; de Lanerolle, P.; Eils, R.; Rohr, K.; Knipe, D.M. Herpesviral Replication Compartments Move and Coalesce at Nuclear Speckles to Enhance Export of Viral Late MRNA. Proc. Natl. Acad Sci. USA 2011, 108, E136–E144. [Google Scholar] [CrossRef] [Green Version]
- Seyffert, M.; Georgi, F.; Tobler, K.; Bourqui, L.; Anfossi, M.; Michaelsen, K.; Vogt, B.; Greber, U.F.; Fraefel, C. The HSV-1 Transcription Factor ICP4 Confers Liquid-Like Properties to Viral Replication Compartments. Int. J. Mol. Sci. 2021, 22, 4447. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Wang, L.; Qin, Z.; Wang, J.; Zheng, X.; Wei, L.; Zhang, X.; Zhang, X.; Liu, C.; Li, Z.; et al. Phase Separation of Epstein-Barr Virus EBNA2 and Its Coactivator EBNALP Controls Gene Expression. J. Virol. 2020, 94, e01771-19. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular Condensates: Organizers of Cellular Biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Wang, Y.-Q.; Zhao, X.-Y. Human Cytomegalovirus Primary Infection and Reactivation: Insights From Virion-Carried Molecules. Front. Microbiol. 2020, 11, 1511. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Fang, C.; Su, Y.; Liu, H.; Lang, F.; Li, X.; Chen, G.; Lu, D.; Zhou, J. Visualizing the Replicating HSV-1 Virus Using STED Super-Resolution Microscopy. Virol. J. 2016, 13, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapa, T.J.; Du, Y.; Sun, R.; Yu, D.; French, A.R. Proteomic and Phylogenetic Coevolution Analyses of PM79 and PM92 Identify Interactions with RNA Polymerase II and Delineate the Murine Cytomegalovirus Late Transcription Complex. J. Gen. Virol. 2017, 98, 242–250. [Google Scholar] [CrossRef]
- Pan, D.; Han, T.; Tang, S.; Xu, W.; Bao, Q.; Sun, Y.; Xuan, B.; Qian, Z. Murine Cytomegalovirus Protein PM91 Interacts with PM79 and Is Critical for Viral Late Gene Expression. J. Virol. 2018, 92, e00675-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Shah, S.; Atanasov, I.; Lo, P.; Liu, F.; Britt, W.J.; Zhou, Z.H. Three-Dimensional Localization of the Smallest Capsid Protein in the Human Cytomegalovirus Capsid. J. Virol. 2005, 79, 1327–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draganova, E.B.; Valentin, J.; Heldwein, E.E. The Ins and Outs of Herpesviral Capsids: Divergent Structures and Assembly Mechanisms across the Three Subfamilies. Viruses 2021, 13, 1913. [Google Scholar] [CrossRef]
- Brocchieri, L.; Kledal, T.N.; Karlin, S.; Mocarski, E.S. Predicting Coding Potential from Genome Sequence: Application to Betaherpesviruses Infecting Rats and Mice. J. Virol. 2005, 79, 7570–7596. [Google Scholar] [CrossRef] [Green Version]
- Sampaio, K.L.; Cavignac, Y.; Stierhof, Y.-D.; Sinzger, C. Human Cytomegalovirus Labeled with Green Fluorescent Protein for Live Analysis of Intracellular Particle Movements. J. Virol. 2005, 79, 2754–2767. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmutefendić Lučin, H.; Lukanović Jurić, S.; Marcelić, M.; Štimac, I.; Viduka, I.; Blagojević Zagorac, G.; Lisnić, B.; Ruzsics, Z.; Lučin, P. Membraneless Compartmentalization of Nuclear Assembly Sites during Murine Cytomegalovirus Infection. Viruses 2023, 15, 766. https://doi.org/10.3390/v15030766
Mahmutefendić Lučin H, Lukanović Jurić S, Marcelić M, Štimac I, Viduka I, Blagojević Zagorac G, Lisnić B, Ruzsics Z, Lučin P. Membraneless Compartmentalization of Nuclear Assembly Sites during Murine Cytomegalovirus Infection. Viruses. 2023; 15(3):766. https://doi.org/10.3390/v15030766
Chicago/Turabian StyleMahmutefendić Lučin, Hana, Silvija Lukanović Jurić, Marina Marcelić, Igor Štimac, Ivona Viduka, Gordana Blagojević Zagorac, Berislav Lisnić, Zsolt Ruzsics, and Pero Lučin. 2023. "Membraneless Compartmentalization of Nuclear Assembly Sites during Murine Cytomegalovirus Infection" Viruses 15, no. 3: 766. https://doi.org/10.3390/v15030766
APA StyleMahmutefendić Lučin, H., Lukanović Jurić, S., Marcelić, M., Štimac, I., Viduka, I., Blagojević Zagorac, G., Lisnić, B., Ruzsics, Z., & Lučin, P. (2023). Membraneless Compartmentalization of Nuclear Assembly Sites during Murine Cytomegalovirus Infection. Viruses, 15(3), 766. https://doi.org/10.3390/v15030766