
BAK-Mediated Pyroptosis Promotes Japanese Encephalitis Virus Proliferation in Porcine Kidney 15 Cells
 ,
,
Abstract
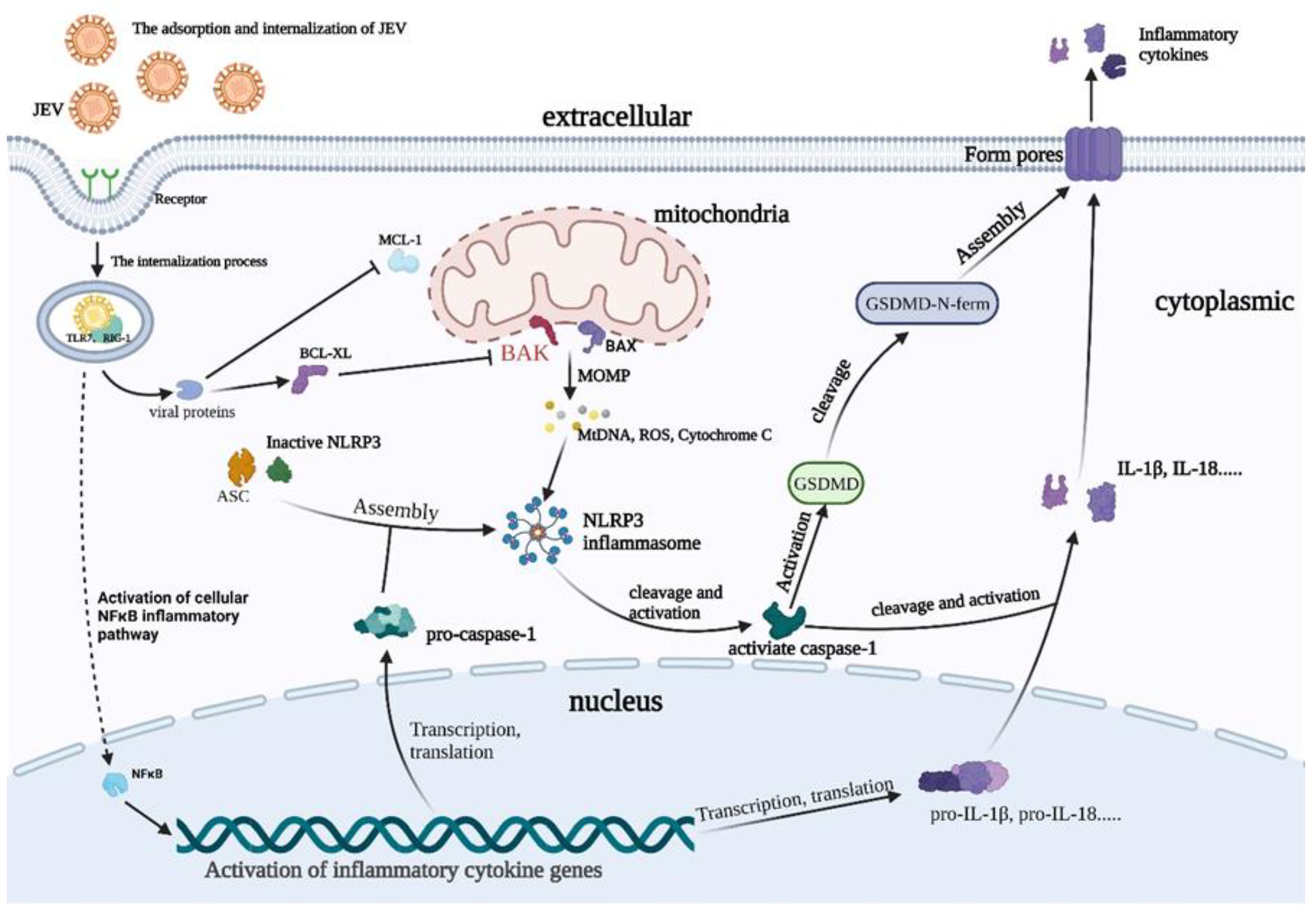
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus Infection
2.2. Plasmids and Antibodies
2.3. Construction and Characterization of BAK Knockdown PK15 Cell Line by CRISPR/Cas9
2.4. Reactive Oxygen Species Assay
2.5. Complementation and Overexpression Experiment of BAK Gene
2.6. EDU Cell Proliferation Assay
2.7. Detection and Analysis of the Apoptosis and Necrosis of YO-PRO-1/PI Cells
2.8. RNA Extraction and Quantitative Real-Time RT-PCR
2.9. Western Blot
2.10. Indirect Immunofluorescence (IF) Assay for the Detection of GSDMD and JEV E
2.11. Pyroptosis Inhibitor Disulfiram
2.12. Quantification and Statistical Analysis
3. Results
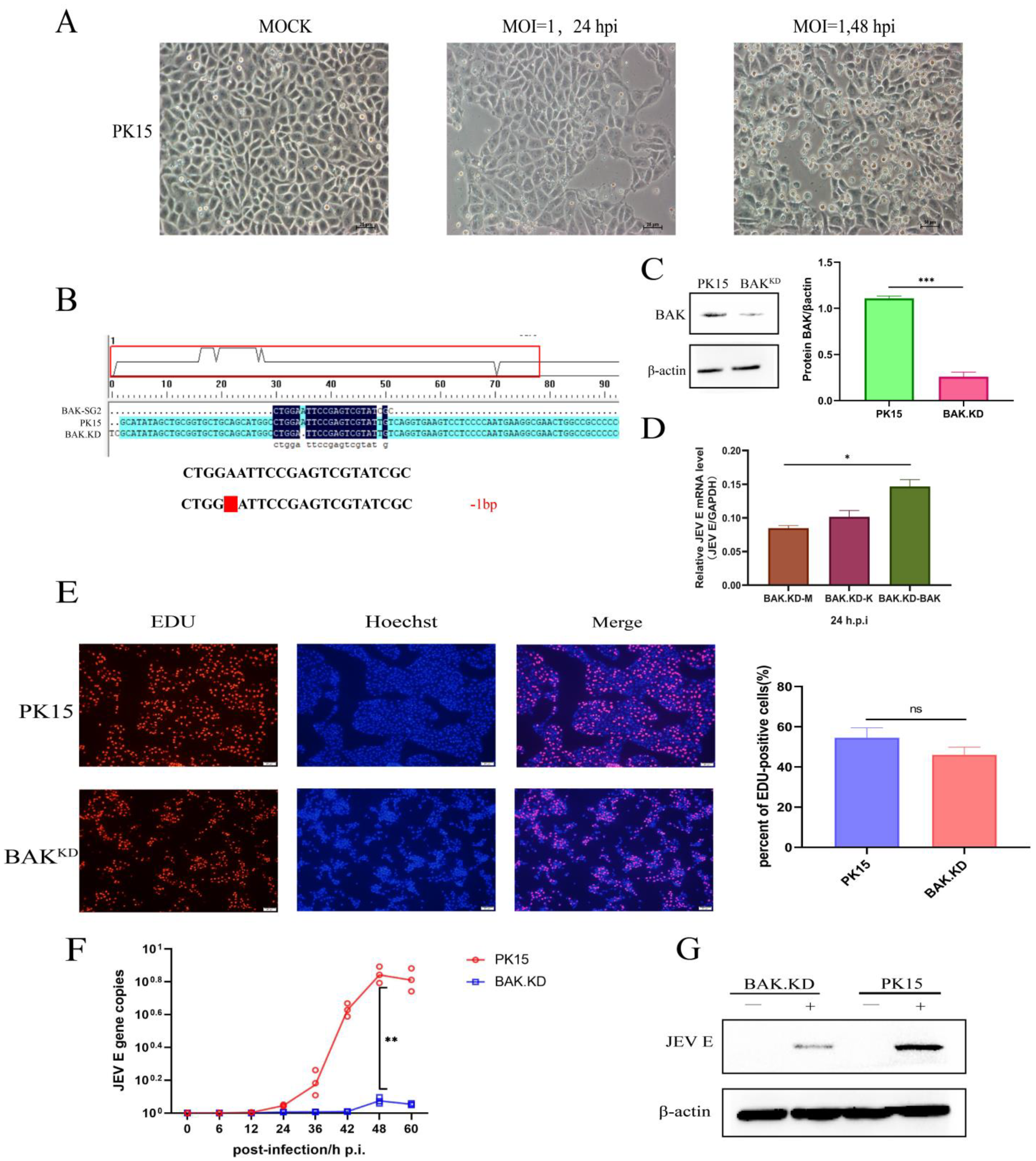
3.1. Proliferation of JEV Is Inhibited in BAK.KD Cells
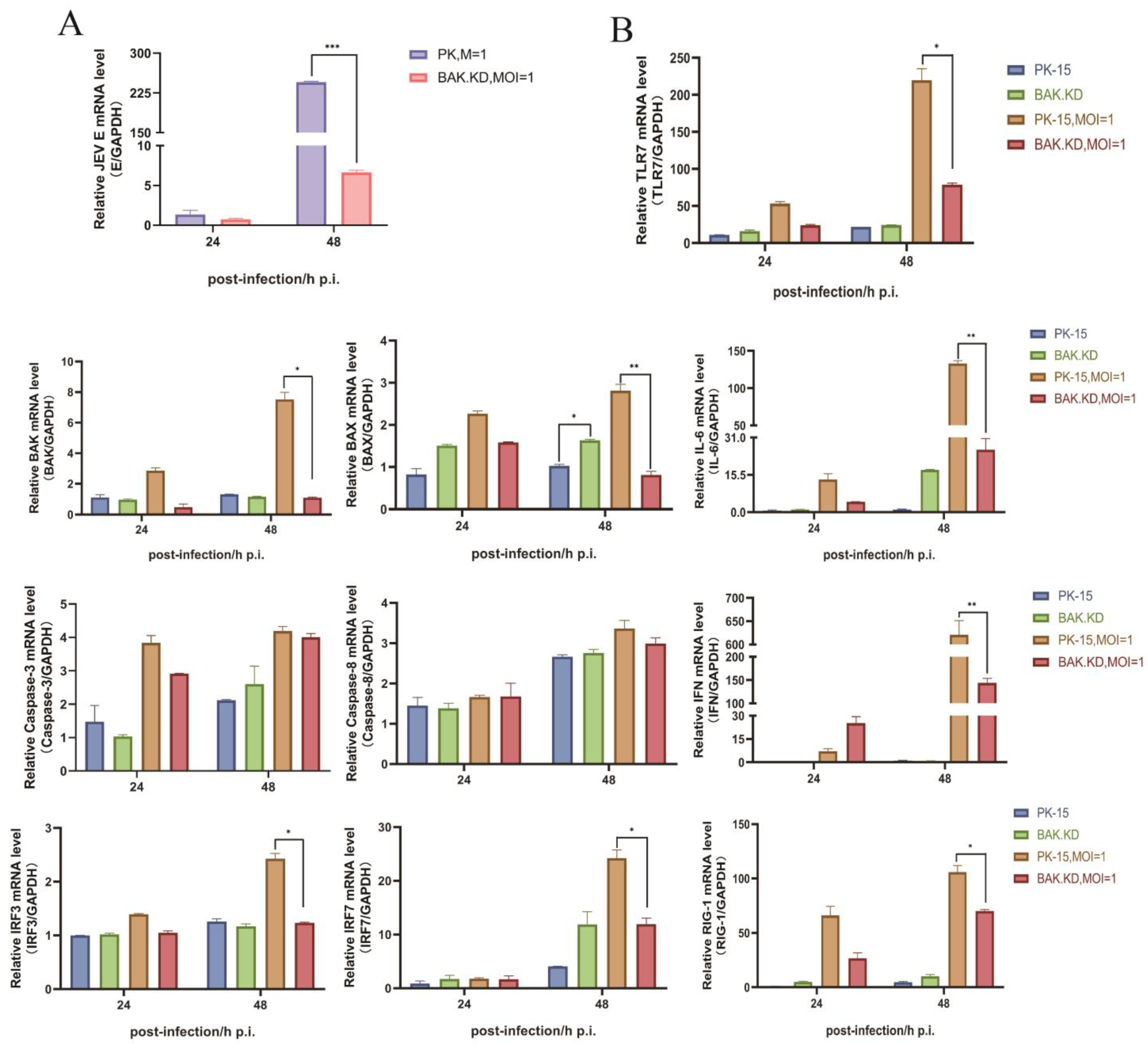
3.2. BAK-Mediated Cytokine Transcription Levels Significantly Increased in the Late Stage of JEV Proliferation
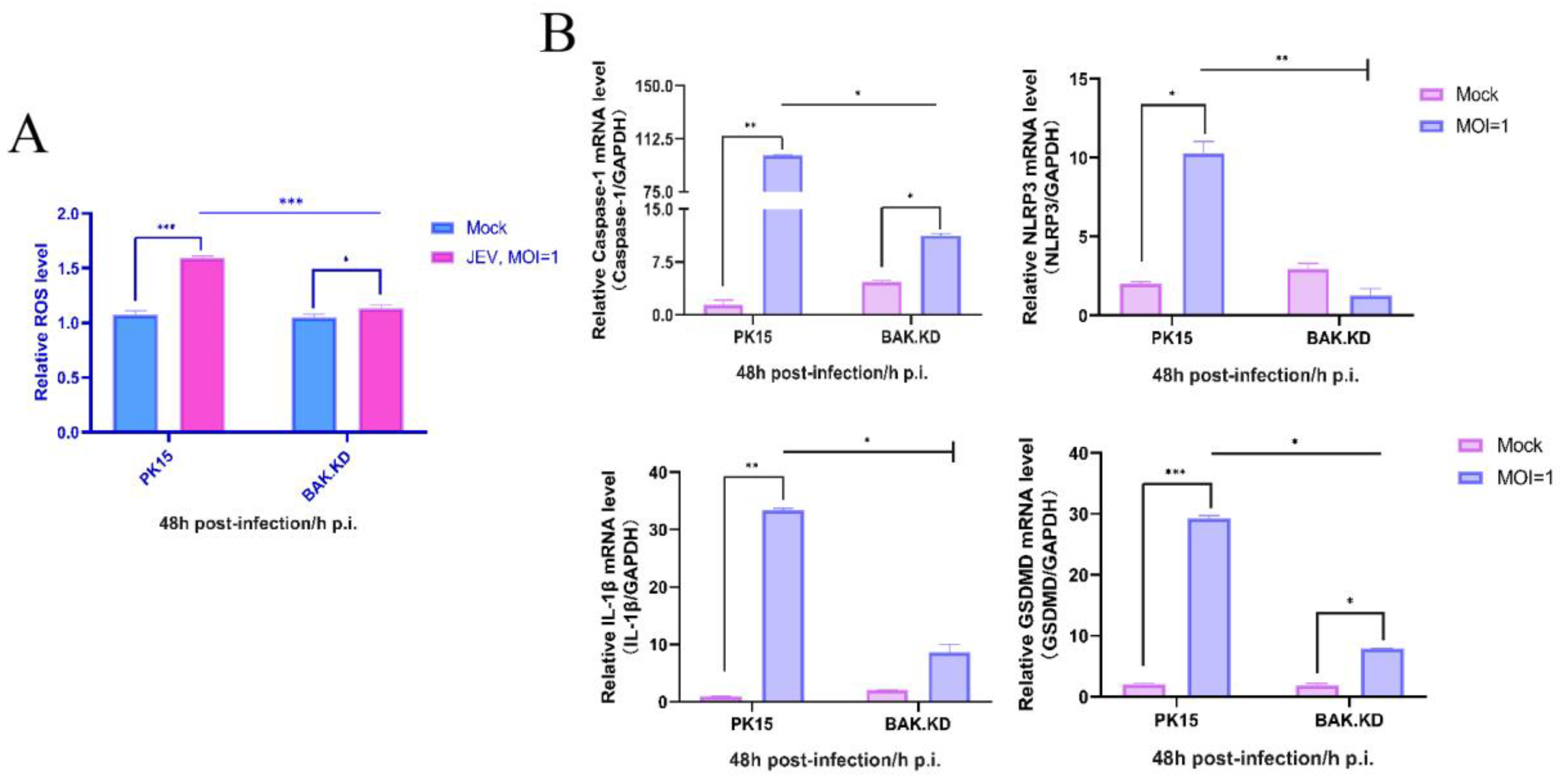
3.3. Knockdown of BAK Function Inhibits JEV-Activated ROS and Other Cytokines
3.4. The Activation of NLRP3 Inflammatory Pathway Proteins Induced by JEV Is Significantly Inhibited in BAK.KD Cells
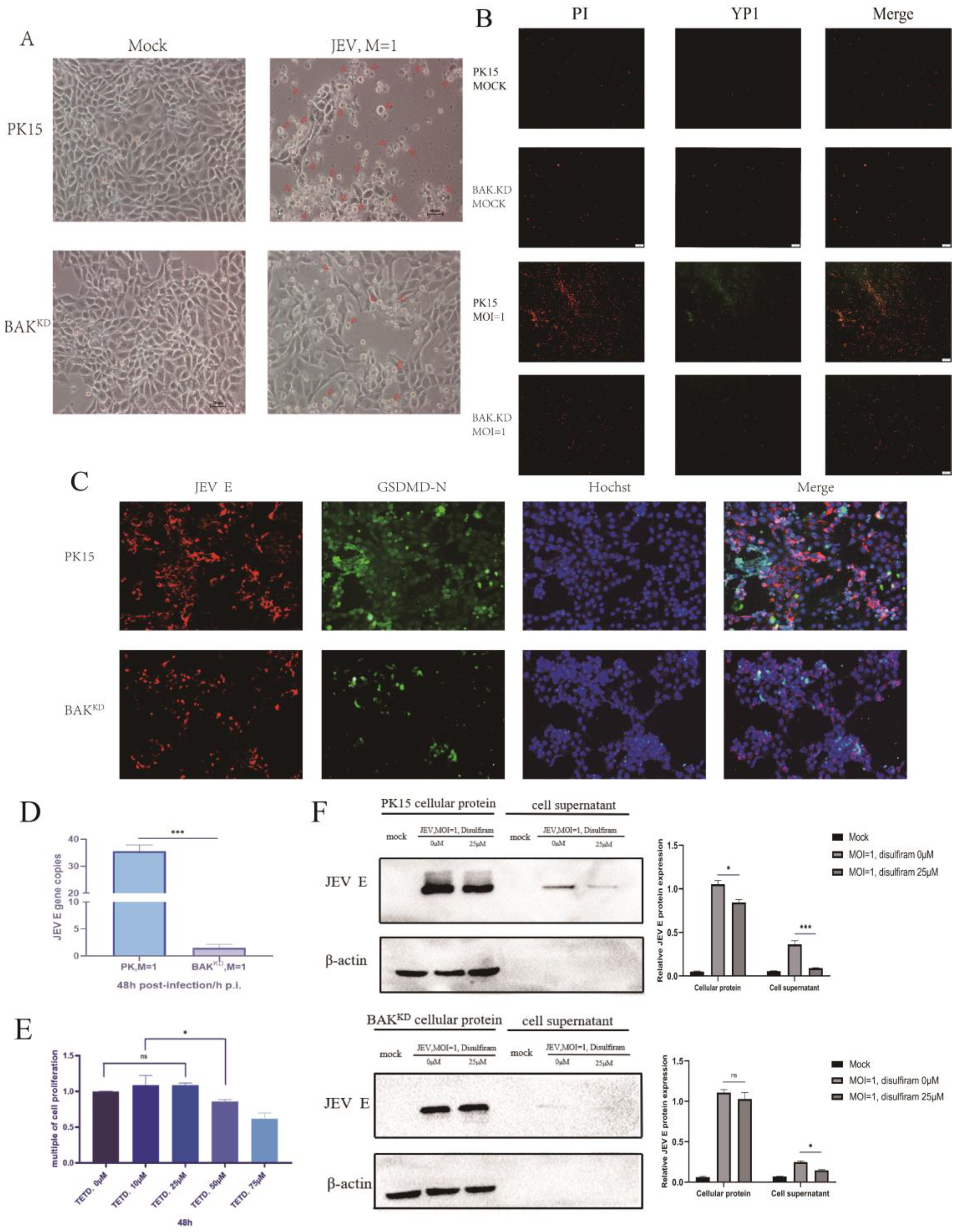
3.5. Activation of the Pyroptotic Pathway May Promote the Release of JEV
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chong, H.Y.; Leow, C.Y.; Abdul Majeed, A.B.; Leow, C.H. Flavivirus infection-A review of immunopathogenesis, immunological response, and immunodiagnosis. Virus Res. 2019, 274, 197770. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Singh, R.; Mani, V.E.; Poddar, B. Japanese B Encephalitis. Indian J. Crit. Care Med. 2021, 25, S171–S174. [Google Scholar] [CrossRef]
- Banerjee, A.; Tripathi, A. Recent advances in understanding Japanese encephalitis. F1000Res 2019, 8, Faculty Rev-1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulvey, P.; Duong, V.; Boyer, S.; Burgess, G.; Williams, D.T.; Dussart, P.; Horwood, P.F. The Ecology and Evolution of Japanese Encephalitis Virus. Pathogens 2021, 10, 1534. [Google Scholar] [CrossRef]
- Mansfield, K.L.; Hernández-Triana, L.M.; Banyard, A.C.; Fooks, A.R.; Johnson, N. Japanese encephalitis virus infection, diagnosis and control in domestic animals. Vet. Microbiol. 2017, 201, 85–92. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; An, S.; Chen, J.; Zhang, S.; Tan, C.; Yu, J.; Ye, H.; Wu, Y.; Yuan, J.; Wu, J.; et al. Neural progenitor cell pyroptosis contributes to Zika virus-induced brain atrophy and represents a therapeutic target. Proc. Natl. Acad. Sci. USA 2020, 117, 23869–23878. [Google Scholar] [CrossRef] [PubMed]
- Nie, M.; Zhou, Y.; Li, F.; Deng, H.; Zhao, M.; Huang, Y.; Jiang, C.; Sun, X.; Xu, Z.; Zhu, L. Epidemiological investigation of swine Japanese encephalitis virus based on RT-RAA detection method. Sci. Rep. 2022, 12, 9392. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Wang, X.; Liu, Y.; Li, Y.; Long, S.; Gu, C.; Ye, J.; Xie, S.; Cao, S. Japanese Encephalitis Virus infection induces inflammation of swine testis through RIG-I-NF-ĸB signaling pathway. Vet. Microbiol. 2019, 238, 108430. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zhang, M.; Stoilov, P.; Chen, L.; Zheng, S. Developmental Attenuation of Neuronal Apoptosis by Neural-Specific Splicing of Bak1 Microexon. Neuron 2020, 107, 1180–1196.e1188. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Kvansakul, M.; Caria, S.; Hinds, M.G. The Bcl-2 Family in Host-Virus Interactions. Viruses 2017, 9, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, V.H.; Bartolo, R.; Westphal, D.; Alsop, A.; Dewson, G.; Kluck, R.M. Bak apoptotic function is not directly regulated by phosphorylation. Cell Death Dis. 2013, 4, e452. [Google Scholar] [CrossRef] [Green Version]
- Burdette, B.E.; Esparza, A.N.; Zhu, H.; Wang, S. Gasdermin D in pyroptosis. Acta Pharm. Sin. B 2021, 11, 2768–2782. [Google Scholar] [CrossRef]
- Lindsten, T.; Ross, A.J.; King, A.; Zong, W.X.; Rathmell, J.C.; Shiels, H.A.; Ulrich, E.; Waymire, K.G.; Mahar, P.; Frauwirth, K.; et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 2000, 6, 1389–1399. [Google Scholar] [CrossRef] [Green Version]
- Kesavardhana, S.; Malireddi, R.K.S.; Kanneganti, T.D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Nie, G.; Yang, F.; Pi, S.; Wang, C.; Cao, H.; Guo, X.; Liu, P.; Li, G.; Hu, G.; et al. Inhibition of ROS/NLRP3/Caspase-1 mediated pyroptosis attenuates cadmium-induced apoptosis in duck renal tubular epithelial cells. Environ. Pollut. 2020, 273, 115919. [Google Scholar] [CrossRef]
- Kaushik, D.K.; Gupta, M.; Kumawat, K.L.; Basu, A. NLRP3 inflammasome: Key mediator of neuroinflammation in murine Japanese encephalitis. PLoS ONE 2012, 7, e32270. [Google Scholar] [CrossRef]
- Li, S.; Li, H.; Zhang, Y.L.; Xin, Q.L.; Guan, Z.Q.; Chen, X.; Zhang, X.A.; Li, X.K.; Xiao, G.F.; Lozach, P.Y.; et al. SFTSV Infection Induces BAK/BAX-Dependent Mitochondrial DNA Release to Trigger NLRP3 Inflammasome Activation. Cell Rep. 2020, 30, 4370–4385.e4377. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Dong, X.R.; Zhang, B.; Zhang, X.T.; Liu, J.Z.; Ma, D.S.; Ma, L. Molecular mechanism and therapeutic targeting of necrosis, apoptosis, pyroptosis, and autophagy in cardiovascular disease. Chin. Med. J. 2021, 134, 2647–2655. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zhang, M.X.; Zhang, L.; Zhang, D.; Li, C.; Li, Y.L. Autophagy, Pyroptosis, and Ferroptosis: New Regulatory Mechanisms for Atherosclerosis. Front. Cell Dev. Biol. 2021, 9, 809955. [Google Scholar] [CrossRef]
- Zheng, X.; Chen, W.; Gong, F.; Chen, Y.; Chen, E. The Role and Mechanism of Pyroptosis and Potential Therapeutic Targets in Sepsis: A Review. Front. Immunol. 2021, 12, 711939. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Park, S.L.; Huang, Y.S.; Vanlandingham, D.L. Re-Examining the Importance of Pigs in the Transmission of Japanese Encephalitis Virus. Pathogens 2022, 11, 575. [Google Scholar] [CrossRef]
- Wang, Y.; Kanneganti, T.D. From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways. Comput. Struct. Biotechnol. J. 2021, 19, 4641–4657. [Google Scholar] [CrossRef] [PubMed]
- Caine, E.A.; Scheaffer, S.M.; Broughton, D.E.; Salazar, V.; Govero, J.; Poddar, S.; Osula, A.; Halabi, J.; Skaznik-Wikiel, M.E.; Diamond, M.S.; et al. Zika Virus Causes Acute Infection and Inflammation in the Ovary of Mice Without Apparent Defects in Fertility. J. Infect. Dis. 2019, 220, 1904–1914. [Google Scholar] [CrossRef]
- Adams, J.M. BAX and BAK become killers without a BH3 trigger. Cell Res. 2019, 29, 967–968. [Google Scholar] [CrossRef] [PubMed]
- Leshchiner, E.S.; Braun, C.R.; Bird, G.H.; Walensky, L.D. Direct activation of full-length proapoptotic BAK. Proc. Natl. Acad. Sci. USA 2013, 110, E986–E995. [Google Scholar] [CrossRef] [Green Version]
- Riley, J.S.; Tait, S.W. Mitochondria and pathogen immunity: From killer to firestarter. Embo J. 2019, 38, e102325. [Google Scholar] [CrossRef]
- Suzuki, T.; Okamoto, T.; Katoh, H.; Sugiyama, Y.; Kusakabe, S.; Tokunaga, M.; Hirano, J.; Miyata, Y.; Fukuhara, T.; Ikawa, M.; et al. Infection with flaviviruses requires BCLXL for cell survival. PLoS Pathog. 2018, 14, e1007299. [Google Scholar] [CrossRef]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond—mitochondrial performance in apoptosis. Febs J. 2018, 285, 416–431. [Google Scholar] [CrossRef] [Green Version]
- Karmakar, M.; Minns, M.; Greenberg, E.N.; Diaz-Aponte, J.; Pestonjamasp, K.; Johnson, J.L.; Rathkey, J.K.; Abbott, D.W.; Wang, K.; Shao, F.; et al. N-GSDMD trafficking to neutrophil organelles facilitates IL-1beta release independently of plasma membrane pores and pyroptosis. Nat. Commun. 2020, 11, 2212. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, Y.; Miyata, N.; Mukai, S.; Okumoto, K.; Cheng, E.H. BAK regulates catalase release from peroxisomes. Mol. Cell Oncol. 2017, 4, e1306610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redant, V.; Favoreel, H.W.; Dallmeier, K.; Van Campe, W.; De Regge, N. Efficient control of Japanese encephalitis virus in the central nervous system of infected pigs occurs in the absence of a pronounced inflammatory immune response. J. Neuroinflammation 2020, 17, 315. [Google Scholar] [CrossRef]
- Hsu, S.K.; Li, C.Y.; Lin, I.L.; Syue, W.J.; Chen, Y.F.; Cheng, K.C.; Teng, Y.N.; Lin, Y.H.; Yen, C.H.; Chiu, C.C. Inflammation-related pyroptosis, a novel programmed cell death pathway, and its crosstalk with immune therapy in cancer treatment. Theranostics 2021, 11, 8813–8835. [Google Scholar] [CrossRef]
- Deng, W.; Bai, Y.; Deng, F.; Pan, Y.; Mei, S.; Zheng, Z.; Min, R.; Wu, Z.; Li, W.; Miao, R.; et al. Streptococcal pyrogenic exotoxin B cleaves GSDMA and triggers pyroptosis. Nature 2022, 602, 496–502. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid and Antibody | Source | Identifier |
|---|---|---|
| pCDNA-MYC-BAK | Research Center of Swine Disease | N/A |
| FITC Goat Anti-rabbit IgG | ABclonal Technology | Cat#AS011 |
| Cy3 Goat Anti-Mouse IgG | ABclonal Technology | Cat#AS008 |
| Anti-BAK Rabbit polyclonal antibody | ABclonal Technology | Cat#A0498 |
| Anti-ASC Rabbit polyclonal antibody | ABclonal Technology | Cat#A11433 |
| Anti-cleaved N-terminal GSDMD antibody | Abcam | Cat#Ab215203 |
| Anti-NLRP3 Rabbit polyclonal antibody | ABclonal Technology | Cat#A5652 |
| Anti-IL-1β Rabbit polyclonal antibody | ABclonal Technology | Cat#A11369 |
| Anti-Caspase-1 Rabbit polyclonal antibody | ABclonal Technology | Cat#A0964 |
| Anti-β-actin Rabbit polyclonal antibody | ABclonal Technology | Cat#AC038 |
| Anti-JEV E Mouse polyclonal antibodies | Research Center of Swine Disease | N/A |
| Gene | Sequence (5′-3′) | Length (bp) |
|---|---|---|
| JEV E | F: GGGAAGGGAAGCATTGAC R: AAGGAGCATTGGGTGTTA | 231 bp |
| BAK | F: TCAACCGGCGATACGACT R: GTAGCCAAAGCCCAGAAGAG | 155 bp |
| BAX | F: GCAGCCGATCTCGAAGGA R: GCCGAAATGTTTGCTGACG | 154 bp |
| IRF3 | F: AAGAAGCATTGCGTTTAGC R: AGGTACTGTATCTGCCATGAG | 138 bp |
| IRF7 | F: CATGGGGCGTGGATCTGA R: GCACCGTTCGACCTTTGT | 234 bp |
| TLR7 | F: TAACCTCAGTCAACCGCAAGT R: CCATCTTTGGGGCACATAC | 190 bp |
| TRANS | F: AGACGACTGCTGCCATGGA R: ACACCCTGCTGTTTTTCTACCT | 155 bp |
| RIG-1 | F: TCCCAGCAACGAGAACCCT R: TTCGTTTTGCCACGTCCAG | 195 bp |
| TNF | F: GGCATTGGCATACCCACTCT R: GCCCAAGGACTCAGATCATC | 178 bp |
| IFNα | F: GGACTCCATCCTGGCTGTGA R: GACTTCTGCCCTGATGATCTCC | 103 bp |
| IL-1β | F: ATGTGGACCTCTGGGTATGG R: ACAAAAGCCCGTCTTCCTG | 188 bp |
| IL-6 | F: GGCATCACCTTTGGCATCTT R: CTGCTTCTGGTGATGGTACTG | 204 bp |
| GSDMD | F: ACATGGCCTCCAAGGTAAGA R: GATCGAGTTGGGGCTGTGACT | 120 bp |
| NLRP3 | F: AATGGATGGGTTTACTGG R: GGTGAAGCGTTTGTTGAG | 189 bp |
| Caspase-1 | F: GTCCGAAGCGGTGAGATT R: CCCAGACATAGCCCAAAG | 220 bp |
| Caspase-3 | F: ACCCGAGTAAGAATGTGC R: GACTGTGGGATTGAGACG | 212 bp |
| Caspase-8 | F: CTCCTCCTCATTGGTTTCC R: TCCTGAGCCTGGACTACAT | 196 bp |
| GAPDH | F: ACATGGCCTCCAAGGTAAGA R: GATCGAGTTGGGGCTGTGACT | 202 bp |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, W.; Yang, K.; Zheng, Y.; Cao, S.; Yan, Q.; Huang, X.; Wen, Y.; Zhao, Q.; Du, S.; Lang, Y.; et al. BAK-Mediated Pyroptosis Promotes Japanese Encephalitis Virus Proliferation in Porcine Kidney 15 Cells. Viruses 2023, 15, 974. https://doi.org/10.3390/v15040974
Xu W, Yang K, Zheng Y, Cao S, Yan Q, Huang X, Wen Y, Zhao Q, Du S, Lang Y, et al. BAK-Mediated Pyroptosis Promotes Japanese Encephalitis Virus Proliferation in Porcine Kidney 15 Cells. Viruses. 2023; 15(4):974. https://doi.org/10.3390/v15040974
Chicago/Turabian StyleXu, Weimin, Ke Yang, Yi Zheng, Sanjie Cao, Qigui Yan, Xiaobo Huang, Yiping Wen, Qin Zhao, Senyan Du, Yifei Lang, and et al. 2023. "BAK-Mediated Pyroptosis Promotes Japanese Encephalitis Virus Proliferation in Porcine Kidney 15 Cells" Viruses 15, no. 4: 974. https://doi.org/10.3390/v15040974