
BET Inhibitor JQ1 Attenuates Feline Leukemia Virus DNA, Provirus, and Antigen Production in Domestic Cat Cell Lines

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Virus
2.2. Cell Counts
2.3. Treatments
2.4. Creation of Virus Stocks
2.5. Treatment and Virus Challenge of Cell Lines
2.6. FeLV p27 Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. Normalization of FeLV p27 Concentration
2.8. DNA Extraction and Measurement
2.9. Creation of Plasmid Standards for Absolute Quantification and Normalization of Total FeLV DNA
2.10. Quantitative PCR (qPCR) for Absolute Quantification and Normalization of Total FeLV DNA Load
2.11. Relative Quantification of FeLV Proviral Load
2.12. Statistical Analyses
3. Results
3.1. Effects of (+)-JQ1 on 81C Cell Cultures Challenged with FeLV
3.1.1. Effect on Viable Adherent Cell Counts
3.1.2. Effect on FeLV p27 Capsid Protein Expression
3.1.3. Effect on Total FeLV DNA Load
3.1.4. Effect on Proviral Integration
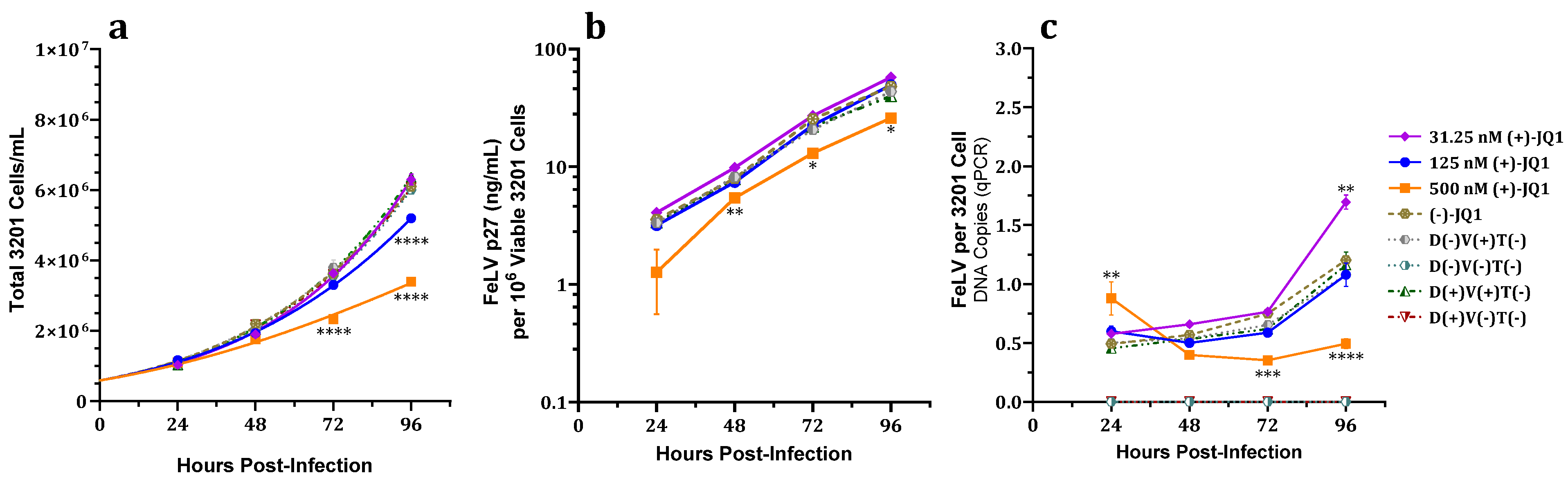
3.2. Effects of (+)-JQ1 on 3201 Cell Cultures Challenged with FeLV
3.2.1. Effects of DMSO Vehicle
3.2.2. Effect on Total and Viable Cell Counts
3.2.3. Effect on FeLV p27 Capsid Protein Expression
3.2.4. Effect on Total FeLV DNA Load
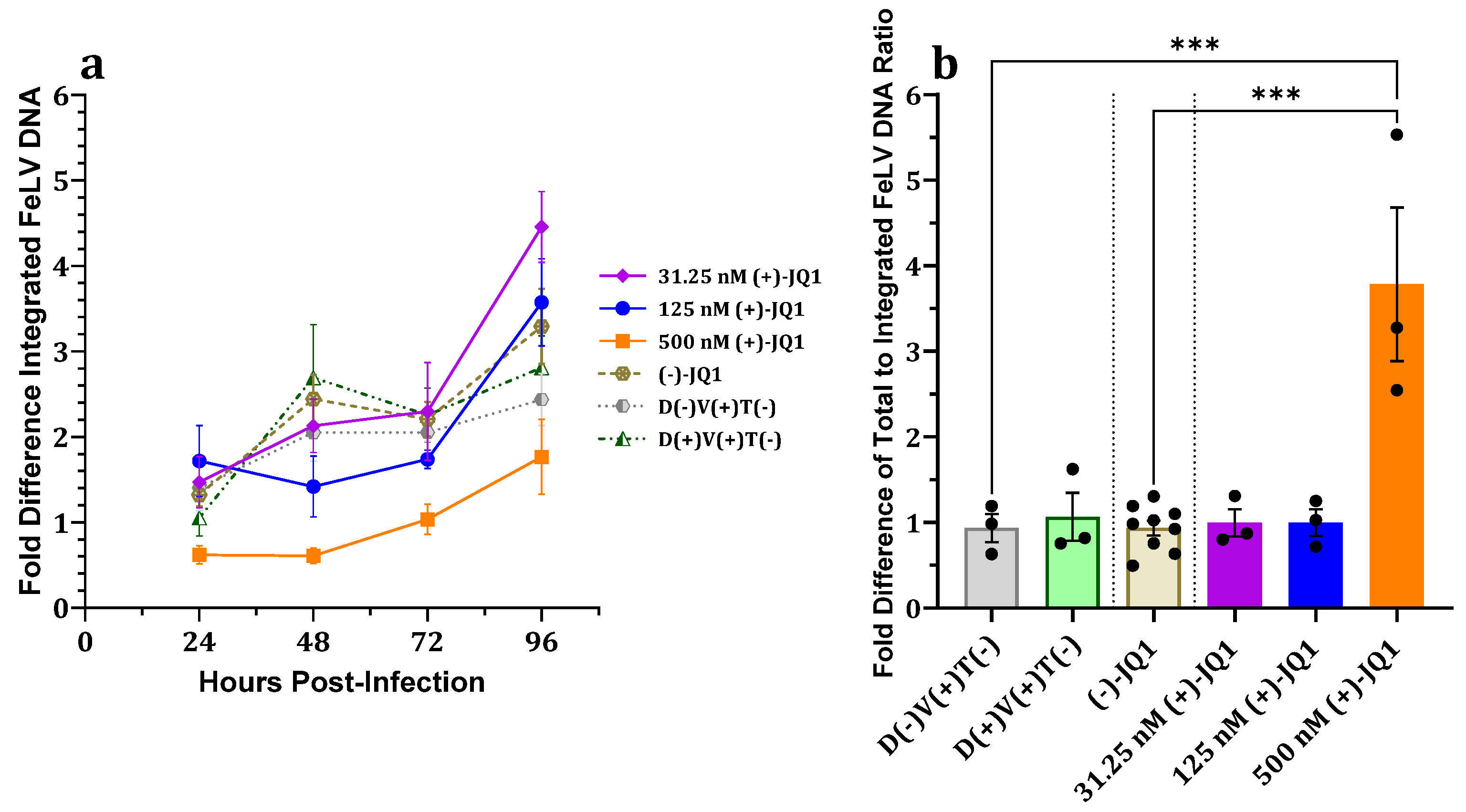
3.2.5. Effect on Proviral Integration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BET | bromodomain and extra-terminal domain |
| BETi | BET protein (bromodomain) inhibitor |
| DMSO | dimethyl sulfoxide |
| ELISA | enzyme-linked immunosorbent assay |
| ET | extra-terminal |
| fALB | feline albumin |
| FeLV | feline leukemia virus |
| HIV | human immunodeficiency virus |
| hpi | hours post-infection |
| JQ1 | archetypal BETi |
| LTR | long terminal repeat |
| MuLV | murine leukemia virus |
| ‘NOISE’ | nested qPCR using λ-bacteriophage-specific sequence as a first-round primer binding site |
| p27 | FeLV capsid protein |
| PCR | polymerase chain reaction |
| qPCR | quantitative PCR |
| SINE | short interspersed nuclear elements |
| ‘SINE’ | nested qPCR utilizing SINEs as a first-round primer binding site |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligomer | Sequence (5′-3′) |
|---|---|
| FeLVU3-Fo | AACAGCAGAAGTTTCAAGGCC |
| FeLVU3-Re | TTATAGCAGAAAGCGCGCG |
| FeLVU3-Pr | CCAGCGGTCTCCAGGCTCCCCA 1 |
| fALB-Fo | GATGGCTGATTGCTGTGAGA |
| fALB-Re | CCCAGGAACCTCTGTTCATT |
| fALB-Pr | ATCCCGGCTTCGGTCAGCTG 2 |
| Oligomer | Sequence (5′-3′) |
|---|---|
| SINE1-Fo | ATGCCACGTAAGCGAAACATTATAGCAGAAAGCGCGCG 1 |
| SINE1-Re | AGAAGCCGAAGCAGGCTC 2 |
| NOISE-Fo | ATGCCACGTAAGCGAAACATTATAGCAGAAAGCGCGCG 1 |
| NOISE-Re | ATGCCACGTAAGCGAAACATT 1 |
| IntFeLV-Fo | ATGCCACGTAAGCGAAACATT 1 |
| IntFeLV-Re | AACAGCAGAAGTTTCAAGGCC 3 |
| FeLVU3-Pr | CCAGCGGTCTCCAGGCTCCCCA 4 |
References
- Levy, J.K.; Lorentzen, L.; Shields, J.; Lewis, H. Long-term outcome of cats with natural FeLV and FIV infection. In Proceedings of the 8th International Feline Retrovirus Research Symposium, Washington, DC, USA, 8–11 October 2006. [Google Scholar]
- Flynn, J.N.; Dunham, S.P.; Watson, V.; Jarrett, O. Longitudinal analysis of feline leukemia virus-specific cytotoxic T lymphocytes: Correlation with recovery from infection. J. Virol. 2002, 76, 2306–2315. [Google Scholar] [CrossRef]
- Pepin, A.C.; Tandon, R.; Cattori, V.; Niederer, E.; Riond, B.; Willi, B.; Lutz, H.; Hofmann-Lehmann, R. Cellular segregation of feline leukemia provirus and viral RNA in leukocyte subsets of long-term experimentally infected cats. Virus Res. 2007, 127, 9–16. [Google Scholar] [CrossRef]
- Cattori, V.; Tandon, R.; Pepin, A.; Lutz, H.; Hofmann-Lehmann, R. Rapid detection of feline leukemia virus provirus integration into feline genomic DNA. Mol. Cell. Probes 2006, 20, 172–181. [Google Scholar] [CrossRef]
- Torres, A.N.; Mathiason, C.K.; Hoover, E.A. Re-examination of feline leukemia virus: Host relationships using real-time PCR. Virology 2005, 332, 272–283. [Google Scholar] [CrossRef]
- Powers, J.A.; Chiu, E.S.; Kraberger, S.J.; Roelke-Parker, M.; Lowery, I.; Erbeck, K.; Troyer, R.; Carver, S.; VandeWoude, S. Feline leukemia virus (FeLV) disease outcomes in a domestic cat breeding colony: Relationship to endogenous FeLV and other chronic viral infections. J. Virol. 2018, 92, e00649-18. [Google Scholar] [CrossRef]
- Helfer-Hungerbuehler, A.K.; Widmer, S.; Kessler, Y.; Riond, B.; Boretti, F.S.; Grest, P.; Lutz, H.; Hofmann-Lehmann, R. Long-term follow up of feline leukemia virus infection and characterization of viral RNA loads using molecular methods in tissues of cats with different infection outcomes. Virus Res. 2015, 197, 137–150. [Google Scholar] [CrossRef]
- Beall, M.J.; Buch, J.; Cahill, R.J.; Clark, G.; Hanscom, J.; Estrada, M.; Leutenegger, C.M.; Chandrashekar, R. Evaluation of a quantitative enzyme-linked immunosorbent assay for feline leukemia virus p27 antigen and comparison to proviral DNA loads by real-time polymerase chain reaction. Comp. Immunol. Microbiol. Infect. Dis. 2019, 67, 101348. [Google Scholar] [CrossRef]
- Beall, M.J.; Buch, J.; Clark, G.; Estrada, M.; Rakitin, A.; Hamman, N.T.; Frenden, M.K.; Jefferson, E.P.; Amirian, E.S.; Levy, J.K. Feline leukemia virus p27 antigen concentration and proviral DNA load are associated with survival in naturally infected cats. Viruses 2021, 13, 302. [Google Scholar] [CrossRef]
- Francis, D.P.; Essex, M.; Cotter, S.M.; Gutensohn, N.; Jakowski, R.; Hardy, W.D., Jr. Epidemiologic association between virus-negative feline leukemia and the horizontally transmitted feline leukemia virus. Cancer Lett. 1981, 12, 37–42. [Google Scholar] [CrossRef]
- Boesch, A.; Cattori, V.; Riond, B.; Willi, B.; Meli, M.L.; Rentsch, K.M.; Hosie, M.J.; Hofmann-Lehmann, R.; Lutz, H. Evaluation of the effect of short-term treatment with the integrase inhibitor raltegravir (Isentress) on the course of progressive feline leukemia virus infection. Vet. Microbiol. 2015, 175, 167–178. [Google Scholar] [CrossRef]
- Lutz, H.; Pedersen, N.; Higgins, J.; Hubscher, U.; Troy, F.A.; Theilen, G.H. Humoral immune reactivity to feline leukemia virus and associated antigens in cats naturally infected with feline leukemia virus. Cancer Res. 1980, 40, 3642–3651. [Google Scholar] [PubMed]
- Nicastri, E.; Tommasi, C.; Abbate, I.; Bonora, S.; Tempestilli, M.; Bellagamba, R.; Viscione, M.; Rozera, G.; Gallo, A.L.; Ivanovic, J.; et al. Effect of raltegravir on the total and unintegrated proviral HIV DNA during raltegravir-based HAART. Antivir. Ther. 2011, 16, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Raffi, F.; Rachlis, A.; Stellbrink, H.J.; Hardy, W.D.; Torti, C.; Orkin, C.; Bloch, M.; Podzamczer, D.; Pokrovsky, V.; Pulido, F.; et al. Once-daily dolutegravir versus raltegravir in antiretroviral-naive adults with HIV-1 infection: 48 week results from the randomised, double-blind, non-inferiority SPRING-2 study. Lancet 2013, 381, 735–743. [Google Scholar] [CrossRef]
- De Rijck, J.; de Kogel, C.; Demeulemeester, J.; Vets, S.; El Ashkar, S.; Malani, N.; Bushman, F.D.; Landuyt, B.; Husson, S.J.; Busschots, K.; et al. The BET family of proteins targets Moloney murine leukemia virus integration near transcription start sites. Cell Rep. 2013, 5, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.S.; Maetzig, T.; Maertens, G.N.; Sharif, A.; Rothe, M.; Weidner-Glunde, M.; Galla, M.; Schambach, A.; Cherepanov, P.; Schulz, T.F. Bromo- and extraterminal domain chromatin regulators serve as cofactors for murine leukemia virus integration. J. Virol. 2013, 87, 12721–12736. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Larue, R.C.; Plumb, M.R.; Malani, N.; Male, F.; Slaughter, A.; Kessl, J.J.; Shkriabai, N.; Coward, E.; Aiyer, S.S.; et al. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc. Natl. Acad. Sci. USA 2013, 110, 12036–12041. [Google Scholar] [CrossRef] [PubMed]
- Aiyer, S.; Swapna, G.V.; Malani, N.; Aramini, J.M.; Schneider, W.M.; Plumb, M.R.; Ghanem, M.; Larue, R.C.; Sharma, A.; Studamire, B.; et al. Altering murine leukemia virus integration through disruption of the integrase and BET protein family interaction. Nucleic Acids Res. 2014, 42, 5917–5928. [Google Scholar] [CrossRef]
- Crowe, B.L.; Larue, R.C.; Yuan, C.; Hess, S.; Kvaratskhelia, M.; Foster, M.P. Structure of the Brd4 ET domain bound to a C-terminal motif from γ-retroviral integrases reveals a conserved mechanism of interaction. Proc. Natl. Acad. Sci. USA 2016, 113, 2086–2091. [Google Scholar] [CrossRef]
- Larue, R.C.; Plumb, M.R.; Crowe, B.L.; Shkriabai, N.; Sharma, A.; DiFiore, J.; Malani, N.; Aiyer, S.S.; Roth, M.J.; Bushman, F.D.; et al. Bimodal high-affinity association of Brd4 with murine leukemia virus integrase and mononucleosomes. Nucleic Acids Res. 2014, 42, 4868–4881. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Fischinger, P.J.; Blevins, C.S.; Nomura, S. Simple, quantitative assay for both xenotropic murine leukemia and ecotropic feline leukemia viruses. J. Virol. 1974, 14, 177–179. [Google Scholar] [CrossRef]
- Crandell, R.A.; Fabricant, C.G.; Nelson-Rees, W.A. Development, characterization, and viral susceptibility of a feline (Felis catus) renal cell line (CRFK). In Vitro 1973, 9, 176–185. [Google Scholar] [CrossRef]
- Snyder, H.W., Jr.; Hardy, W.D., Jr.; Zuckerman, E.E.; Fleissner, E. Characterisation of a tumour-specific antigen on the surface of feline lymphosarcoma cells. Nature 1978, 275, 656–658. [Google Scholar] [CrossRef]
- Theilen, G.H.; Kawakami, T.G.; Rush, J.D.; Munn, R.J. Replication of cat leukemia virus in cell suspension cultures. Nature 1969, 222, 589–590. [Google Scholar] [CrossRef]
- Sarma, P.S.; Log, T. Subgroup classification of feline leukemia and sarcoma viruses by viral interference and neutralization tests. Virology 1973, 54, 160–169. [Google Scholar] [CrossRef]
- Cattori, V.; Hofmann-Lehmann, R. Absolute quantitation of feline leukemia virus proviral DNA and viral RNA loads by TaqMan real-time PCR and RT-PCR. Methods Mol. Biol. 2008, 429, 73–87. [Google Scholar]
- Tandon, R.; Cattori, V.; Gomes-Keller, M.A.; Meli, M.L.; Golder, M.C.; Lutz, H.; Hofmann-Lehmann, R. Quantitation of feline leukaemia virus viral and proviral loads by TaqMan real-time polymerase chain reaction. J. Virol. Methods 2005, 130, 124–132. [Google Scholar] [CrossRef]
- Helfer-Hungerbuehler, A.K.; Widmer, S.; Hofmann-Lehmann, R. GAPDH pseudogenes and the quantification of feline genomic DNA equivalents. Mol. Biol. Int. 2013, 2013, 587680. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- Aickin, M.; Gensler, H. Adjusting for multiple testing when reporting research results: The Bonferroni vs. Holm methods. Am. J. Public. Health 1996, 86, 726–728. [Google Scholar] [CrossRef]
- Brown, M.B.; Forsythe, A.B. Robust tests for the equality of variances. J. Am. Stat. Assoc. 1974, 69, 364–367. [Google Scholar] [CrossRef]
- Shapiro, S.S.; Wilk, M.B. An analysis of variance test for normality (complete samples). Biometrika 1965, 52, 591–611. [Google Scholar] [CrossRef]
- Brussel, A.; Sonigo, P. Analysis of early human immunodeficiency virus type 1 DNA synthesis by use of a new sensitive assay for quantifying integrated provirus. J. Virol. 2003, 77, 10119–10124. [Google Scholar] [CrossRef]
- Scholtissek, C.; Muller, K. Effect of dimethylsulfoxide (DMSO) on virus replication and maturation. Arch. Virol. 1988, 100, 27–35. [Google Scholar] [CrossRef]
- Seki, J.; Ikeda, R.; Hoshino, H. Dimethyl sulfoxide and related polar compounds enhance infection of human T cells with HIV-1 in vitro. Biochem. Biophys. Res. Commun. 1996, 227, 724–729. [Google Scholar] [CrossRef]
- Warren, J.; Sacksteder, M.R.; Ellis, B.M.; Schwartz, R.D. Enhancement of viral oncogenicity by the prior administration of dimethyl sulfoxide. Cancer Res. 1973, 33, 618–622. [Google Scholar]
- Hartmann, K. Clinical aspects of feline retroviruses: A review. Viruses 2012, 4, 2684–2710. [Google Scholar] [CrossRef]
- Hartmann, K. Efficacy of antiviral chemotherapy for retrovirus-infected cats: What does the current literature tell us? J. Feline Med. Surg. 2015, 17, 925–939. [Google Scholar] [CrossRef]
- Swenson, C.L.; Polas, P.J.; Weisbrode, S.E.; Nagode, L.A.; Kociba, G.J.; Hayes, K.A.; Mathes, L.E. Prophylactic efficacy and bone toxicity associated with phosphonoformate therapy against retrovirus infection. Antivir. Chem. Chemoth 1992, 3, 335–343. [Google Scholar] [CrossRef]
- Cattori, V.; Weibel, B.; Lutz, H. Inhibition of feline leukemia virus replication by the integrase inhibitor raltegravir. Vet. Microbiol. 2011, 152, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lucia, E.; Collado, V.M.; Miro, G.; Martin, S.; Benitez, L.; Domenech, A. Follow-up of viral parameters in FeLV- or FIV-naturally infected cats treated orally with low doses of human interferon alpha. Viruses 2019, 11, 845. [Google Scholar] [CrossRef]
- Gomez-Lucia, E.; Collado, V.M.; Miro, G.; Martin, S.; Benitez, L.; Domenech, A. Clinical and hematological follow-up of long-term oral therapy with type-I interferon in cats naturally infected with feline leukemia virus or feline immunodeficiency virus. Animals 2020, 10, 1464. [Google Scholar] [CrossRef] [PubMed]
- Leal, R.O.; Gil, S.; Sepulveda, N.; McGahie, D.; Duarte, A.; Niza, M.M.; Tavares, L. Monitoring acute phase proteins in retrovirus infected cats undergoing feline interferon-ω therapy. J. Small Anim. Pract. 2014, 55, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Weber, K.; Rauch, G.; Hofmann-Lehmann, R.; Hosie, M.J.; Meli, M.L.; Hartmann, K. Efficacy of siRNA on feline leukemia virus replication in vitro. Berl. Munch. Tierarztl. Wochenschr. 2015, 128, 209–217. [Google Scholar] [PubMed]
- Boenzli, E.; Robert-Tissot, C.; Sabatino, G.; Cattori, V.; Meli, M.L.; Gutte, B.; Rovero, P.; Flynn, N.; Hofmann-Lehmann, R.; Lutz, H. In vitro inhibition of feline leukaemia virus infection by synthetic peptides derived from the transmembrane domain. Antivir. Ther. 2011, 16, 905–913. [Google Scholar] [CrossRef]
- Helfer-Hungerbuehler, A.K.; Shah, J.; Meili, T.; Boenzli, E.; Li, P.; Hofmann-Lehmann, R. Adeno-associated vector-delivered CRISPR/SaCas9 system reduces feline leukemia virus production in vitro. Viruses 2021, 13, 1636. [Google Scholar] [CrossRef]
- Xing, E.; Surendranathan, N.; Kong, X.; Cyberski, N.; Garcia, J.D.; Cheng, X.; Sharma, A.; Li, P.K.; Larue, R.C. Development of murine leukemia virus integrase-derived peptides that bind Brd4 extra-terminal domain as candidates for suppression of acute myeloid leukemia. ACS Pharmacol. Transl. Sci. 2021, 4, 1628–1638. [Google Scholar] [CrossRef]
- Loyola, L.; Achuthan, V.; Gilroy, K.; Borland, G.; Kilbey, A.; Mackay, N.; Bell, M.; Hay, J.; Aiyer, S.; Fingerman, D.; et al. Disrupting MLV integrase:BET protein interaction biases integration into quiescent chromatin and delays but does not eliminate tumor activation in a MYC/Runx2 mouse model. PLoS Pathog. 2019, 15, e1008154. [Google Scholar] [CrossRef]
- Chen, I.P.; Ott, M. Viral hijacking of BET proteins. Viruses 2022, 14, 2274. [Google Scholar] [CrossRef]
- Lara-Urena, N.; Garcia-Dominguez, M. Relevance of BET family proteins in SARS-CoV-2 infection. Biomolecules 2021, 11, 1126. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, G.L.; Ming, S.L.; Wang, C.F.; Shi, L.J.; Su, B.Q.; Wu, H.T.; Zeng, L.; Han, Y.Q.; Liu, Z.H.; et al. BRD4 inhibition exerts anti-viral activity through DNA damage-dependent innate immune responses. PLoS Pathog. 2020, 16, e1008429. [Google Scholar] [CrossRef]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef]
- Sheppard, G.S.; Wang, L.; Fidanze, S.D.; Hasvold, L.A.; Liu, D.; Pratt, J.K.; Park, C.H.; Longenecker, K.; Qiu, W.; Torrent, M.; et al. Discovery of N-ethyl-4-[2-(4-fluoro-2,6-dimethyl-phenoxy)-5-(1-hydroxy-1-methyl-ethyl)phenyl]-6-methyl-7-oxo-1H-pyrrolo[2,3-c]pyridine-2-carboxamide (ABBV-744), a BET bromodomain inhibitor with selectivity for the second bromodomain. J. Med. Chem. 2020, 63, 5585–5623. [Google Scholar] [CrossRef]
- Gilan, O.; Rioja, I.; Knezevic, K.; Bell, M.J.; Yeung, M.M.; Harker, N.R.; Lam, E.Y.N.; Chung, C.W.; Bamborough, P.; Petretich, M.; et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020, 368, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Menotti-Raymond, M.; David, V.; Wachter, L.; Yuhki, N.; O’Brien, S.J. Quantitative polymerase chain reaction-based assay for estimating DNA yield extracted from domestic cat specimens. Croat. Med. J. 2003, 44, 327–331. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moll, G.M.; Swenson, C.L.; Yuzbasiyan-Gurkan, V. BET Inhibitor JQ1 Attenuates Feline Leukemia Virus DNA, Provirus, and Antigen Production in Domestic Cat Cell Lines. Viruses 2023, 15, 1853. https://doi.org/10.3390/v15091853
Moll GM, Swenson CL, Yuzbasiyan-Gurkan V. BET Inhibitor JQ1 Attenuates Feline Leukemia Virus DNA, Provirus, and Antigen Production in Domestic Cat Cell Lines. Viruses. 2023; 15(9):1853. https://doi.org/10.3390/v15091853
Chicago/Turabian StyleMoll, Garrick M., Cheryl L. Swenson, and Vilma Yuzbasiyan-Gurkan. 2023. "BET Inhibitor JQ1 Attenuates Feline Leukemia Virus DNA, Provirus, and Antigen Production in Domestic Cat Cell Lines" Viruses 15, no. 9: 1853. https://doi.org/10.3390/v15091853