Abstract
Phages possess the ability to selectively eliminate pathogenic bacteria by recognizing bacterial surface receptors. Since their discovery, phages have been recognized for their potent bactericidal properties, making them a promising alternative to antibiotics in the context of rising antibiotic resistance. However, the rapid emergence of phage-resistant strains (generally involving temperature phage) and the limited host range of most phage strains have hindered their antibacterial efficacy, impeding their full potential. In recent years, advancements in genetic engineering and biosynthesis technology have facilitated the precise engineering of phages, thereby unleashing their potential as a novel source of antibacterial agents. In this review, we present a comprehensive overview of the diverse strategies employed for phage genetic engineering, as well as discuss their benefits and drawbacks in terms of bactericidal effect.
1. Introduction
The World Health Organization (WHO) has proposed that antimicrobial resistance is one of the greatest threats to global public health in the 21st century. According to WHO, approximately 700,000 individuals succumb to antibiotic resistance annually [1,2]. Furthermore, it is estimated that by 2050, ten million deaths will be attributed to drug-resistant bacterial infections [3]. Meanwhile, the increasing prevalence of antibiotic resistance suggests the end of the golden era of antibiotics and the possibility of entering a post-antibiotic era, where we face a world lacking efficient antimicrobial agents [4,5,6].
Bacteriophages (phages), also known as bacterial viruses, exhibit a high specificity towards particular bacterial species, with fewer off-target effects on intestinal microbiota compared to antibiotics [7,8,9]. Although the first successful application of phage therapy was reported nearly a century ago, it has recently regained attention due to the escalating threat of antibiotic resistance. Despite the potential of phages as antimicrobial agents [10], several major concerns remain with regard to their clinical applications, such as the emergence of phage-resistant bacteria [11], the detrimental inflammatory response [12], and the physical barrier formed by biofilm [13]. However, genetic engineering and biosynthesis technology allow for the modification of phages to expand their host range and enhance their therapeutic potential, offering promising solutions to these challenges [14]. For example, phages can be engineered to express enzymes that degrade bacterial biofilms, which are often resistant to antibiotics and phages. Biosynthesis technology enables the production of phages on a large scale, which is essential for clinical applications. In addition, genetic engineering and biosynthesis technology can be combined to create synthetic phages with tailored properties. For instance, synthetic phages can be designed to target specific bacterial strains or to carry therapeutic payloads such as antibiotics or immunomodulators. Moreover, the growing availability of complete phage genome sequences in public databases, coupled with advances in understanding the structure of phage components and phage-host bacterial interactions, has facilitated the targeted engineering of phages [15].
Genetic engineering and biosynthesis technology hold great promise for advancing phage therapy and overcoming the challenges associated with bacterial infections. This paper aims to present a comprehensive review of the latest advancements in phage engineering and biosynthesis technology, as well as their diverse applications in the treatment of bacterial infections.
2. Phage Engineering and Biosynthesis Strategies
Recent advances in phage engineering and biosynthesis have led to the development of phage-based therapeutics with has improved efficacy and safety profiles. These include host-mediated homologous recombination, in vivo recombineering, BRED, yeast-based assembly of phage genomes, L-form bacteria, and CRISPR-Cas. Phage engineering and biosynthesis strategies hold great promise for the development of novel antibacterial agents to combat antibiotic-resistant bacteria.
2.1. Host-Mediated Homologous Recombination
Homologous recombination (HR) is a highly versatile strategy for phage genome engineering, which can occur between two DNA fragments that share only limited regions of homology (Figure 1). To execute HR, exogenous DNA segments containing flanking regions homologous to the phage genome sequence are initially cloned into a vector [16]. The homologous regions dictate the integration site of exogenous DNA segments within the phage genome. The host strain carrying the donor vector is subsequently infected with the engineered phage. In vivo, homologous recombination (HR) occurs between the vector and the phage genome, facilitated by host recombinases, such as RecA. This process enables the integration of foreign DNA fragments into the phage genome, which are ultimately packaged within the phage particles [17]. Recent studies have demonstrated that retrons possess the ability to function as a recombination template, without the need for lengthy homologous flanking sequences. This feature facilitates a rapid and uncomplicated cloning procedure, although the insertion of large fragments may be limited by the restricted size of the homologous domain in the retron, which typically spans 75 base pairs (75 bp) [18,19]. However, one significant drawback of engineering methods based on homologous recombination is the relatively low efficiency of recombination, which necessitates time-consuming and labor-intensive screening methods to identify recombinant phages. This limitation arises from the inability to employ selectable markers, such as antibiotic resistance genes, during the lytic growth phase of phages. To streamline the screening process, it is feasible to integrate marker genes that enable a targeted selection for mutated phages or to employ a subsequent counter-selection technique to remove the wild-type phages (refer to the subsequent sections for more information).
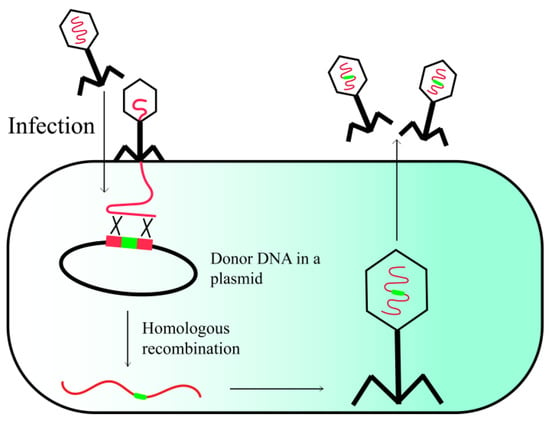
Figure 1.
Phage engineering via homologous recombination.
2.2. In Vivo Recombineering
HR is a rare occurrence in most organisms, and attaining the desired recombinant through endogenous recombination machinery is frequently challenging. To circumvent this issue, an improved editing technology termed recombineering has been devised. It employs the recombination system harbored by phage genomes to increase the frequency of HR, thereby enabling the generation of gene knockouts, deletions, and point mutations (Figure 2) [20].
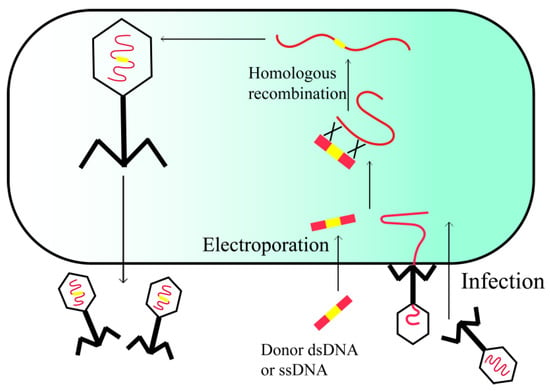
Figure 2.
In vivo recombineering of phage.
Recombineering relies on linear donor DNA and heterologous proteins, including Gam, Bet, and Exo, which are expressed from the phage genome. These proteins protect linear dsDNA from intracellular degradation and promote recombination between the linear donor DNA and the injected phage genome. This process increases the frequency of recombination and reduces the length of homologous regions to as little as 50 bp [21]. The enhanced accessibility of phage genome sequences has greatly facilitated the convenience of recombineering, as it is only viable to recombine a phage with a known genome sequence [22].
2.3. BRED
Bacteriophage recombineering with electroporated DNA (BRED) is a valuable technique for genetically modifying lytic phages. This method entails the electroporation of linearized phage genome fragments and homologous DNA into phage-sensitive bacterial cells harboring plasmids that express HR-enhancing proteins, such as RecE/RecT-like proteins (Figure 3) [23]. Because the success rate of BRED is relatively high (10% in M. smegmatis), recombinant phages can be retrieved through PCR-based plaque recovery following cell lysis and infection of susceptible hosts. The benefits of BRED include the absence of a need for constructing intricate cloning systems and a selectable marker, as well as the ability to introduce mutations into any region of the phage genome [24]. BRED offers the potential to produce phages that have undergone editing without recombination, thereby excluding them from the classification of genetically modified organisms. This enables regulatory agencies to more readily approve the use of modified phages for therapeutic purposes, as evidenced by the recent authorization of the first engineered phage therapy in the United Kingdom [25].
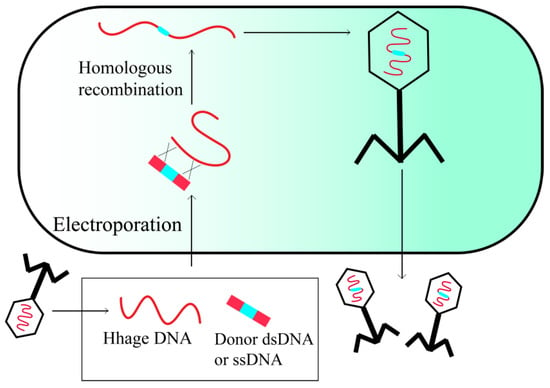
Figure 3.
Phage recombineering of electroporated DNA.
At present, the progress of BRED is primarily hindered by challenges in attaining sufficiently high transformation efficiencies when working with the extensive genomes of phages in various host organisms [26]. In order to address these constraints, alternative approaches to BRED have been utilized, such as the utilization of bacteriophage recombineering with infectious particles (BRIP) [27]. In this method, the synthetic DNA substrate containing the desired modification is introduced into the bacteria through electroporation followed by infection of the bacteria with the phage, as opposed to the conventional approach of electroporating the bacteria with phage DNA.
2.4. Yeast-Based Assembly of Phage Genomes
Phage genome modification can be achieved by utilizing a yeast cell as an alternative host for phage assembly. The intrinsic capacity of Saccharomyces cerevisiae, a yeast species, to recombine linear double-stranded DNA fragments into a single genome facilitates genetic modifications and empowers the generation of engineered phages [28]. To assemble the complete phage genome in yeast, PCR amplification is conducted with homologous termini that are retained with at least 30 bp in length. The initial and final genome fragments are amplified using primers that contain homologous sequences with a yeast artificial chromosome (YAC). All amplified segments of the genome and YAC are subsequently transformed into S. cerevisiae, where gap repair enables a recombination-mediated joining of all phage genome segments (Figure 4). After purifying the recombinant vector, the phage genome can be assembled to generate phage particles upon transformation into the host bacteria. The resulting plaques are subsequently selected and sequenced to confirm the successful incorporation of the desired mutations [29]. This phage engineering strategy has been utilized to genetically modify multiple phage genomes, such as T3 and φX174 coliphages, as well as Klebsiella phage K11 [28,30,31].
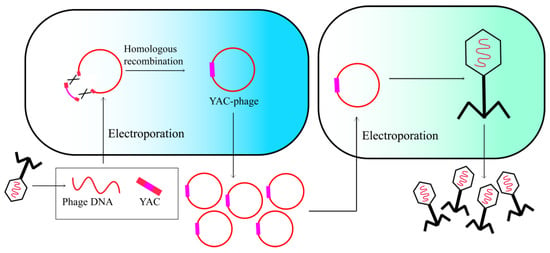
Figure 4.
Yeast-based assembly of phage genomes.
The utilization of YAC-assisted assembly for modified genomes has proven to be highly efficient. However, this approach is not devoid of limitations, as certain phages may possess repetitive sequences at their termini that could result in a phage excision from the vector during recombination [32]. Nevertheless, this challenge can be overcome by incorporating selective markers for yeast within the phage genome, which is a promising approach. In addition, the process of rebooting phage genomes has traditionally relied on the transformation of host bacteria, which limits the potential application of this engineering strategy to bacteria that are highly transformable [33]. As an alternative, it may be possible to reboot assembled phages through alternative methods such as the use of bacterial L-forms and cell-free techniques.
2.5. L-Form Bacteria
The limited ability of Gram-positive bacteria to undergo transformation poses a significant obstacle in the reactivation of engineered phages that target these hosts. However, this limitation can be overcome by substituting the robust cell walls of Gram-positive hosts with L-form bacteria, thereby enabling the reactivation of the synthetically constructed phage genome. L-form bacteria are cell-wall-deficient organisms that are capable of undergoing transformation with macromolecular DNA, as well as replicating and transcribing newly introduced genetic molecules. Drawing on these advantages, L-form bacteria were assessed as potential hosts for rebooting the synthetic genomes of phages (Figure 5). Furthermore, Kilcher et al. demonstrated the ability to reboot various Gibson assembled or wild-type phage genomes in L-form Listeria monocytogenes or related Gram-positive hosts. L-type bacteria release phage particles via hypoosmotic lysis, which enables the phages to infect specific bacterial hosts for reproduction. The L-forms of Listeria monocytogenes have been found to possess the potential to serve as a platform for the reactivation of Bacillus and Staphylococcus phages, indicating the intergeneric applicability of L-form cells in phage genome engineering [34]. This discovery is of significant interest and may have important implications for the development of novel strategies for phage-based therapies.
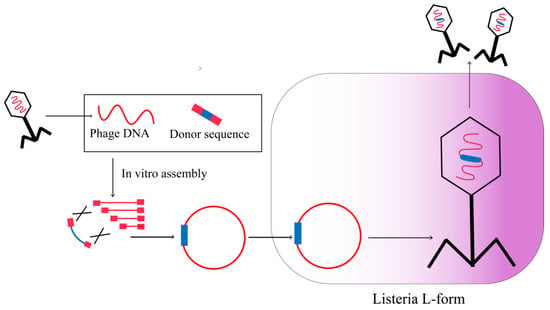
Figure 5.
Synthetic phage genomes in L-form bacteria.
The ability to reboot phage genomes in bacterial L-forms has been observed to be largely unaffected by various factors such as the viral lifestyle, morphology, DNA packaging strategy, genome ends (including cohesive ends, terminally redundant, and all double-stranded DNA), and size [34]. This finding provides a valuable engineering platform for a diverse array of phages. Although the rebooting of engineered phage genomes in L-forms has thus far only been observed in Gram-positive cells, the potential for generating L-forms of Gram-negative bacteria implies that they could also serve as phage rebooting mechanisms [35]. However, it remains uncertain whether L-forms can be effectively produced for all types of bacteria.
2.6. CRISPR-Cas
The CRISPR-Cas system is a naturally occurring adaptive defense mechanism present in numerous prokaryotes, which confers sequence-specific protection against invasive nucleic acids [36,37,38]. The CRISPR-Cas system comprises two genetic components: Cas proteins and the CRISPR array [39,40,41]. The type I-E CRISPR-Cas system has recently been utilized as a counter-selection mechanism for engineered T7 phages (Figure 6). The phage underwent homologous recombination-mediated editing to excise the dispensable gene 1.7 [42]. The strategy selectively eliminated nonrecombinant phage genomes harboring gene 1.7, while sparing the recombinant phage genomes devoid of this gene. Similarly, the CRISPR/Cas II-A system can also be utilized for in vivo editing of the phage 2972 genome, encompassing point mutations, gene deletions, and DNA exchange [37]. Given the successful genetic manipulation demonstrated in the above results, the CRISPR/Cas strategy holds promise for application to other phage genomes.
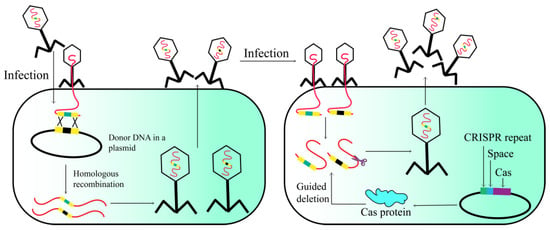
Figure 6.
Phage genome engineering with CRISPR-Cas13a.
The utilization of CRISPR-Cas-based phage engineering techniques is constrained to bacteria that possess a well-defined native CRISPR-Cas system or have the ability to undergo transformation in order to facilitate the expression of an operational heterologous CRISPR-Cas system. This can pose a significant constraint for the manipulation of bacteriophages targeting bacteria that lack genetic manipulability. Furthermore, phages have developed various mechanisms to resist targeting by CRISPR-Cas, including the concealment of their DNA through covalent modifications of nucleotides or the utilization of anti-CRISPR proteins (Acrs) [43]. The extensively studied phage T4 of E. coli has been found to exhibit glucosyl DNA hypermodifications as well as DNA recombination and repair mechanisms. These mechanisms serve to safeguard the phage against the DNA-targeting type I and II CRISPR-Cas systems, thereby leading to a diminished efficacy in counter-selecting the wild-type T4 [44,45].
3. Application of Engineering Phage
Phage therapy has emerged as a promising approach to address the global rise of antibiotic resistance among bacterial pathogens. Advancements in biosynthesis and genetic engineering technologies have greatly facilitated phage genome engineering. Significant progress has been made in engineering phages to restore the sensitivity of drug-resistant bacteria, reduce the minimum inhibitory concentration (MIC) of antibiotics, target the deletion of essential genes of host bacteria, and provide crucial therapies for patient treatment (Table 1). Notably, these advancements hold immense potential in combating antibiotic resistance and improving patient prognosis.
3.1. Enhancing Bactericidal Activity
The bactericidal efficacy of natural phages varies depending on the specific phage and can be enhanced through the development of genetically modified phages. A phage K1F was genetically modified using the homologous recombination technique to incorporate fluorescence and facilitate the expression of epidermal growth factor (EGF) derived from the ErbB family of tyrosine kinases [46]. The modified phage K1F bearing EGF, demonstrated an increased ability to enter human cells and exhibited improved effectiveness in eradicating intracellular E. coli EV36. Distinctive trafficking pathways between the two phages were also observed: K1F-GFP-EGF entered cells through the endolysosomal pathway by inducing the EGF receptor (EGFR), while K1F-GFP entered cells and underwent degradation through LC3-assisted phagocytosis. This enabled K1F-GFP-EGF to accumulate rapidly within different human cell lines, thereby enhancing its efficiency in locating its intracellular host.
3.2. Restoring the Sensitivity of Drug-Resistant Bacteria to Antibiotics
RNA-guided nucleases (RGNs), derived from Type II CRISPR-Cas systems, are programmable endonucleases that enable precise genome editing. RGNs comprise two essential components: a guide RNA (gRNA) of approximately 100 nucleotides, which utilizes 20 variable nucleotides at its 5′ end to form base pairs with a specific genomic DNA sequence, and a nuclease (e.g., Cas9 endonuclease) that cleaves the target DNA [47]. Several studies have demonstrated that modified phages overexpressing RGNs can restore the sensitivity of drug-resistant bacteria to antibiotics [48,49,50]. For instance, Ido Yosef and colleagues designed a temperate phage to transfer the effective RGNs into the genome of antibiotic-resistant bacteria [51]. The gRNA sequences were initially designed to target conserved regions of the resistance genes ndm-1 and ctx-M-15. Subsequently, the designed gRNA and Cas9 endonuclease encoding gene were introduced into λ prophage via homologous recombination. As a result, the resistance gene in E. coli lysogenized with the engineered λ prophage was deleted, resulting in a restored antibiotic sensitivity. In another study, Edgar R.’s group employed homologous recombination to introduce a streptomycin-sensitive gene into a phage genome [52]. This engineered phage treatment effectively restored antibiotic sensitivity in streptomycin-resistant E. coli, resulting in a significant reduction of the MIC of antibiotics from 100 to 12.5 mg/mL. The same strategy restored the sensitivity of E. coli to naphthyridic acid, resulting in a twofold reduction in the MIC value.
These strategies could be utilized to treat hospital surfaces and hand sanitizers, specifically targeting the skin microbiota of medical personnel. Unlike antibiotics and disinfectants, which promote the growth of resistant pathogens, this proposed treatment enriches and selects for susceptible pathogens. Additionally, these strategies promote the growth of pathogens that are unable to acquire or transfer resistance determinants horizontally, potentially reducing the spread of antibiotic resistance. The enriched susceptible population may also prevent the establishment of newly introduced resistant pathogens by outcompeting them for their ecological niche.
3.3. Altering Phages’ Host Range
One major limitation of phage-based applications is their narrow and specific host range. The natural host range of phages is insufficient to cover all pathogenic microbial strains. To circumvent this challenge, genetic engineering techniques have been adopted to expand or alter the host range of phages [53]. Several reports have demonstrated that the host specificity of phages is determined by their tail fiber [54]. To broaden the host range of phages, researchers employed a homologous recombination technique to rationally replace the tail fiber encoding genes of phage T2 with their homologous counterparts from phage IP008 [55]. The chimeric phage generated exhibited an expanded host range compared to the original T2 phage, while maintaining an equivalent lytic activity [56]. In a similar study, the gene encoding the tail fiber (gp69) in phage PaP1 was substituted with the homologous gene (gp84) from phage JG004 via HR, resulting in the creation of an engineered phage that could induce plaque formation in the bacterial host of phage JG004 [57]. In another study, the host range of the filamentous phage fd was expanded through the incorporation of a receptor-binding domain from the phage IKe into the N-terminus of protein G3p [58]. Similarly, phage fd could be engineered to recognize Vibrio cholerae by fusing the minor coat-encoding gene pIII from fd with a sequence of the orfU gene from CTXφ, another filamentous phage [59]. However, these strategies are constrained by the need to modify tail components that recognize host receptors, which are specific to known phages. These methods offer a versatile tool for rapidly altering and broadening the host range of bacteriophages. With the acquisition of a greater number of suitable homologous sequences, future clinical applications for screening phages of specific clinical isolates can be developed. Furthermore, these methods may enable a direct modification of bacteriophage genomes to expand or alter their host ranges.
3.4. Increasing the Cleavage of Biofilm
Bacterial growth within biofilms is frequently associated with the pathogenesis of numerous clinically important infections [60]. Biofilms pose a formidable challenge for eradication due to their inherent resistance to antimicrobial agents, such as phages and antibiotics. This phenomenon is often attributed to the extracellular polymeric substances (EPS) of biofilm, which limit the diffusion of molecules. To address this issue, E. coli-specific phage T7 was genetically modified to incorporate the biofilm-degrading enzyme dispersin B (DspB) during phage infection [61]. DspB exerts its action via the aminohydrolysis of β-1, 6-Nacetyl-D-glucosamine, thereby disrupting the biofilm’s formation and integrity. This engineered T7 phage exhibited a significant increase of 4.5 orders of magnitude in biofilm reduction after a 24 h treatment compared to its nonengineered counterpart, while the control group only showed a reduction of 2.5 orders of magnitude [61]. These findings indicated that the expression of DspB was crucial for elevating the efficacy of biofilm removal in the engineered virus, while the control group without DspB expression failed to achieve the same effect.
Acyl-homoserine lactone (AHL) is a well-known quorum-sensing signal that plays a crucial role in the formation of biofilm. The activity of AHLs can be effectively inhibited by AHL-lactonases, which catalyze the cleavage of the lactone bond in AHLs, thereby preventing biofilm formation [62]. Accordingly, the gene encoding AHL-lactonase (aiiA) from Bacillus anthracis was cloned into the T7 phage, resulting in the creation of an engineered phage named T7aiiA. The biofilm formation was significantly inhibited by phage T7aiiA, resulting in a remarkable reduction in biomass by 74.9%, whereas the T7control phage only caused a reduction of 23.8% [63].
This design obviates the necessity of expressing, purifying, and delivering high doses of enzymes to hard-to-reach infection sites, thereby enhancing the effectiveness of phage therapy in biofilm removal. The cost-effectiveness of genome sequencing and synthetic biology technologies, such as phage genome refactoring and large-scale DNA synthesis, should facilitate the production of engineered enzymatic phages and expand the limited range of biofilm-degrading phages isolated from the environment.
3.5. Increasing the Half-Life of Phage In Vivo
Despite the clinical success of some antimicrobial treatments, the widespread application of phage therapies has been hindered by several severe physiochemical obstacles that phages encounter in the digestive or circulatory systems [64]. It is noteworthy that the efficacy of phages in vivo is decreased due to the action of phagocytes, antibodies, and gastric protease. In order to prevent the threat of phage abolition by the gastric acidic protease, an engineered phage T7 was constructed by fusing a membrane phosphoprotein (PhoE) signal peptide to the major capsid protein [65]. It interacted with the phospholipids of E. coli through the PhoE signal peptide, thus resulting in the formation of a lipid coating on the surface of T7 phage. The lipid-coated engineered T7 phage exhibited a 100-fold increase in stability compared to the wild-type phage T7 in the gastrointestinal tract of animals, representing a promising candidate for orally delivered phage therapy. Compared to other methods, such as microencapsulation, this approach offers the advantages of process simplicity, requiring significantly fewer optimization steps and a straightforward scale-up process (only the phage amplification is necessary). Importantly, this research demonstrates the feasibility, simplicity, and cost-effectiveness of phage engineering as a means of enhancing phage properties for oral administration in animals.
3.6. Reducing Endotoxin Release
Treatment with lytic phages can elicit rapid cell lysis, which may subsequently lead to the release of cell debris and toxins, thereby triggering an adverse immune response. To address this limitation, phages have been modified as nonreplicative variants. For instance, an engineered nonlytic phage (Pf3R) was constructed to minimize endotoxin release by replacing ORF40 with an endonuclease gene in the phage genome. The bactericidal efficacy of Pf3R remained unchanged, and it could effectively reduce the colony-forming units (CFUs) of Pseudomonas aeruginosa strain PAO1 by 99% upon infection, comparable to that of the parental phage [66]. However, the OD600 value of PAO1 infected with Pf3R remained constant for 7 h, and endotoxin levels in the supernatant were not significantly increased. In an animal trial, the survival rate of mice treated with a Pf3R phage was significantly higher than that of mice treated with a lytic phage. The improved survival rate observed in that study was associated with the attenuation of the inflammatory response induced by the Pf3R treatment, rather than the lytic phage itself.
The phage P954, which targets Staphylococcus aureus, was genetically modified to create a lysis-deficient variant (known as P954R) by replacing an endolysin-encoding gene with the chloramphenicol acetyl transferase (cat) gene through HR [1]. The phage P954R retained a genotype lacking endolysin and was capable of generating plaques by utilizing a heterologous endolysin that was expressed in the propagation host. The bactericidal efficacy of P954 and P954R was similar, both of which resulted in a nearly 90% reduction in colony-forming units (CFU). In addition, the engineered phage exhibited the ability to effectively treat lethal methicillin-resistant Staphylococcus aureus (MRSA) infections in mice, highlighting its potential as a promising therapeutic intervention.
These studies highlight the potential of endolysin gene disruption in reducing the number of phages released by their lytic parent phage following infection. In clinical settings, this approach offers the benefit of a precise dosage, addressing a key concern regarding phage therapy. Additionally, it may result in a reduced immune response and endotoxin release when targeting Gram-negative bacteria.
3.7. Engineering Phage as a Nanocarrier
Phages can also be utilized as nanocarriers for the targeted elimination of pathogenic microorganisms and tumor cells through genetic manipulation or chemical modification [67]. These modified phages exhibit a specific recognition of targeted cells, triggering the controlled release of payloads. Peng et al. [68] engineered the filamentous phage M13 by exchanging its receptor-binding domain with that of another phage naturally targeting a different bacterial genus, resulting in the creation of a chimeric phage, M13KE. Subsequently, the pVIII shell proteins of the M13KE phage was chemically modified using n-succinimidyl S-acetylthioacetate (SATP) to introduce thiol groups, enabling the phage to conjugate with gold nanorods. Upon excitation with near-infrared light, the chimeric phage M13KE conjugated with gold nanorods released energy through nonradiative decay pathways, resulting in a localized heat generation that effectively eradicated the targeted bacterial cells. In addition, the irradiation of the gold nanorods resulted in the destruction of phages, reducing the potential toxicity compared to traditional phage therapy, while achieving a precise dosage control [69,70].
This approach proposed herein is most appropriate for treating tissues or surfaces that are directly accessible. In the short term, this technique could be used for localized topical therapy, particularly for wound infections or the colonization of medical devices, where engineered phages can be directly applied to the biofilm. For instance, P. aeruginosa is recognized as a pulmonary pathogen, but drug-resistant P. aeruginosa is also a significant pathogen in chronic wounds [46], surgical site infections, and burns [71]. These wounds are readily accessible for the application of therapeutic nanomaterials and NIR irradiation.
3.8. Clinical Application of Engineering Bacteriophages
In 2019, the application of genetically modified phages in clinical treatment was initially observed. In this particular instance, a 15-year-old recipient of a bilateral lung transplant was afflicted with an infection caused by M. abscessus, resulting in the development of cystic fibrosis in the transplanted lungs [25]. Three mycobacterophages, namely, Muddy, ZoeJ, and Bps, were administered intravenously to the patient as a therapeutic intervention. Two mycobacteriophages, ZoeJ and Bps, were genetically modified using the BRED strategy to eliminate the suppressor gene, resulting in their transformation into lytic bacteriophages. The administration of phages intravenously was well tolerated and resulted in a significant clinical improvement, such as the closure of sternal wounds, enhanced liver function, and a substantial resolution of infected skin nodules.
In a similar study, two types of mycobacteriophages, namely, D29_HRMGD40 and BPsΔ33HTH_HRM10, were administered intravenously to a 26-year-old patient suffering from severe cystic fibrosis caused by drug-resistant M. abscessus in pulmonary infections [72]. BPsΔ33HTH_HRM10 is a variant of the host range mutans (HRMs) derived from an engineered lytic derivative of BPs, and D29_ HRMGD40 is an HRM of D29 isolated on an M. abscessus strain GD40, which does not resemble the D29 parent’s effects on severe M. abscessus clinical isolates. The M. abscessus isolate was successfully eradicated through incubation with BPsΔ33HTH_HRM10 and D29_HRMGD40, alone or in combination, across a diverse range of bacterial and phage concentrations. Following the administration of the treatment, the patient’s lungs were effectively cleared of the drug-resistant mycobacterium abscesses, leading to the successful completion of a lung transplantation procedure. This case represents a significant milestone in the field of medical research as it marks the initial application of engineered phage therapy in the treatment of drug-resistant M. abscessus.
However, the phages Muddy, BPs, and ZoeJ exhibited limited efficacy in eradicating other clinical isolates of M. abscessus, suggesting that the three-phage cocktail may not be universally effective. The intricate nature of cases such as this one poses challenges in accurately evaluating the efficacy of phage therapy. As there have been no prior reports of successfully treating pulmonary M. abscessus infection with phages, it is challenging to ascertain whether the outcomes observed in this singular case can be extrapolated to other patients undergoing this therapy. Additionally, there may be unidentified factors that have differentially influenced the treatment response.

Table 1.
Bactericidal effect of genetically engineered phage.
Table 1.
Bactericidal effect of genetically engineered phage.
| Strain | Phage | Technology | Result | Ref. |
|---|---|---|---|---|
| E. coli | T3, T7 | Engineering phage genomes in Saccharomyces cerevisiae | Expanding phage host range | [28] |
| P. aeruginosa | P793 | Recombining with pGhost8 | Expanding phage host range | [73] |
| E. coli | T2 | Recombining with tail fiber gene | Expanding phage host range | [56] |
| P. aeruginosa | PaP1 | Recombining with ORF84 | Expanding phage host range | [74] |
| E. coli | T3 | Phage tail fiber mutagenesis | Expanding phage host range | [75] |
| E. coli | Fd | Recombining with tail fiber gene | Expanding phage host range | [58] |
| E. coli | T2, Fd | Recombining with tail fiber gene | Expanding phage host range | [76] |
| E. coli | PSA | Recombining with receptor binding proteins (RBPs) | Expanding phage host range | [77] |
| E. coli | T4 | Generating gp37 and gp38 variants | Expanding phage host range | [78] |
| E. coli | fd | Recombining with OrfU | Expanding phage host range | [59] |
| E. coli | T7 | Recombining with aiiA | Reducing biofilm formation | [63] |
| E. coli | T7 | Recombining with DspB | Reducing biofilm formation | [61] |
| E. coli | T7 | Recombining with peptide 1018 | Reducing biofilm formation | [79] |
| P. aeruginosa | Pf3 | Recombining with endonuclease BglII | Reducing endotoxin production | [66] |
| S. aureus | P954 | Recombining with chloramphenicol acetyl transferase (cat) gene | Reducing endotoxin production | [80] |
| E. coli | M13 | Recombining with antimicrobial peptides (AMPs) and protein toxins | Reducing endotoxin production | [81] |
| E. coli | λ | Integrating with Ndm-1 and Ctx-M-15 using CRISPR/Cas | Restoring antibiotic sensitivity | [51] |
| E. coli | M13 | Recombining with streptomycin sensitive genes | Restoring antibiotic sensitivity | [52] |
| L. monocytogenes | B025 | Removing lysogen module | Improving lytic ability | [34] |
| S. aureus | φMN1 | Integrating with CRISPR/Cas | Improving lytic ability | [48] |
| S. aureus | ØSaBov | Integrating with CRISPR/Cas | Improving lytic ability | [82] |
| E. amylovora | Y2 | Recombining with Depolymerase | Improving lytic ability | [83] |
| E. coli | M13 | CRISPR-cas9 target resistance genes and virulent genes | Improving lytic ability | [47] |
| E. coli | M13 | Recombining with peptide RGD and PmpD | Improving lytic ability | [84] |
| E. coli | T4 | HIV antigen was fused to outer capsid proteins | HIV vaccine | [85] |
| E. coli | T4 | Anthrax toxin proteins was fused to outer capsid proteins | Anthrax vaccine | [86] |
| E. coli | T4 | FMDV p1 protein was fused to outer capsid proteins | FMDV vaccine | [87] |
| E. coli | MS2 | Capsids radiolabeled with 64Cu | Targeted drug carriers | [88] |
| E. coli | λ | Recombining with integrin-binding peptide | Phage-mediated gene delivery and expression | [89] |
| S. typhimurium | P22 | Chemical modification by DTPA | Gd (III) carrier | [90] |
| E. coli | T7 | Recombining with gold-binding peptide | Gold nanorods carrier | [91] |
| E. coli | fd–tet | Self-assembled siRNA−nanophages | siRNA carrier | [92] |
| E. coli | M13 | Chemical modification to form Au-S bonds | Gold nanorods carrier | [68] |
| E. coli | M13 | Recombining with a biotin acceptor peptide (BAP) | Targeted drug carriers | [93] |
| E. coli | M13 | Recombining with a biotin acceptor peptide (BAP) | Targeted drug carriers | [94] |
| E. coli | fUSE5-ZZ | Recombining with IgG Fc-binding ZZ domain of protein A | Targeted drug carriers | [95] |
| E. coli | fUSE5-ZZ | Recombining with IgG Fc-binding ZZ domain of protein A | Antibacterial drug carriers | [96,97] |
| E. coli | f88 | Recombining with myelin oligodendrocyte glycoprotein (MOG) | Vector-mediated antigen delivery | [98] |
| E. coli | T4 | Recombining with GFP | Luciferase reporter phage | [99] |
| E. coli | T7 | Recombining with biotinylation peptide | Streptavidin-coated quantum dots reporter phage | [100] |
| B. anthracis | Wβ | Recombining with luxAB-2 | Bioluminescent reporter phage | [101,102] |
| E. coli | phiV10 | Recombining with luxCDABE operon | Luciferase reporter phage | [103] |
| L. monocytogenes | A511 | Recombining with nanoluciferase | Nanoluciferase (NLuc) reporter phage | [104] |
| E. coli | T7 | Recombining with nanoluciferase | Nanoluciferase (NLuc) reporter phage | [105] |
| E. coli | ΦV10 | Recombining with nanoluciferase | Nanoluciferase (NLuc) reporter phage | [106] |
| E. coli | K1E | Recombining with nanoluciferase | Nanoluciferase (NLuc) reporter phage | [107] |
| E. coli | T7 | Recombining with β-galactosidase | β-galactosidase reporter phage | [108] |
| M. smegmatis | TM4 | Recombining with GFP or ZsYellow | Fluorescent reporter phage | [109] |
| M. smegmatis | D29 | Recombining with Phsp60-egfp cassette using BRED | EGFP reporter phage | [24] |
| E. amylovora | Y2 | Homologous recombination with LuxAB | Luciferase reporter phage | [83] |
| E. coli | T7 | Homologous recombination with PhoE | Enhancing the half-life of phage | [65] |
| L. monocytogenes | A511 | Bacteriophages PEGylation | Enhancing the half-life of phage | [110] |
| S. typhi | Felix-O1 | Bacteriophages PEGylation | Enhancing the half-life of phage | [110] |
| E. faecalis | fEf11 | Recombining with defective prophage | Improving lytic ability and expanding phage host range | [111] |
4. Discussion
Compared to natural phages, genetically modified phages offer stronger advantages in combating bacterial infections. As stated in Section 3, genetically modified phages have demonstrated an enhanced efficacy in combating bacterial infections through various mechanisms, including an improved bactericidal activity, restored antibiotic susceptibility, decreased endotoxin secretion, and other related effects. Additionally, genetically modified phages have the ability to display antigenic peptides on their capsid proteins, thereby effectively activating the immune system and eliciting immune responses. For instance, phage T4 has been demonstrated to effectively present HIV and anthrax toxin antigens on its surface, thereby eliciting an optimal immune response in mice [86,112]. Finally, genetically modified phages are eligible for patent protection, whereas natural phages are not. This enables companies that have utilized phages as products to attract investment in the capital market, thereby promoting the advancement of engineered phage research [4].
While engineered phages offer numerous benefits, there are also potential consequences that need to be considered. Firstly, there is no doubt that the safety of utilizing engineered phages in human applications is of paramount concern. While phages are generally regarded as safe, rigorous testing is imperative to ensure that engineered phages do not elicit any deleterious effects on the overall health of patients. Additionally, like antibiotics, phages can also develop tolerance in host bacteria. Furthermore, there are concerns regarding the potential for phages to trigger bacterial tolerance similar to that observed with antibiotics. While certain treatment approaches, such as cocktail therapy, have been shown to mitigate bacterial tolerance to phages, the sustained efficacy of phage therapy may still be constrained by the existence of phage tolerance [113]. Finally, in comparison to conventional antibiotics, the regulatory framework for phage therapy remains imperfect. The approval process for phage therapy may be more intricate and time-consuming, potentially impeding patients in need from accessing this treatment modality.
While there are numerous unresolved issues surrounding the application of engineered phages, it is indisputable that, given the pervasive issue of antibiotic resistance, they continue to represent a crucial area of current research. Engineered phages possess immense potential for diverse applications, owing to their ability to be designed and modified to meet specific requirements for each application. Hence, they are emerging as a novel category of biological agents with extensive potential.
Author Contributions
Conceptualization, T.T. and Y.J.; writing—original draft preparation, S.L.; Y.W. and K.J.; writing—review and editing, F.Z., X.G. and J.Z.; visualization, T.T. and Q.L.; supervision, C.Y. and S.L.; funding acquisition, T.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by The Key R&D and Promotion Projects of Henan Province (232102311139); China Postdoctoral Science Foundation (2021m690095); National Innovation and entrepreneurship training program for college students (202210475005; 202210475112; 20221022001; 202210475113; 202210475134); Natural Science Foundation of Henan Province (202300410052).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The study did not report any data.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hansson, K.; Brenthel, A. Imagining a post-antibiotic era: A cultural analysis of crisis and antibiotic resistance. Med. Humanit. 2022, 48, 381–388. [Google Scholar] [CrossRef]
- Tarín-Pelló, A.; Suay-García, B.; Pérez-Gracia, M.T. Antibiotic resistant bacteria: Current situation and treatment options to accelerate the development of a new antimicrobial arsenal. Expert Rev. Anti Infect. Ther. 2022, 20, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Tagliabue, A.; Rappuoli, R. Changing Priorities in Vaccinology: Antibiotic Resistance Moving to the Top. Front. Immunol. 2018, 9, 1068. [Google Scholar] [CrossRef]
- Gordillo Altamirano, F.L.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev. 2019, 32, e00066-18. [Google Scholar] [CrossRef] [PubMed]
- Bragg, R.; van der Westhuizen, W.; Lee, J.Y.; Coetsee, E.; Boucher, C. Bacteriophages as potential treatment option for antibiotic resistant bacteria. Adv. Exp. Med. Biol. 2014, 807, 97–110. [Google Scholar] [CrossRef]
- Royer, S.; Morais, A.P.; da Fonseca Batistão, D.W. Phage therapy as strategy to face post-antibiotic era: A guide to beginners and experts. Arch. Microbiol. 2021, 203, 1271–1279. [Google Scholar] [CrossRef]
- Hatfull, G.F.; Dedrick, R.M.; Schooley, R.T. Phage Therapy for Antibiotic-Resistant Bacterial Infections. Annu. Rev. Med. 2022, 73, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Aslam, S.; Lampley, E.; Wooten, D.; Karris, M.; Benson, C.; Strathdee, S.; Schooley, R.T. Lessons Learned From the First 10 Consecutive Cases of Intravenous Bacteriophage Therapy to Treat Multidrug-Resistant Bacterial Infections at a Single Center in the United States. Open Forum Infect. Dis. 2020, 7, ofaa389. [Google Scholar] [CrossRef]
- Suh, G.A.; Lodise, T.P.; Van Tyne, D.; Tamma, P.D.; Sund, Z.; van Duin, D.; Rybak, M.J.; Patel, R.; Maresso, A.; Nussenblatt, V.; et al. Considerations for the Use of Phage Therapy in Clinical Practice. Antimicrob. Agents Chemother. 2022, 66, e0207121. [Google Scholar] [CrossRef]
- Gurney, J.; Brown, S.P.; Kaltz, O.; Hochberg, M.E. Steering Phages to Combat Bacterial Pathogens. Trends Microbiol. 2020, 28, 85–94. [Google Scholar] [CrossRef]
- Torres-Barceló, C.; Turner, P.E.; Buckling, A. Mitigation of evolved bacterial resistance to phage therapy. Curr. Opin. Virol. 2022, 53, 101201. [Google Scholar] [CrossRef]
- Stacey, H.J.; De Soir, S.; Jones, J.D. The Safety and Efficacy of Phage Therapy: A Systematic Review of Clinical and Safety Trials. Antibiotics 2022, 11, 1340. [Google Scholar] [CrossRef]
- Pires, D.P.; Meneses, L.; Brandão, A.C.; Azeredo, J. An overview of the current state of phage therapy for the treatment of biofilm-related infections. Curr. Opin. Virol. 2022, 53, 101209. [Google Scholar] [CrossRef] [PubMed]
- Kortright, K.E.; Chan, B.K.; Koff, J.L.; Turner, P.E. Phage Therapy: A Renewed Approach to Combat Antibiotic-Resistant Bacteria. Cell Host Microbe 2019, 25, 219–232. [Google Scholar] [CrossRef]
- Kilcher, S.; Loessner, M.J. Engineering Bacteriophages as Versatile Biologics. Trends Microbiol. 2019, 27, 355–367. [Google Scholar] [CrossRef]
- Marinelli, L.J.; Hatfull, G.F.; Piuri, M. Recombineering: A powerful tool for modification of bacteriophage genomes. Bacteriophage 2012, 2, 5–14. [Google Scholar] [CrossRef]
- Del Val, E.; Nasser, W.; Abaibou, H.; Reverchon, S. RecA and DNA recombination: A review of molecular mechanisms. Biochem. Soc. Trans. 2019, 47, 1511–1531. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T.K. Genetically Engineered Phages: A Review of Advances over the Last Decade. Microbiol. Mol. Biol. Rev. MMBR 2016, 80, 523–543. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Chamorro, L.; Boulanger, P.; Rossier, O. Strategies for Bacteriophage T5 Mutagenesis: Expanding the Toolbox for Phage Genome Engineering. Front. Microbiol. 2021, 12, 667332. [Google Scholar] [CrossRef]
- Sharan, S.K.; Thomason, L.C.; Kuznetsov, S.G.; Court, D.L. Recombineering: A homologous recombination-based method of genetic engineering. Nat. Protoc. 2009, 4, 206–223. [Google Scholar] [CrossRef]
- Murphy, K.C. λ Recombination and Recombineering. EcoSal Plus 2016, 7, e0011-2015. [Google Scholar] [CrossRef] [PubMed]
- Łobocka, M.; Dąbrowska, K.; Górski, A. Engineered Bacteriophage Therapeutics: Rationale, Challenges and Future. BioDrugs 2021, 35, 255–280. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, L.J.; Piuri, M.; Swigonová, Z.; Balachandran, A.; Oldfield, L.M.; van Kessel, J.C.; Hatfull, G.F. BRED: A simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS ONE 2008, 3, e3957. [Google Scholar] [CrossRef] [PubMed]
- da Silva, J.L.; Piuri, M.; Broussard, G.; Marinelli, L.J.; Bastos, G.M.; Hirata, R.D.; Hatfull, G.F.; Hirata, M.H. Application of BRED technology to construct recombinant D29 reporter phage expressing EGFP. FEMS Microbiol. Lett. 2013, 344, 166–172. [Google Scholar] [CrossRef]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Fehér, T.; Karcagi, I.; Blattner, F.R.; Pósfai, G. Bacteriophage recombineering in the lytic state using the lambda red recombinases. Microb. Biotechnol. 2012, 5, 466–476. [Google Scholar] [CrossRef]
- Wetzel, K.S.; Guerrero-Bustamante, C.A.; Dedrick, R.M.; Ko, C.C.; Freeman, K.G.; Aull, H.G.; Divens, A.M.; Rock, J.M.; Zack, K.M.; Hatfull, G.F. CRISPY-BRED and CRISPY-BRIP: Efficient bacteriophage engineering. Sci. Rep. 2021, 11, 6796. [Google Scholar] [CrossRef]
- Ando, H.; Lemire, S.; Pires, D.P.; Lu, T.K. Engineering Modular Viral Scaffolds for Targeted Bacterial Population Editing. Cell Syst. 2015, 1, 187–196. [Google Scholar] [CrossRef]
- Gibson, D.G. Oligonucleotide assembly in yeast to produce synthetic DNA fragments. Methods Mol. Biol. 2012, 852, 11–21. [Google Scholar] [CrossRef]
- Jaschke, P.R.; Lieberman, E.K.; Rodriguez, J.; Sierra, A.; Endy, D. A fully decompressed synthetic bacteriophage øX174 genome assembled and archived in yeast. Virology 2012, 434, 278–284. [Google Scholar] [CrossRef]
- Assad-Garcia, N.; D’Souza, R.; Vashee, S.; Fouts, D.E.; Buzzeo, R.; Tripathi, A.; Oldfield, L.M. Cross-Genus “Boot-Up” of Synthetic Bacteriophage in Staphylococcus aureus by Using a New and Efficient DNA Transformation Method. Appl. Environ. Microbiol. 2022, 88, e0148621. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Deng, Z.; Tao, H.; Song, W.; Xing, B.; Liu, W.; Kong, L.; Yuan, S.; Ma, Y.; Wu, Y.; et al. Harnessing stepping-stone hosts to engineer, select, and reboot synthetic bacteriophages in one pot. Cell Rep. Methods 2022, 2, 100217. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Monteiro, R.; Mil-Homens, D.; Fialho, A.; Lu, T.K.; Azeredo, J. Designing P. aeruginosa synthetic phages with reduced genomes. Sci. Rep. 2021, 11, 2164. [Google Scholar] [CrossRef] [PubMed]
- Kilcher, S.; Studer, P.; Muessner, C.; Klumpp, J.; Loessner, M.J. Cross-genus rebooting of custom-made, synthetic bacteriophage genomes in L-form bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Chikada, T.; Kanai, T.; Hayashi, M.; Kasai, T.; Oshima, T.; Shiomi, D. Direct Observation of Conversion From Walled Cells to Wall-Deficient L-Form and Vice Versa in Escherichia coli Indicates the Essentiality of the Outer Membrane for Proliferation of L-Form Cells. Front. Microbiol. 2021, 12, 645965. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinform. 2007, 8, 172. [Google Scholar] [CrossRef]
- Martel, B.; Moineau, S. CRISPR-Cas: An efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res. 2014, 42, 9504–9513. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Oromí-Bosch, A.; Mendoza, S.D.; Karambelkar, S.; Berry, J.D.; Bondy-Denomy, J. Bacteriophage genome engineering with CRISPR-Cas13a. Nat. Microbiol. 2022, 7, 1956–1966. [Google Scholar] [CrossRef]
- Haft, D.H.; Selengut, J.; Mongodin, E.F.; Nelson, K.E. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 2005, 1, e60. [Google Scholar] [CrossRef]
- Shivram, H.; Cress, B.F.; Knott, G.J.; Doudna, J.A. Controlling and enhancing CRISPR systems. Nat. Chem. Biol. 2021, 17, 10–19. [Google Scholar] [CrossRef]
- Lemay, M.L.; Tremblay, D.M.; Moineau, S. Genome Engineering of Virulent Lactococcal Phages Using CRISPR-Cas9. ACS Synth. Biol. 2017, 6, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Kiro, R.; Shitrit, D.; Qimron, U. Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 2014, 11, 42–44. [Google Scholar] [CrossRef]
- Malone, L.M.; Birkholz, N.; Fineran, P.C. Conquering CRISPR: How phages overcome bacterial adaptive immunity. Curr. Opin. Biotechnol. 2021, 68, 30–36. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, L.; Dong, J.; Chen, C.; Zhu, J.; Rao, V.B.; Tao, P. Covalent Modifications of the Bacteriophage Genome Confer a Degree of Resistance to Bacterial CRISPR Systems. J. Virol. 2020, 94, e01630-20. [Google Scholar] [CrossRef]
- Wu, X.; Zhu, J.; Tao, P.; Rao, V.B. Bacteriophage T4 Escapes CRISPR Attack by Minihomology Recombination and Repair. mBio 2021, 12, e0136121. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.; Kerven, J.; Chen, Y.; Sagona, A.P. Genetic Engineering of Bacteriophage K1F with Human Epidermal Growth Factor to Enhance Killing of Intracellular E. coli K1. ACS Synth. Biol. 2023, 12, 2094–2106. [Google Scholar] [CrossRef] [PubMed]
- Citorik, R.J.; Mimee, M.; Lu, T.K. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 2014, 32, 1141–1145. [Google Scholar] [CrossRef]
- Bikard, D.; Euler, C.W.; Jiang, W.; Nussenzweig, P.M.; Goldberg, G.W.; Duportet, X.; Fischetti, V.A.; Marraffini, L.A. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014, 32, 1146–1150. [Google Scholar] [CrossRef]
- Azam, A.H.; Suzuki, M.; Penadés, J.R.; Cui, L.; Kiga, K.; Tan, X.E.; Ibarra-Chávez, R.; Watanabe, S.; Aiba, Y.; Sato’o, Y.; et al. Development of CRISPR-Cas13a-based antimicrobials capable of sequence-specific killing of target bacteria. Nat. Commun. 2020, 11, 2934. [Google Scholar] [CrossRef]
- Meile, S.; Du, J.; Dunne, M.; Kilcher, S.; Loessner, M.J. Engineering therapeutic phages for enhanced antibacterial efficacy. Curr. Opin. Virol. 2022, 52, 182–191. [Google Scholar] [CrossRef]
- Yosef, I.; Manor, M.; Kiro, R.; Qimron, U. Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 7267–7272. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Friedman, N.; Molshanski-Mor, S.; Qimron, U. Reversing bacterial resistance to antibiotics by phage-mediated delivery of dominant sensitive genes. Appl. Environ. Microbiol. 2012, 78, 744–751. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, P.A.; Nobrega, F.L.; Brouns, S.J.J.; Dutilh, B.E. Molecular and Evolutionary Determinants of Bacteriophage Host Range. Trends Microbiol. 2019, 27, 51–63. [Google Scholar] [CrossRef]
- Yosef, I.; Goren, M.G.; Globus, R.; Molshanski-Mor, S.; Qimron, U. Extending the Host Range of Bacteriophage Particles for DNA Transduction. Mol. Cell 2017, 66, 721–728.e3. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, L.; Abdelgader, S.A.; Yu, L.; Xu, J.; Yao, H.; Lu, C.; Zhang, W. Alterations in gp37 Expand the Host Range of a T4-Like Phage. Appl. Environ. Microbiol. 2017, 83, e01576-17. [Google Scholar] [CrossRef] [PubMed]
- Mahichi, F.; Synnott, A.J.; Yamamichi, K.; Osada, T.; Tanji, Y. Site-specific recombination of T2 phage using IP008 long tail fiber genes provides a targeted method for expanding host range while retaining lytic activity. FEMS Microbiol. Lett. 2009, 295, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Le, S.; He, X.; Tan, Y.; Huang, G.; Zhang, L.; Lux, R.; Shi, W.; Hu, F. Mapping the tail fiber as the receptor binding protein responsible for differential host specificity of Pseudomonas aeruginosa bacteriophages PaP1 and JG004. PLoS ONE 2013, 8, e68562. [Google Scholar] [CrossRef]
- Marzari, R.; Sblattero, D.; Righi, M.; Bradbury, A. Extending filamentous phage host range by the grafting of a heterologous receptor binding domain. Gene 1997, 185, 27–33. [Google Scholar] [CrossRef]
- Heilpern, A.J.; Waldor, M.K. pIIICTX, a predicted CTXphi minor coat protein, can expand the host range of coliphage fd to include Vibrio cholerae. J. Bacteriol. 2003, 185, 1037–1044. [Google Scholar] [CrossRef]
- Azeredo, J.; Sutherland, I.W. The use of phages for the removal of infectious biofilms. Curr. Pharm. Biotechnol. 2008, 9, 261–266. [Google Scholar] [CrossRef]
- Lu, T.K.; Collins, J.J. Dispersing biofilms with engineered enzymatic bacteriophage. Proc. Natl. Acad. Sci. USA 2007, 104, 11197–11202. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.H.; Wang, L.H.; Xu, J.L.; Zhang, H.B.; Zhang, X.F.; Zhang, L.H. Quenching quorum-sensing-dependent bacterial infection by an N-acyl homoserine lactonase. Nature 2001, 411, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Pei, R.; Lamas-Samanamud, G.R. Inhibition of biofilm formation by T7 bacteriophages producing quorum-quenching enzymes. Appl. Environ. Microbiol. 2014, 80, 5340–5348. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Wang, L.; Sha, R.; Liu, L.; Qian, J.; Ishimwe, N.; Zhang, W.; Qian, J.; Zhang, Y.; Wen, L. A blood circulation-prolonging peptide anchored biomimetic phage-platelet hybrid nanoparticle system for prolonged blood circulation and optimized anti-bacterial performance. Theranostics 2021, 11, 2278–2296. [Google Scholar] [CrossRef]
- Nobrega, F.L.; Costa, A.R.; Santos, J.F.; Siliakus, M.F.; van Lent, J.W.; Kengen, S.W.; Azeredo, J.; Kluskens, L.D. Genetically manipulated phages with improved pH resistance for oral administration in veterinary medicine. Sci. Rep. 2016, 6, 39235. [Google Scholar] [CrossRef]
- Hagens, S.; Habel, A.; von Ahsen, U.; von Gabain, A.; Bläsi, U. Therapy of experimental pseudomonas infections with a nonreplicating genetically modified phage. Antimicrob. Agents Chemother. 2004, 48, 3817–3822. [Google Scholar] [CrossRef]
- Kaur, S.; Kumari, A.; Kumari Negi, A.; Galav, V.; Thakur, S.; Agrawal, M.; Sharma, V. Nanotechnology Based Approaches in Phage Therapy: Overcoming the Pharmacological Barriers. Front. Pharmacol. 2021, 12, 699054. [Google Scholar] [CrossRef]
- Chen, I.A.; Peng, H.; Borg, R.E.; Dow, L.P.; Pruitt, B.L. Controlled phage therapy by photothermal ablation of specific bacterial species using gold nanorods targeted by chimeric phages. Proc. Natl. Acad. Sci. USA 2020, 117, 1951–1961. [Google Scholar] [CrossRef]
- Wang, P.; Yu, G.; Wei, J.; Liao, X.; Zhang, Y.; Ren, Y.; Zhang, C.; Wang, Y.; Zhang, D.; Wang, J.; et al. A single thiolated-phage displayed nanobody-based biosensor for label-free detection of foodborne pathogen. J. Hazard. Mater. 2023, 443, 130157. [Google Scholar] [CrossRef]
- Peng, H.; Chen, I.A. Rapid Colorimetric Detection of Bacterial Species through the Capture of Gold Nanoparticles by Chimeric Phages. ACS Nano 2019, 13, 1244–1252. [Google Scholar] [CrossRef]
- Turner, K.H.; Everett, J.; Trivedi, U.; Rumbaugh, K.P.; Whiteley, M. Requirements for Pseudomonas aeruginosa acute burn and chronic surgical wound infection. PLoS Genet. 2014, 10, e1004518. [Google Scholar] [CrossRef] [PubMed]
- Nick, J.A.; Dedrick, R.M.; Gray, A.L.; Vladar, E.K.; Smith, B.E.; Freeman, K.G.; Malcolm, K.C.; Epperson, L.E.; Hasan, N.A.; Hendrix, J.; et al. Host and pathogen response to bacteriophage engineered against Mycobacterium abscessus lung infection. Cell 2022, 185, 1860–1874.e12. [Google Scholar] [CrossRef] [PubMed]
- Kot, W.; Hammer, K.; Neve, H.; Vogensen, F.K. Identification of the receptor-binding protein in lytic Leuconostoc pseudomesenteroides bacteriophages. Appl. Environ. Microbiol. 2013, 79, 3311–3314. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, L.G.; Slade, S.E.; Seyffert, N.; Santos, A.R.; Castro, T.L.; Silva, W.M.; Santos, A.V.; Santos, S.G.; Farias, L.M.; Carvalho, M.A.; et al. A combined approach for comparative exoproteome analysis of Corynebacterium pseudotuberculosis. BMC Microbiol. 2011, 11, 12. [Google Scholar] [CrossRef]
- Yehl, K.; Lemire, S.; Yang, A.C.; Ando, H.; Mimee, M.; Torres, M.T.; de la Fuente-Nunez, C.; Lu, T.K. Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail Fiber Mutagenesis. Cell 2019, 179, 459–469.e9. [Google Scholar] [CrossRef]
- Lin, T.Y.; Lo, Y.H.; Tseng, P.W.; Chang, S.F.; Lin, Y.T.; Chen, T.S. A T3 and T7 recombinant phage acquires efficient adsorption and a broader host range. PLoS ONE 2012, 7, e30954. [Google Scholar] [CrossRef]
- Dunne, M.; Rupf, B.; Tala, M.; Qabrati, X.; Ernst, P.; Shen, Y.; Sumrall, E.; Heeb, L.; Plückthun, A.; Loessner, M.J.; et al. Reprogramming Bacteriophage Host Range through Structure-Guided Design of Chimeric Receptor Binding Proteins. Cell Rep. 2019, 29, 1336–1350.e4. [Google Scholar] [CrossRef]
- Pouillot, F.; Blois, H.; Iris, F. Genetically engineered virulent phage banks in the detection and control of emergent pathogenic bacteria. Biosecurity Bioterrorism Biodefense Strategy Pract. Sci. 2010, 8, 155–169. [Google Scholar] [CrossRef]
- Lemon, D.J.; Kay, M.K.; Titus, J.K.; Ford, A.A.; Chen, W.; Hamlin, N.J.; Hwang, Y.Y. Construction of a genetically modified T7Select phage system to express the antimicrobial peptide 1018. J. Microbiol. 2019, 57, 532–538. [Google Scholar] [CrossRef]
- Paul, V.D.; Sundarrajan, S.; Rajagopalan, S.S.; Hariharan, S.; Kempashanaiah, N.; Padmanabhan, S.; Sriram, B.; Ramachandran, J. Lysis-deficient phages as novel therapeutic agents for controlling bacterial infection. BMC Microbiol. 2011, 11, 195. [Google Scholar] [CrossRef]
- Krom, R.J.; Bhargava, P.; Lobritz, M.A.; Collins, J.J. Engineered Phagemids for Nonlytic, Targeted Antibacterial Therapies. Nano Lett. 2015, 15, 4808–4813. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Moon, B.Y.; Park, J.W.; Thornton, J.A.; Park, Y.H.; Seo, K.S. Genetic engineering of a temperate phage-based delivery system for CRISPR/Cas9 antimicrobials against Staphylococcus aureus. Sci. Rep. 2017, 7, 44929. [Google Scholar] [CrossRef] [PubMed]
- Born, Y.; Fieseler, L.; Thöny, V.; Leimer, N.; Duffy, B.; Loessner, M.J. Engineering of Bacteriophages Y2::dpoL1-C and Y2::luxAB for Efficient Control and Rapid Detection of the Fire Blight Pathogen, Erwinia amylovora. Appl. Environ. Microbiol. 2017, 83, e00341-17. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, S.R.; Yoo, S.Y.; Lee, S.W.; Dean, D. Engineered phage-based therapeutic materials inhibit Chlamydia trachomatis intracellular infection. Biomaterials 2012, 33, 5166–5174. [Google Scholar] [CrossRef]
- Sathaliyawala, T.; Rao, M.; Maclean, D.M.; Birx, D.L.; Alving, C.R.; Rao, V.B. Assembly of human immunodeficiency virus (HIV) antigens on bacteriophage T4: A novel in vitro approach to construct multicomponent HIV vaccines. J. Virol. 2006, 80, 7688–7698. [Google Scholar] [CrossRef] [PubMed]
- Shivachandra, S.B.; Li, Q.; Peachman, K.K.; Matyas, G.R.; Leppla, S.H.; Alving, C.R.; Rao, M.; Rao, V.B. Multicomponent anthrax toxin display and delivery using bacteriophage T4. Vaccine 2007, 25, 1225–1235. [Google Scholar] [CrossRef]
- Ren, Z.J.; Tian, C.J.; Zhu, Q.S.; Zhao, M.Y.; Xin, A.G.; Nie, W.X.; Ling, S.R.; Zhu, M.W.; Wu, J.Y.; Lan, H.Y.; et al. Orally delivered foot-and-mouth disease virus capsid protomer vaccine displayed on T4 bacteriophage surface: 100% protection from potency challenge in mice. Vaccine 2008, 26, 1471–1481. [Google Scholar] [CrossRef]
- Aanei, I.L.; ElSohly, A.M.; Farkas, M.E.; Netirojjanakul, C.; Regan, M.; Taylor Murphy, S.; O’Neil, J.P.; Seo, Y.; Francis, M.B. Biodistribution of Antibody-MS2 Viral Capsid Conjugates in Breast Cancer Models. Mol. Pharm. 2016, 13, 3764–3772. [Google Scholar] [CrossRef]
- Lankes, H.A.; Zanghi, C.N.; Santos, K.; Capella, C.; Duke, C.M.; Dewhurst, S. In vivo gene delivery and expression by bacteriophage lambda vectors. J. Appl. Microbiol. 2007, 102, 1337–1349. [Google Scholar] [CrossRef]
- Min, J.; Jung, H.; Shin, H.H.; Cho, G.; Cho, H.; Kang, S. Implementation of P22 viral capsids as intravascular magnetic resonance T1 contrast conjugates via site-selective attachment of Gd(III)-chelating agents. Biomacromolecules 2013, 14, 2332–2339. [Google Scholar] [CrossRef]
- Zhou, X.; Cao, P.; Zhu, Y.; Lu, W.; Gu, N.; Mao, C. Phage-mediated counting by the naked eye of miRNA molecules at attomolar concentrations in a Petri dish. Nat. Mater. 2015, 14, 1058–1064. [Google Scholar] [CrossRef]
- Bedi, D.; Gillespie, J.W.; Petrenko, V.A., Jr.; Ebner, A.; Leitner, M.; Hinterdorfer, P.; Petrenko, V.A. Targeted delivery of siRNA into breast cancer cells via phage fusion proteins. Mol. Pharm. 2013, 10, 551–559. [Google Scholar] [CrossRef] [PubMed]
- DePorter, S.M.; McNaughton, B.R. Engineered M13 bacteriophage nanocarriers for intracellular delivery of exogenous proteins to human prostate cancer cells. Bioconjugate Chem. 2014, 25, 1620–1625. [Google Scholar] [CrossRef]
- Ghosh, D.; Kohli, A.G.; Moser, F.; Endy, D.; Belcher, A.M. Refactored M13 bacteriophage as a platform for tumor cell imaging and drug delivery. ACS Synth. Biol. 2012, 1, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Bar, H.; Yacoby, I.; Benhar, I. Killing cancer cells by targeted drug-carrying phage nanomedicines. BMC Biotechnol. 2008, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Yacoby, I.; Bar, H.; Benhar, I. Targeted drug-carrying bacteriophages as antibacterial nanomedicines. Antimicrob. Agents Chemother. 2007, 51, 2156–2163. [Google Scholar] [CrossRef]
- Yacoby, I.; Shamis, M.; Bar, H.; Shabat, D.; Benhar, I. Targeting antibacterial agents by using drug-carrying filamentous bacteriophages. Antimicrob. Agents Chemother. 2006, 50, 2087–2097. [Google Scholar] [CrossRef]
- Rakover, I.S.; Zabavnik, N.; Kopel, R.; Paz-Rozner, M.; Solomon, B. Antigen-specific therapy of EAE via intranasal delivery of filamentous phage displaying a myelin immunodominant epitope. J. Neuroimmunol. 2010, 225, 68–76. [Google Scholar] [CrossRef]
- Namura, M.; Hijikata, T.; Miyanaga, K.; Tanji, Y. Detection of Escherichia coli with fluorescent labeled phages that have a broad host range to E. coli in sewage water. Biotechnol. Prog. 2008, 24, 481–486. [Google Scholar] [CrossRef]
- Edgar, R.; McKinstry, M.; Hwang, J.; Oppenheim, A.B.; Fekete, R.A.; Giulian, G.; Merril, C.; Nagashima, K.; Adhya, S. High-sensitivity bacterial detection using biotin-tagged phage and quantum-dot nanocomplexes. Proc. Natl. Acad. Sci. USA 2006, 103, 4841–4845. [Google Scholar] [CrossRef]
- Nguyen, C.; Makkar, R.; Sharp, N.J.; Page, M.A.; Molineux, I.J.; Schofield, D.A. Detection of Bacillus anthracis spores from environmental water using bioluminescent reporter phage. J. Appl. Microbiol. 2017, 123, 1184–1193. [Google Scholar] [CrossRef]
- Sharp, N.J.; Molineux, I.J.; Page, M.A.; Schofield, D.A. Rapid Detection of Viable Bacillus anthracis Spores in Environmental Samples by Using Engineered Reporter Phages. Appl. Environ. Microbiol. 2016, 82, 2380–2387. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, M.; Kim, S.; Ryu, S. Sensitive detection of viable Escherichia coli O157:H7 from foods using a luciferase-reporter phage phiV10lux. Int. J. Food Microbiol. 2017, 254, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Meile, S.; Sarbach, A.; Du, J.; Schuppler, M.; Saez, C.; Loessner, M.J. Engineered Reporter Phages for Rapid Bioluminescence-Based Detection and Differentiation of Viable Listeria Cells. Appl. Environ. Microbiol. 2020, 86, e00442-20. [Google Scholar] [CrossRef] [PubMed]
- Hinkley, T.C.; Garing, S.; Singh, S.; Le Ny, A.M.; Nichols, K.P.; Peters, J.E.; Talbert, J.N.; Nugen, S.R. Reporter bacteriophage T7(NLC) utilizes a novel NanoLuc::CBM fusion for the ultrasensitive detection of Escherichia coli in water. Analyst 2018, 143, 4074–4082. [Google Scholar] [CrossRef]
- Zhang, D.; Coronel-Aguilera, C.P.; Romero, P.L.; Perry, L.; Minocha, U.; Rosenfield, C.; Gehring, A.G.; Paoli, G.C.; Bhunia, A.K.; Applegate, B. The Use of a Novel NanoLuc-Based Reporter Phage for the Detection of Escherichia coli O157:H7. Sci. Rep. 2016, 6, 33235. [Google Scholar] [CrossRef]
- Dow, P.; Kotz, K.; Gruszka, S.; Holder, J.; Fiering, J. Acoustic separation in plastic microfluidics for rapid detection of bacteria in blood using engineered bacteriophage. Lab A Chip 2018, 18, 923–932. [Google Scholar] [CrossRef]
- Chen, J.; Picard, R.A.; Wang, D. Lyophilized Engineered Phages for Escherichia coli Detection in Food Matrices. ACS Sens. 2017, 2, 1573–1577. [Google Scholar] [CrossRef]
- Piuri, M.; Jacobs, W.R., Jr.; Hatfull, G.F. Fluoromycobacteriophages for rapid, specific, and sensitive antibiotic susceptibility testing of Mycobacterium tuberculosis. PLoS ONE 2009, 4, e4870. [Google Scholar] [CrossRef]
- Kim, K.P.; Cha, J.D.; Jang, E.H.; Klumpp, J.; Hagens, S.; Hardt, W.D.; Lee, K.Y.; Loessner, M.J. PEGylation of bacteriophages increases blood circulation time and reduces T-helper type 1 immune response. Microb. Biotechnol. 2008, 1, 247–257. [Google Scholar] [CrossRef]
- Zhang, H.; Fouts, D.E.; DePew, J.; Stevens, R.H. Genetic modifications to temperate Enterococcus faecalis phage Ef11 that abolish the establishment of lysogeny and sensitivity to repressor, and increase host range and productivity of lytic infection. Microbiology 2013, 159, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Zalewska-Piątek, B.; Piątek, R. Bacteriophages as Potential Tools for Use in Antimicrobial Therapy and Vaccine Development. Pharmaceuticals 2021, 14, 331. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Costa, A.R.; Pinto, G.; Meneses, L.; Azeredo, J. Current challenges and future opportunities of phage therapy. FEMS Microbiol. Rev. 2020, 44, 684–700. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).