
An In Silico Design of Peptides Targeting the S1/S2 Cleavage Site of the SARS-CoV-2 Spike Protein


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Data Mining and Homology Modelling
2.2. Molecular Docking and Structural Analysis
2.3. Peptides Design
2.4. Physicochemical and Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Analyses
2.5. Molecular Dynamic Simulation
2.6. Analysis with The Omicron Variant S Protein
3. Results and Discussion
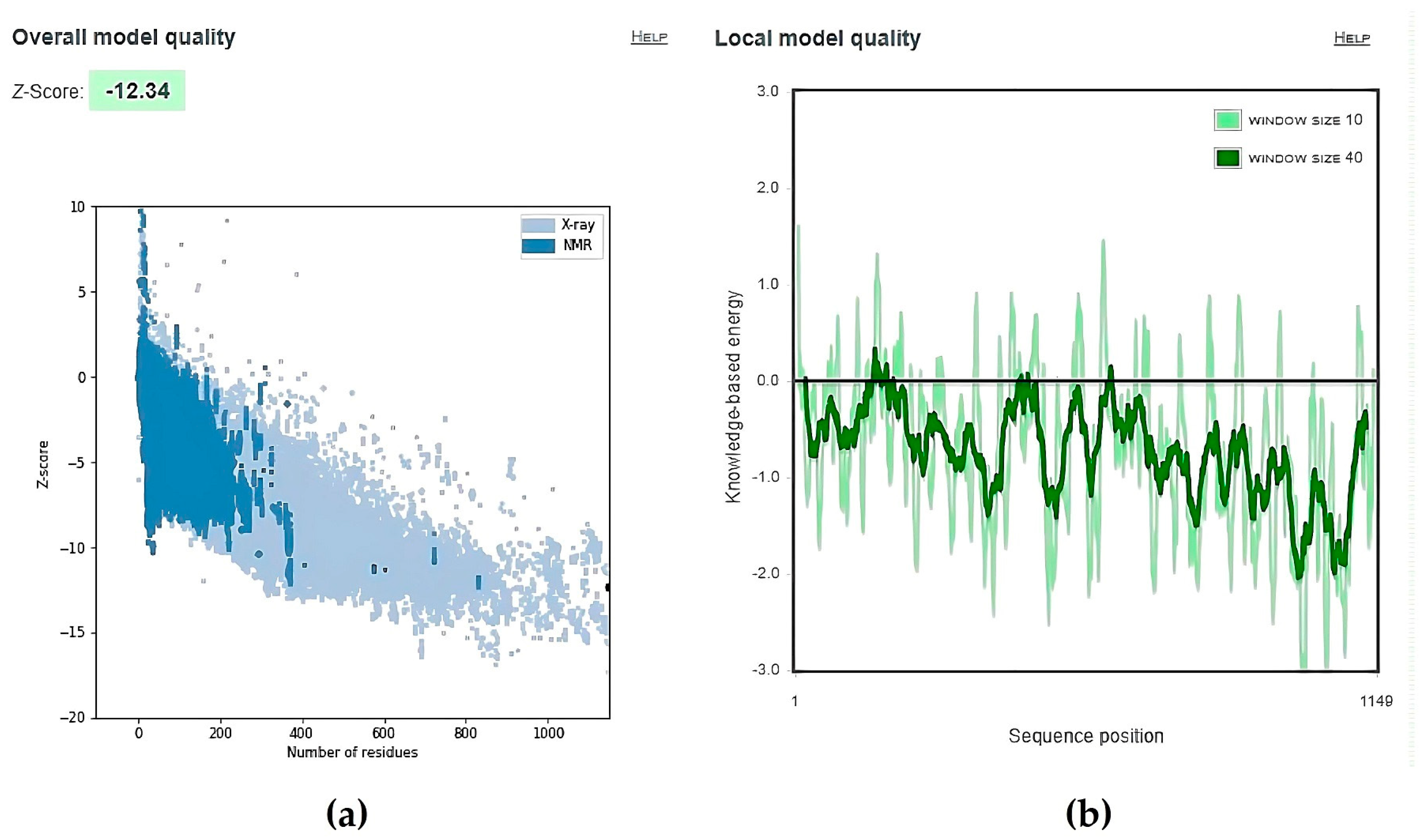
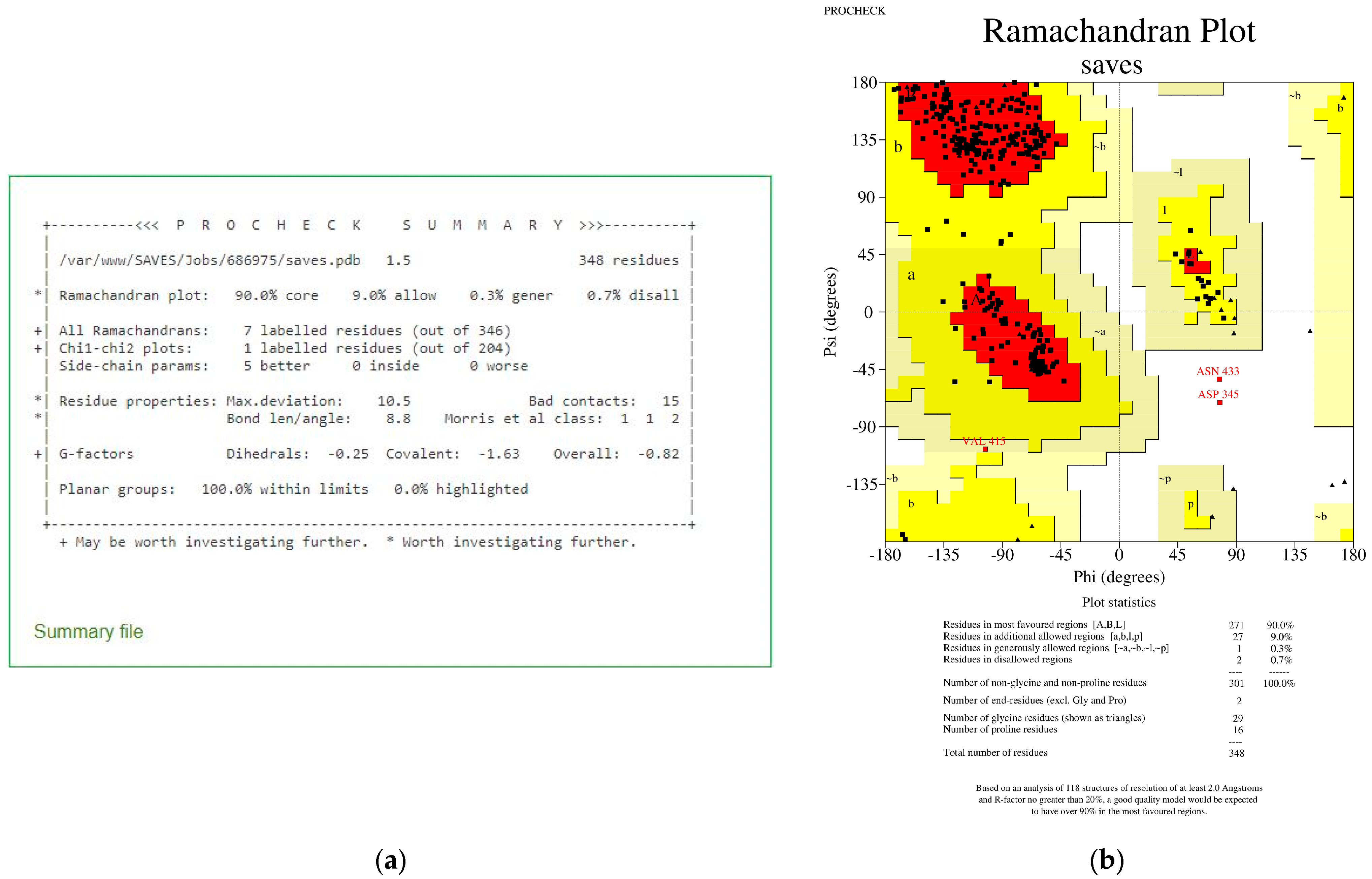
3.1. Homology Modelling of S Protein Chain A and TMPRSS2
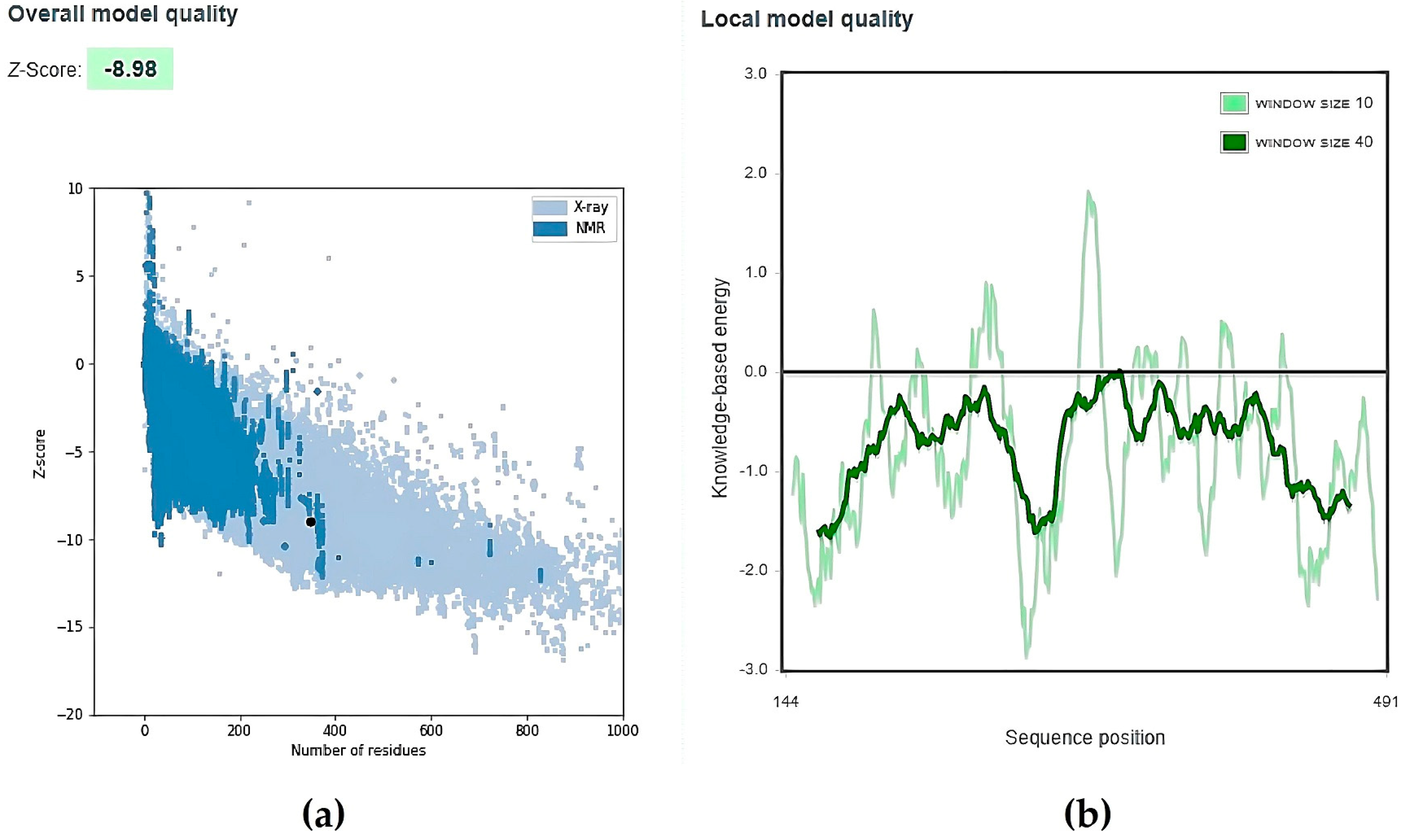
3.2. Molecular Docking and Peptides Design
3.3. Physicochemical and Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Analyses
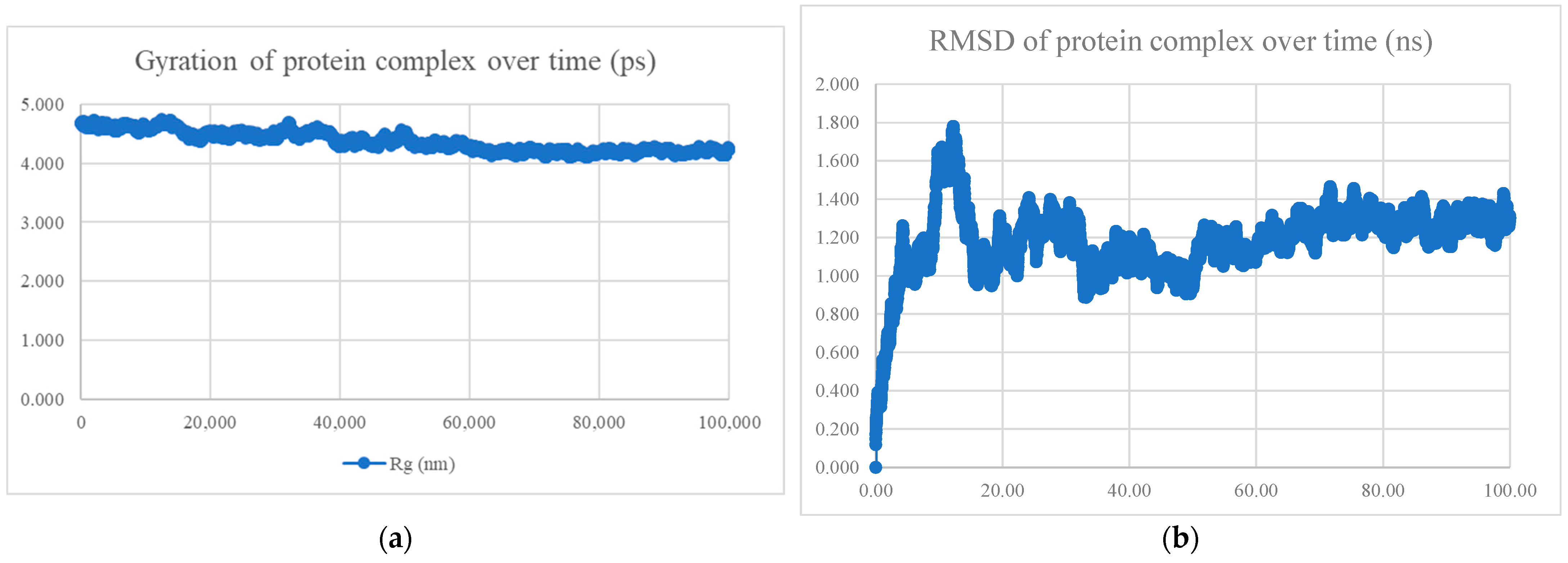
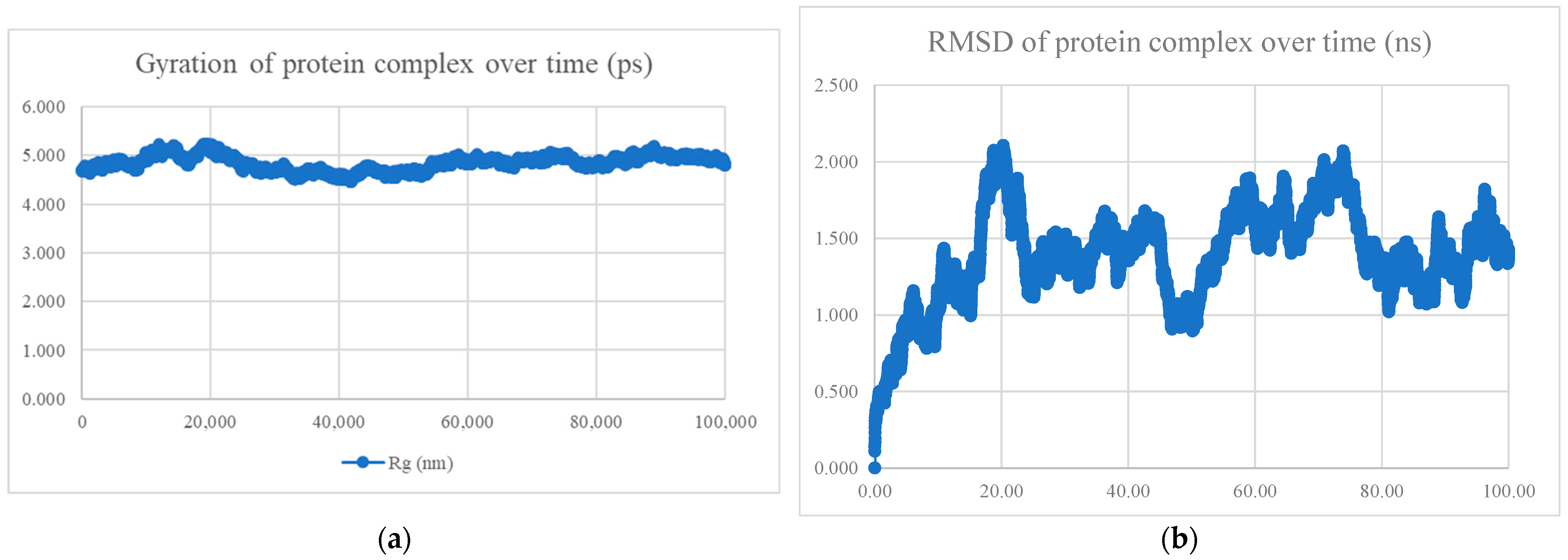
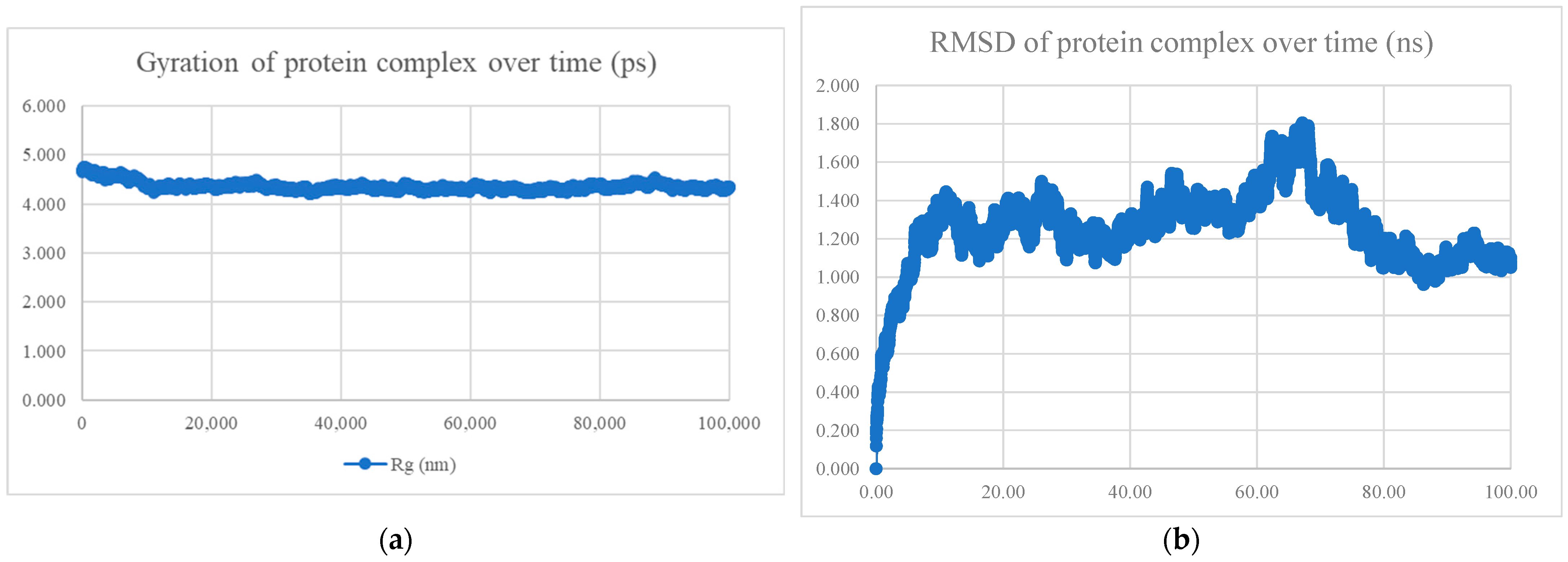
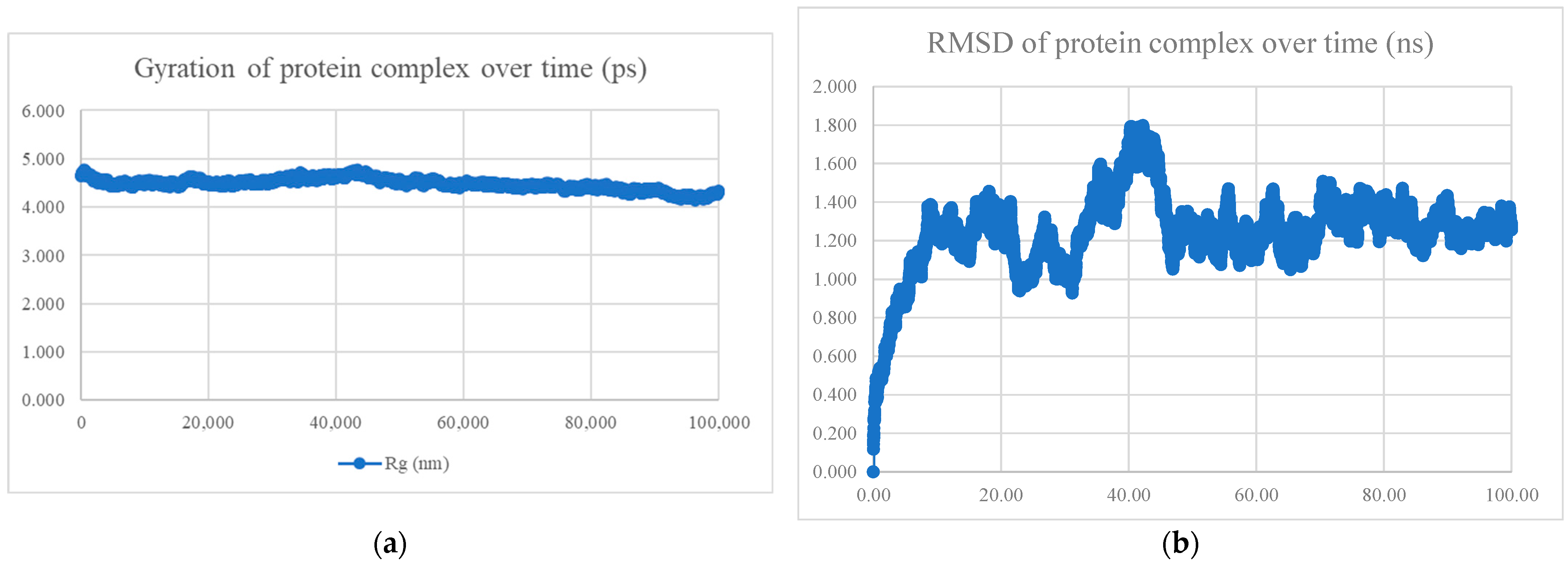
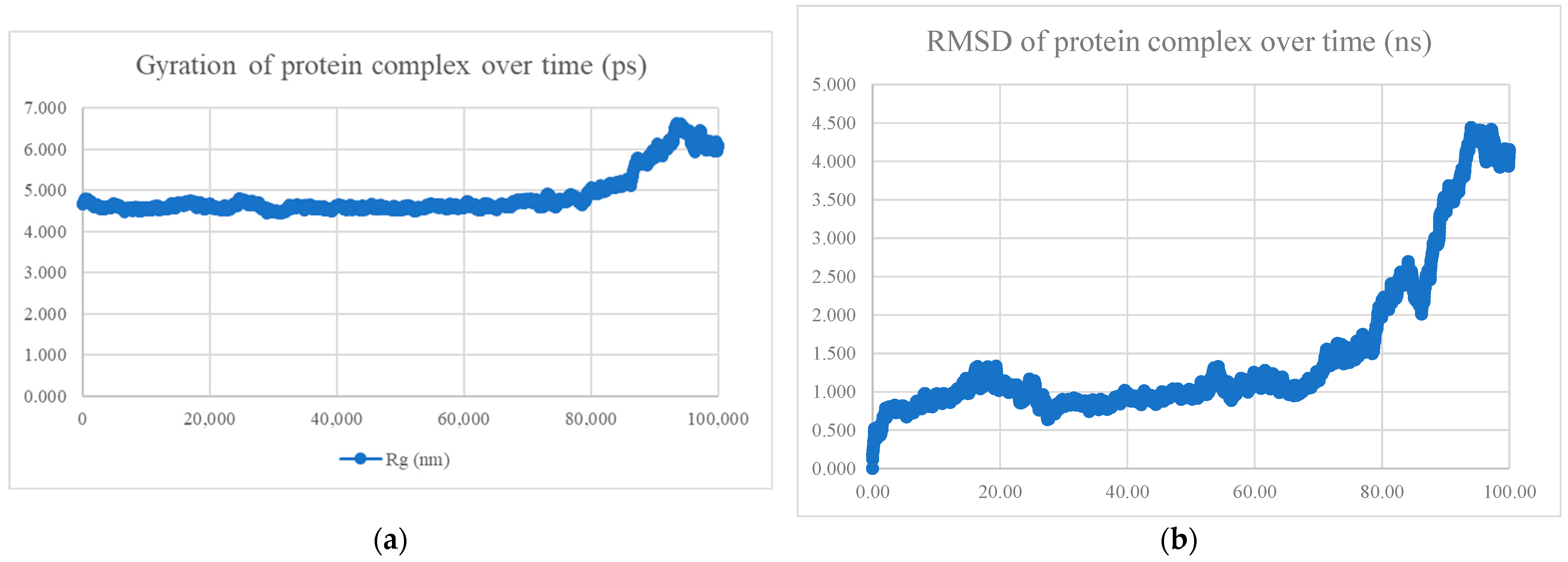
3.4. Molecular Dynamics Simulation
3.5. Analysis of Peptides BR582 and BR599 with The Omicron Variant S Protein
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, Y.; Chen, C.; Chan, Y. The outbreak of COVID-19. J. Chin. Med. Assoc. 2020, 83, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.W.; Chong, E.T.J.; Tan, Y.A.; Lee, H.G.; Chan, L.L.; Lee, Q.Z.; Saw, Y.T.; Wong, Y.; Bin Zakaria, A.A.; Amin, Z.B.; et al. Prevalence of Coronavirus Disease 2019 (COVID-19) in Different Clinical Stages before the National COVID-19 Vaccination Programme in Malaysia: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2022, 19, 2216. [Google Scholar] [CrossRef] [PubMed]
- Tortorici, M.A.; Veesler, D. Structural insights into coronavirus entry. Adv. Virus Res. 2019, 105, 93–116. [Google Scholar] [PubMed]
- Han, Y.; Král, P. Computational design of ACE2-based peptide inhibitors of SARS-CoV-2. ACS Nano 2020, 14, 5143–5147. [Google Scholar] [CrossRef] [PubMed]
- Ling, R.; Dai, Y.; Huang, B.; Huang, W.; Yu, J.; Lu, X.; Jiang, Y. In silico design of antiviral peptides targeting the spike protein of SARS-CoV-2. Peptides 2020, 130, 17038. [Google Scholar] [CrossRef]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 infects cells after viral entry via clathrin-mediated endocytosis. J. Biol. Chem. 2021, 296, 100306. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Hu, B.; Chan, J.F.W.; Liu, H.; Liu, Y.; Chai, Y.; Shi, J.; Shuai, H.; Hou, Y.; Huang, X.; Yuan, T.T.-T.; et al. Spike mutations contributing to the altered entry preference of SARS-CoV-2 omicron BA.1 and BA.2. Emerg. Microbes Infect. 2022, 11, 2275–2287. [Google Scholar] [CrossRef]
- Ahmad, A.; Fawaz, M.A.M.; Aisha, A. A comparative overview of SARS-CoV-2 and its variants of concern. Infez. Med. 2022, 30, 328–343. [Google Scholar]
- Carabelli, A.M.; Peacock, T.P.; Thorne, L.G.; Harvey, W.T.; Hughes, J.; COVID-19 Genomics UK Consortium; Peacock, S.J.; Barclay, W.S.; da Silva, T.I.; Towers, G.J.; et al. SARS-CoV-2 variant biology: Immune escape, transmission and fitness. Nat. Rev. Microbiol. 2023, 21, 162–177. [Google Scholar]
- Coronavirus (COVID-19)|Drugs. Available online: https://www.fda.gov/drugs/emergency-preparedness-drugs/coronavirus-covid-19-drugs (accessed on 19 May 2023).
- Dong, Y.; Shamsuddin, A.; Campbell, H.; Theodoratou, E. Current COVID-19 treatments: Rapid review of the literature. J. Glob. Health. 2021, 11, 10003. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, V.; Finn, A.; Lal, A.; Khan, M.S.; Dapaah-Afriyie, K.; Carino, G.P. Baricitinib in hospitalized patients with COVID-19: A meta-analysis of randomized controlled trials. EClinicalMedicine. 2022, 49, 101489. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021, 371, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Woo, H.G. Omicron: A Heavily Mutated SARS-CoV-2 Variant Exhibits Stronger Binding to ACE2 and Potently Escapes Approved COVID-19 Therapeutic Antibodies. Front. Immunol. 2022, 12, 830527. [Google Scholar] [CrossRef]
- Syed, A.M.; Ciling, A.; Taha, T.Y.; Chen, I.P.; Khalid, M.M.; Sreekumar, B.; Chen, P.-Y.; Kumar, G.R.; Suryawanshi, R.; Silva, I.; et al. Omicron mutations enhance infectivity and reduce antibody neutralization of SARS-CoV-2 virus-like particles. Proc. Natl. Acad. Sci. USA 2022, 119, e2200592119. [Google Scholar] [CrossRef]
- Choudhary, M.C.; Chew, K.W.; Deo, R.; Flynn, J.P.; Regan, J.; Crain, C.R.; Moser, C.; Hughes, M.D.; Ritz, J.; Ribeiro, R.M.; et al. Emergence of SARS-CoV-2 escape mutations during Bamlanivimab therapy in a phase II randomized clinical trial. Nat. Microbiol. 2022, 7, 1906–1917. [Google Scholar] [CrossRef]
- Marqus, S.; Pirogova, E.; Piva, R.J. Evaluation of The Use of Therapeutic Peptides for Cancer Treatment. J. Biomed. Sci. 2017, 24, 21. [Google Scholar] [CrossRef]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The Future of Peptide-Based Drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide Therapeutics: Current Status and Future Directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Schütz, D.; Ruiz-Blanco, Y.B.; Münch, J.; Kirchhoff, F.; Sanchez-Garcia, E.; Müller, J.A. Peptide and peptide-based inhibitors of SARS-CoV-2 entry. Adv. Drug Deliv. Rev. 2020, 167, 47–65. [Google Scholar] [CrossRef]
- Ho, C.; Lee, P.-C. COVID-19 Treatment—Current Status, Advances, and Gap. Pathogens 2022, 11, 1201. [Google Scholar] [CrossRef] [PubMed]
- Jaimes, J.A.; Millet, J.K.; Whittaker, G.R. Proteolytic cleavage of the SARS-CoV-2 spike protein and the role of the novel S1/S2 site. iScience 2020, 23, 101212. [Google Scholar] [CrossRef] [PubMed]
- Dodson, G.; Wlodawer, A. Catalytic triads and their relatives. Trends Biochem. Sci. 1998, 23, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesier, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Zardecki, C.; Di Costanzo, L.; Duarte, J.M.; Hudson, B.P.; Persikova, I.; Segura, J.; Shao, C.; Voigt, M.; Westbrook, J.D.; et al. RCSB Protein Data Bank: Enabling biomedical research and drug discovery. Protein Sci. 2020, 29, 52–65. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; Rohde, C.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef]
- Tang, T.; Jaimes, J.A.; Bidon, M.K.; Straus, M.R.; Daniel, S.; Whittaker, G.R. Proteolytic Activation of SARS-CoV-2 Spike at the S1/S2 Boundary: Potential Role of Proteases beyond Furin. ACS Infect. Dis. 2021, 7, 264–272. [Google Scholar] [CrossRef]
- Dahms, S.O.; Hardes, K.; Becker, G.L.; Steinmetzer, T.; Brandstetter, H.; Than, M.E. X-ray structures of human furin in complex with competitive inhibitors. ACS Chem. Biol. 2014, 9, 1113–1118. [Google Scholar] [CrossRef]
- Salameh, M.A.; Soares, A.S.; Hockla, A.; Radisky, E.S. Structural basis for accelerated cleavage of bovine pancreatic trypsin inhibitor (BPTI) by human mesotrypsin. J. Biol. Chem. 2008, 283, 4115–4123. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Yuan, C.; Li, R.; Qu, D.; Huang, M.; Ngo, J.C.K. Crystal structures of matriptase in complex with its inhibitor hepatocyte growth factor activator inhibitor-1. J. Biol. Chem. 2013, 288, 11155–11164. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Gunzner-Toste, J.; Yao, H.; Wang, T.; Wang, J.; Xu, Z.; Chen, J.; Wai, J.; Nonomiya, J.; Tsai, S.P.; et al. Discovery of Peptidomimetic Antibody-Drug Conjugate Linkers with Enhanced Protease Specificity. J. Med. Chem. 2018, 61, 989–1000. [Google Scholar] [CrossRef]
- Hardegger, L.A.; Kuhn, B.; Spinnler, B.; Anselm, L.; Ecabert, R.; Stihle, M.; Gsell, B.; Thoma, R.; Diez, J.; Benz, J.; et al. Systematic Investigation of Halogen Bonding in Protein–Ligand Interactions. Angew Chem. Int. Ed. 2011, 50, 314–318. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 webserver: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- PyMOL. Available online: http://www.pymol.org/pymol (accessed on 5 September 2019).
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, 449–454. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Nowotny, J.; Cao, R.; Cheng, J. 3Drefine: An interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016, 44, W406–W409. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.J.J.; Vangone, A. PRODIGY: A web-server for predicting the binding affinity in protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S. In Silico approach for predicting toxicity of peptides and proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press Inc.: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- PepSMI: Convert Peptide to SMILES String. Available online: https://www.novoprolabs.com/tools/convert-peptide-to-smiles-string (accessed on 6 July 2022).
- Pharmacokinetic Properties. Available online: https://biosig.lab.uq.edu.au/pkcsm/prediction (accessed on 6 July 2022).
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; MacKerell, A.D., Jr. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, E.; Abraham, M.J.; Hess, B.; van der Spoel, D. GROMACS, version 2020.1 Manual; Zenodo: Honolulu, HI, USA, 2020. [Google Scholar]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comp. Phys. Comm. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Gilby, N.B.; Wei, G.W. Omicron variant (B.1.1.529): Infectivity, vaccine breakthrough, and antibody resistance. J. Chem. Inf. Model. 2022, 62, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhou, J.; Tian, M.; Huang, M.; Liu, S.; Xie, Y.; Han, P.; Bai, C.; Han, P.; Zheng, A.; et al. Omicron SARS-CoV-2 mutations stabilize spike up-RBD conformation and lead to a non-RBM-binding monoclonal antibody escape. Nat. Commun. 2022, 13, 4958. [Google Scholar] [CrossRef]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo—Distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef]
- Sekijima, M.; Motono, C.; Yamasaki, S.; Kaneko, K.; Akiyama, Y. Molecular dynamics simulation of dimeric and monomeric forms of human prion protein: Insight into dynamics and properties. Biophys. J. 2003, 85, 1176–1185. [Google Scholar] [CrossRef]
- Kalathiya, U.; Padariya, M.; Mayordomo, M.; Lisowska, M.; Nicholson, J.; Singh, A.; Baginski, M.; Fahraeus, R.; Carragher, N.; Ball, K.; et al. Highly Conserved Homotrimer Cavity Formed by the SARS-CoV-2 Spike Glycoprotein: A Novel Binding Site. J. Clin. Med. 2020, 9, 1473. [Google Scholar] [CrossRef]
- Sixto-López, Y.; Correa-Basurto, J.; Bello, M.; Landeros-Rivera, B.; Garzón-Tiznado, J.A.; Montaño, S. Structural insights into SARS-CoV-2 spike protein and its natural mutants found in Mexican population. Sci. Rep. 2021, 11, 4659. [Google Scholar] [CrossRef]
- Deganutti, G.; Prischi, F.; Reynolds, C.A. Supervised molecular dynamics for exploring the druggability of the SARS-CoV-2 spike protein. J. Comput. Aided Mol. Des. 2021, 35, 195–207. [Google Scholar] [CrossRef] [PubMed]
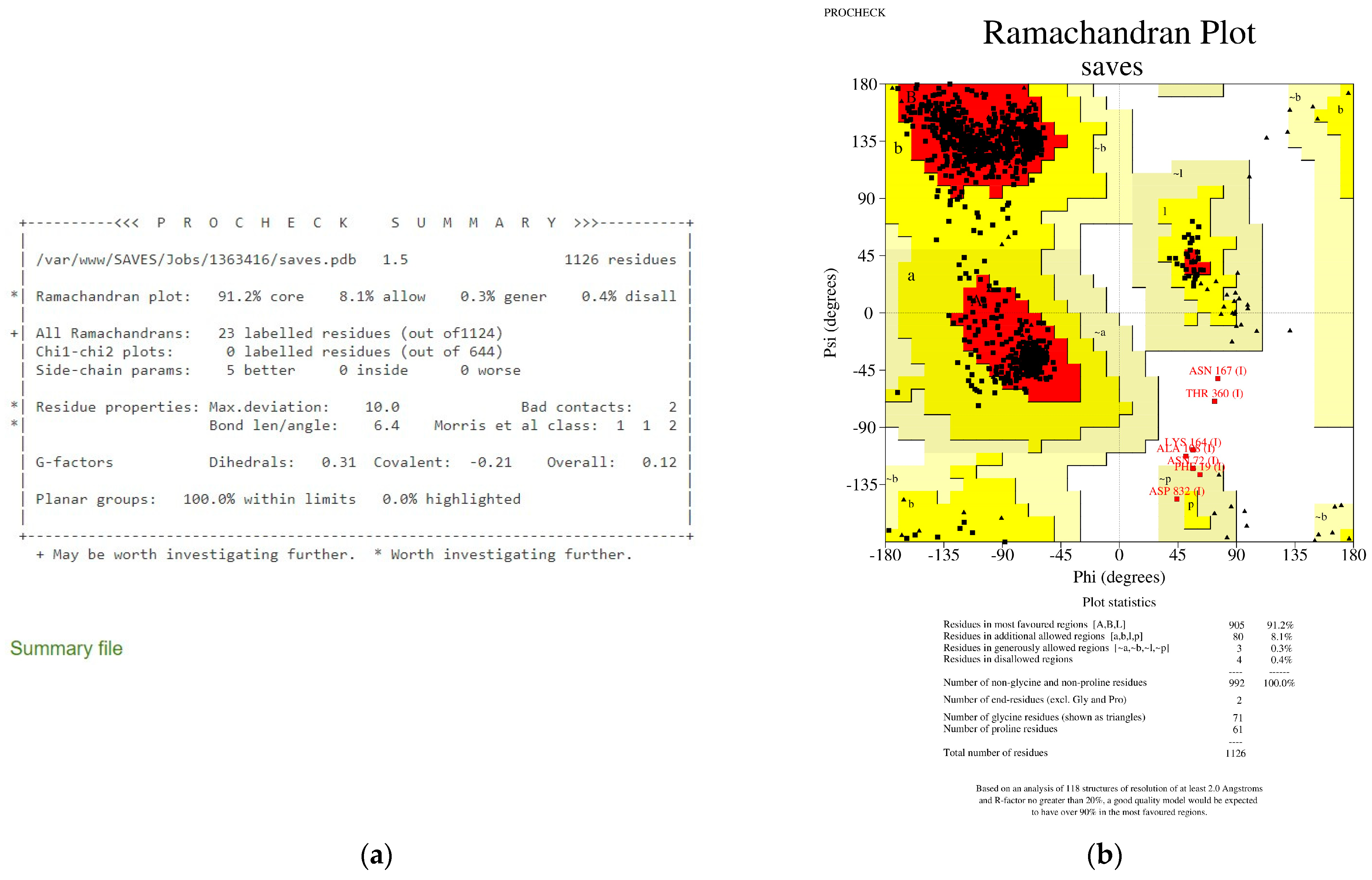
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A. Stereochemistry and Validation of Macromolecular Structures. Methods Mol. Biol. 2017, 1607, 595–610. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Thornton, J.M. PROCHECK: Validation of protein-structure coordinates. Int. Tables Crystallogr. 2012, F, 684–687. [Google Scholar]
- Tran, N.T.; Jakovlić, I.; Wang, W.M. In silico characterization, homology modelling and structure-based functional annotation of blunt snout bream (Megalobrama amblycephala) Hsp70 and Hsc70 proteins. J. Anim. Sci. Technol. 2015, 57, 44. [Google Scholar] [CrossRef]
- Sonawane, K.; Barale, S.S.; Dhanavade, M.J.; Waghmare, S.R.; Nadaf, N.H.; Kamble, S.A.; Mohammed, A.A.; Makandar, A.M.; Fandilolu, P.M.; Dound, A.S.; et al. Homology modeling and docking studies of TMPRSS2 with experimentally known inhibitors Camostat mesylate, Nafamostat and Bromhexine hydrochloride to control SARS-Coronavirus-2. ChemRxiv 2020. [Google Scholar] [CrossRef]
- PROCHECK Operating Manual. Available online: http://www.csb.yale.edu/userguides/datamanip/procheck/manual/manappe.html (accessed on 8 November 2022).
- Ho, B.K.; Brasseur, R. The Ramachandran plots of glycine and pre-proline. BMC Struct. Biol. 2005, 5, 14. [Google Scholar] [CrossRef]
- Mansbach, R.A.; Chakraborty, S.; Travers, T.; Gnanakaran, S. Graph-Directed Approach for Downselecting Toxins for Experimental Structure Determination. Mar. Drugs. 2020, 18, 256. [Google Scholar] [CrossRef]
- Heo, L.; Feig, M. Experimental accuracy in protein structure refinement via molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2018, 115, 13276–13281. [Google Scholar] [CrossRef]
- Karaca, E.; Melquiond, A.S.J.; De Vries, S.J.; Kastritis, P.L.; Bonvin, A.M.J.J. Building macromolecular assemblies by information-driven docking: Introducing the haddock multibody docking server. Mol. Cell Proteomics. 2010, 9, 1784–1794. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Bu, D.; Xu, J.; Li, M. Finding nearly optimal GDT scores. J. Comput. Biol. 2011, 18, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Li, L.; Lu, Y. Selecting near-native protein structures from predicted decoy sets using ordered graphlet degree similarity. Genes 2019, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Zemla, A. LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003, 31, 3370–3374. [Google Scholar] [CrossRef]
- Carlsen, M.; Koehl, P.; Røgen, P. On the importance of the distance measures used to train and test knowledge-based potentials for proteins. PLoS ONE. 2014, 9, e109335. [Google Scholar] [CrossRef]
- Chang, K.Y.; Yang, J.R. Analysis and prediction of highly effective antiviral peptides based on random forests. PloS ONE. 2013, 8, e70166. [Google Scholar] [CrossRef]
- Hu, Y.B.; Dammer, E.B.; Ren, R.J.; Wang, G. The endosomal-lysosomal system: From acidification and cargo sorting to neurodegeneration. Transl. Neurodegener. 2015, 4, 18. [Google Scholar] [CrossRef]
- Zeng, J.; Shirihai, O.S.; Grinstaff, M.W. Modulating lysosomal pH: A molecular and nanoscale materials design perspective. J. Life Sci. 2020, 2, 25–37. [Google Scholar] [CrossRef]
- Ciechanover, A.; Schwartz, A.L. How are substrates recognized by the ubiquitin-mediated proteolytic system? Trends Biochem. Sci. 1989, 14, 483–488. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron. 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Aier, I.; Varadwaj, P.; Raj, U. Structural insights into conformational stability of both wild-type and mutant EZH2 receptor. Sci. Rep. 2016, 6, 34984. [Google Scholar] [CrossRef]
- Ni, D.; Song, K.; Zhang, J.; Lu, S.Y. Molecular dynamics simulations and dynamic network analysis reveal the allosteric unbinding of monobody to H-Ras triggered by R135K mutation. Int. J. Mol. Sci. 2017, 18, 2249. [Google Scholar] [CrossRef] [PubMed]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar]
- Hess, B. Convergence of sampling in protein simulations. Phys. Rev. E. Stat. Nonlin. Soft Matter Phys. 2002, 65, 031910. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domański, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11 July 2016. [Google Scholar]
- Grant, O.C.; Montgomery, D.; Ito, K.; Woods, R.J. Analysis of the SARS-CoV-2 spike protein glycan shield reveals implications for immune recognition. Sci. Rep. 2020, 10, 14991. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, H.; Wang, H. Glycans of SARS-CoV-2 Spike Protein in Virus Infection and Antibody Production. Front. Mol. Biosci. 2021, 8, 629873. [Google Scholar] [CrossRef]
- Pinto, D.; Park, Y.J.; Beltramello, M.; Walls, A.C.; Tortorici, M.A.; Bianchi, S.; Jaconi, S.; Culap, K.; Zatta, F.; De Marco, A.; et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 2020, 583, 290–295. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Qin, S.; Dai, L.; Tian, Z. The glycosylation in SARS-CoV-2 and its receptor ACE2. Sig. Transduct. Target. Ther. 2021, 6, 396. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mann, M.; Syed, Z.A.; Reynolds, H.M.; Tian, E.; Samara, N.L.; Zeldin, D.C.; Tabak, L.A.; Ten Hagen, K.G. Furin cleavage of the SARS-CoV-2 spike is modulated by O-glycosylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2109905118. [Google Scholar] [CrossRef] [PubMed]
- Stanfield, R.L.; Wilson, I.A. Protein-peptide interactions. Curr. Opin. Struct. Biol. 1995, 5, 103–113. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Model | HADDOCK Score | Cluster/Position | Cluster Size | RMSD | Z-Score | i-RMSD | FCC | Average Binding Affinity ΔG (kcal/mol) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | S protein and Furin | −107.1 +/− 7.2 | 10.0/1.0 | 6 | 0.8 +/− 0.5 | −2.5 | 0.0–1.0 | 0.5–1.0 | −12.5 |
| 2 | S protein and Trypsin | −122.0 +/− 9.2 | 1.0/1.0 | 62 | 0.8 +/− 0.6 | −1.1 | 0.0–1.5 | 0.3–1.0 | −12.3 |
| 3 | S protein and TMPRSS2 | −91.7 +/− 9.9 | 3.0/1.0 | 18 | 1.4 +/− 1.1 | −2.4 | 0.0–3.0 | 0.4–1.0 | −10.1 |
| 4 | S protein and Matriptase | −101.5 +/− 5.8 | 2.0/1.0 | 41 | 1.3 +/− 0.8 | −1.3 | 0.0–2.0 | 0.3–1.0 | −11.8 |
| 5 | S protein and Cathepsin B | −109.0 +/− 7.8 | 1.0/1.0 | 122 | 0.8 +/− 0.5 | −1.7 | 0.0–1.2 | 0.4–1.0 | −13.3 |
| 6 | S protein and Cathepsin L | −131.6 +/− 7.7 | 2.0/1.0 | 27 | 0.8 +/− 0.5 | −1.6 | 0.0–2.0 | 0.4–1.0 | −13.8 |
| Furin | Trypsin | TMPRSS2 | Matriptase | Cathepsin B | Cathepsin L | |
|---|---|---|---|---|---|---|
| 660S | - | - | - | 92F | - | 141E |
| 661Y | 119D, 120G, 121E | 22Y, 43K | 301P | 92F, 168Q | 196M | 141E, 145F |
| 662Q | 76R, 118L, 119D, 120G | 22Y | 91D, 92F | 195M, 196M | 141E, 144L | |
| 663T | 76R, 82D, 83N, 118L, 119D | 40H, 174Q | 296H, 299E | 45I, 91D, 94F | 195M, 196M, 197G, 198G | 144L |
| 664Q | 76R, 77Y, 82D | 174Q | - | - | - | - |
| 665T | 80M, 82D | 174Q | - | - | - | - |
| 666N | 80M | 128G | - | - | - | - |
| 668P | 85H | 126S, 128G, 173C, 196G, 197C | 342K, 461W, 463S | 23Q, 47D, 49G, 50F, 52Y | 245E | 65C, 66N |
| 669R | 186N, 219W, 256T, 257G | 193W, 195D, 196G, 199Q | 342K, 417D, 419L | 23Q, 26I, 52Y | 245E | 22C, 23G, 63E, 65C, 66N, 94T, 95E |
| 670R | 148E, 149D, 189R | 193W | 340K | - | 75Y, 245E | 21Q, 23G |
| 671A | 85H | 174Q | 296H | 45I, 47D | 245E | 23G |
| 672R | 44D, 45D, 85H, 118L, 144S, 145W, 146G | 40H, 172S, 173C, 174Q, 175G, 176D, 177S, 191V, 192S, 193W, 194G | 296H, 337Y, 339S, 438Q, 441S, 460S, 461W | 26I, 42H, 43C, 45I, 47D, 52Y, 188Q | 173A, 174F, 175S, 198G, 199H, 240C, 241G, 244S, 245E | 19Q, 23G, 25C, 66N, 67G, 162D, 189W |
| 673S | 148E, 149D | 40H, 193W | 296H, 299E, 339S, 340K | 188Q | 75Y, 76P, 198G, 245E | 19Q, 21Q, 22C, 23G, 189W |
| 674V | 146G, 147P, 148E | 78R | 296H, 299E, 300K, 339S | 141Y, 188Q, 212W, 213G, 214D, 215G | 27G, 29C, 30W, 73G, 74G, 198G, 199H, 200A | 18N, 19Q, 20G, 188S, 189W, 193W |
| 675A | 147P, 148E | - | 339S | 141Y | 74G, 75Y | 20G |
| 676S | - | 299E | - | 74G, 198G | - | |
| 677Q | 118L, 121E | 43K | 299E, 301P | 94F | 196M, 197G, 198G | 144L, 189W, 193W |
| 680I | 76R | - | - | - | - | - |
| No. | Peptide Sequence | Amino Acid Length | Models Observed | HADDOCK Score | Cluster/Position | Cluster Size | RMSD | Z-Score | i-RMSD | FCC | Average Binding Affinity ΔG (kcal/mol) | Average Kd (M) at 25.0 °C | ToxinPred |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BR556 | FDRLYQEFKDDYEDSEER | 18 | 166/200 models are alpha-helix | −125.1 +/− 2.6 | 1.0/1.0 | 71 | 0.9 +/− 0.5 | −2.7 | 0.0–2.0 | 0.5–1.0 | −10.8 | 1.51 × 10−8 | Non-toxin |
| BR563 | FDRLRQEFKDDFSEEEFR | 18 | 141/200 models are alpha-helix | −126.1 +/− 2.1 | 1.0/1.0 | 70 | 0.8 +/− 0.5 | −2.5 | 0.0–1.1 | 0.4–1.0 | −10.1 | 3.80 × 10−8 | Non-toxin |
| BR573 | FDRLRQEFKDDFYESEFR | 18 | All alpha helix | −127.4 +/− 2.7 | 1.0/1.0 | 38 | 0.8 +/− 0.5 | −2.7 | 0.0–1.9 | 0.4–1.0 | −10.4 | 3.30 × 10−8 | Non-toxin |
| BR582 | FDRLRQEFKDDYEDEEERKE | 20 | 110/200 models are alpha-helix | −130.3 +/− 2.3 | 1.0/1.0 | 76 | 0.7 +/− 0.4 | −2.2 | 0.0–3.8 | 0.4–1.0 | −10.9 | 1.04 × 10−8 | Non-toxin |
| BR599 | EKFDRLYQEFKDDYEDSEER | 20 | 156/200 models are alpha-helix | −122.2 +/− 2.3 | 1.0/1.0 | 47 | 0.7 +/− 0.5 | −2.7 | 0.0–2.2 | 0.5–1.0 | −11.0 | 9.38 × 10−9 | Non-toxin |
| BR605 | FDRLRQEFKDDFYEEELREK | 20 | All alpha helix | −131.6 +/− 2.7 | 1.0/1.0 | 62 | 0.9 +/− 0.5 | −2.2 | 0.0–1.8 | 0.4–1.0 | −10.4 | 2.49 × 10−8 | Non-toxin |
| BR606 | FDRLRQEFKDDFYEEELRKE | 20 | All alpha helix | −134.5 +/− 0.6 | 1.0/1.0 | 62 | 0.8 +/− 0.6 | −2.3 | 0.0–1.6 | 0.4–1.0 | −10.2 | 3.48 × 10−8 | Non-toxin |
| BR610 | FDRLRQEFKDDFYESEFRKE | 20 | All alpha helix | −130.4 +/− 4.6 | 1.0/1.0 | 47 | 0.4 +/− 0.3 | −2.3 | 0.0–1.6 | 0.5–1.0 | −10.0 | 4.80 × 10−8 | Non-toxin |
| BR614 | FDRLRQEFKDDFYEEEERKE | 20 | All alpha helix | −129.6 +/− 5.7 | 1.0/1.0 | 54 | 0.8 +/− 0.6 | −1.7 | 0.0–2.4 | 0.3–1.0 | −10.6 | 2.02 × 10−8 | Non-toxin |
| BR556 | BR563 | BR573 | BR582 | BR599 | BR605 | BR606 | BR610 | BR614 | |
|---|---|---|---|---|---|---|---|---|---|
| 641E | - | 8F (v) | - | - | - | - | - | - | - |
| 643V | - | 8F (v) | - | 8F (v) | 10F (v) | 8F (v) | 8F (v) | - | 8F (v) |
| 645N | 8F (v) | - | - | - | 10F (v) | - | - | - | - |
| 646S | L4 (v) | - | - | - | 6L (v) | - | - | - | - |
| 647Y | 1F (v), 4L (v), 8F (v) | 4L (v), 7E (v), 8F (v) | 1F (v), 4L (v), 8F (v) | 1F (v), 4L (v), 8F (v) | 3F (v), 6L (v), 10F (v) | 4L (v), 7E (v), 8F (v) | 4L (v), 7E (v), 8F (v) | 4L (v), 8F (v) | 1F (v), 4L (v) |
| 648E | 1F (H, v), 3R (H, v), 4L (v) | 1F (v), 3R (H, v), 4L (v) | 1F (v), 3R (H, v), 4L (v) | 1F (H, v), 3R (H, v), 4L (v) | 2K (H, v), 3F (v), 6L (v) | 3R (H, v), 4L (v) | 3R (H, v), 4L (v) | 3R (v), 4L (v) | 1F (v), 3R (H, v), 4L (v) |
| 650D | 1F (v) | 1F (v) | - | 1F (v) | 3F (v) | 1F (v) | 1F (v) | - | 1F (v) |
| 660S | - | 1F (v), 4L (v) | 1F (v) | 1F (v) | 3F (v) | 1F (v) | 1F (v) | 1F (v) | 1F (v) |
| 661Y | 1F (v), 2D (H, v), 5Y (v) | 1F (v), 2D (v), 5R (v), | 1F (v), 2D (H, v), 5R (v) | 1F (v), 5R (v) | 7Y (v) | 1F (v) | 1F (v) | 1F (v), 5R (v) | 1F (v), 2D (H, v), 5R (v) |
| 662Q | 1F (v), 5Y (v), 8F (v) | 1F (v), 4L (v), 8F (v) | 1F (v), 5R (v), 8F (v) | 1F (v), 5R (v), 8F (v) | 7Y (v), 10F (v) | 1F (v), 5R (v), 8F (v) | 1F (v), 5R (v), 8F (v) | 5R (v), 8F (v) | 1F (v), 5R (v), 8F (v) |
| 663T | 9K (v), 12Y (v), 13E (H, v) | 8F (v), 9K (H, v), 12F (v) | 8F (v), 9K (H, v), 12F (v) | 8F (v), 9K (v), 12Y (v), 13E (H, v) | 11K (v), 14Y (v), 15E (H, v) | 8F (v), 9K (H, v) | 8F (v), 9K (H, v), 12F (v) | 5R (H, v), 8F (v), 9K (v), 12F (v) | 8F (v), 9K (H, v), 12F (v) |
| 664Q | 12Y (v) | 8F (v) | 12F (v) | 12Y (v) | 14Y (v) | 8F (v), 12F (v) | 12F (v) | 12F (v) | 12F (v) |
| 665T | 12Y (H, v), 16E (H, v) | 12F (v), 16E (H, v), | 12F (v), 16E (v) | 12Y (H, v), 16E (v) | 14Y (H, v), 18E (v) | 12F (v), 16E (v) | 12F (v) | 12F (v) | 12F (v) |
| 666N | - | 16E (v) | 16E (v) | 12Y (v), 16E (v) | 18E (v) | 16E (v), 19K (H, v) | 16E (v), 19K (H, v) | 16E (v), 19K (H, v) | 16E (v) |
| 667S | 16E (H, v) | 16E (H, v) | 16E (H, v) | 16E (H, v) | 18E (H, v) | 16E (v), 20E (H, v) | 16E (v), 20E (H, v) | 16E (v), 20E (H, v) | 16E (v) |
| 668P | 16E (v) | 12F (v), 16E (v), 17F (v) | 13Y (v), 16E (v), 17F (v) | 16E (v), 17E (v), 20E (v) | 18E (v), 19E (v) | 13Y (v), 16E (v), 17L (v), 20E (v) | 13Y (v), 16E (v), 17L (v), 20E (v) | 13Y (v), 16E (v), 17F (v), 19K (v), 20E (v) | 13Y (v), 16E (v), 17E (v), 20E (v) |
| 669R | - | 17F (v) | 13Y (v), 17F (v) | 20E (H, v) | - | 13Y (v), 17L (v), 20E (H, v) | 20E (H, v) | 13Y (v), 17F (v), 20E (H, v) | 13Y (v), 17E (v), 20E (H, v) |
| 670R | - | - | 13Y (v) | - | - | 13Y (v) | - | - | 13Y (v) |
| 671A | 13E (v), 16E (v), E17 (v) | 9K (H, v), 13S (v), 16E (v) | 9K (H, v), 12F (v), 13Y (v), 16E (v) | 13E (v), 16E (v), 17E (v) | 14Y (v), 15E (v), 18E (v) | 9K (H, v), 12F (v), 13Y (v) | 9K (H, v), 13Y (v) | 9K (v), 12F (v), 13Y (v) | 9K (H, v), 12F (v), 13Y (v) |
| 672R | 10D (H, v), 13E (v), 14D (H, v), E17 (H, v) | 9K (v), 10D (H, v), 13S (v), 14E (H, v), 17F (v) | 9K (v), 10D (H, v), 13Y (v), 14E (H, v) | 10D (H, v), 13E (H, v), 14D (H, v), 17E (H, v) | 11K (v), 12D (H, v), 15E (H, v), 16D (v), 19E (H, v) | 9K (H, v), 10D (H, v), 13Y (v), 14E (H, v) | 9K (v), 10D (H, v), 13Y (v), 14E (H, v) | 9K (v), 10D (H, v), 13Y (v), 14E (H, v) | 9K (v), 10D (H, v), 13Y (v), 14E (H, v) |
| 673S | 9K (H, v), 13E (H, v) | 9K (H, v) | 9K (H, v) | 13E (H, v) | - | 9K (H, v) | 9K (H, v) | 9K (H, v) | 9K (H, v) |
| 674V | 9K (v) | 9K (v) | - | - | - | - | - | 9K (v) | 9K (v) |
| 676S | - | 9K (v) | 9K (v) | - | - | 9K (v) | 9K (H, v) | 9K (H, v) | 9K (v) |
| 677Q | 5Y (v), 9K (v) | 5R (H, v), 9K (v) | 5R (H, v), 9K (v) | - | 7Y (H, v) | 5R (v), 9K (v) | 5R (v), 9K (v) | 5R (H, v) | 5R (H, v), 9K (v) |
| 680I | - | 8F (v) | - | - | - | 8F (v) | 8F (v) | - | - |
| 685S | - | - | - | - | 2K (H, v) | - | - | - | - |
| 687G | - | - | - | - | 2K (H, v) | - | - | - | - |
| Peptides | BR556 | BR563 | BR573 | BR582 | BR599 | BR605 | BR606 | BR610 | BR614 |
|---|---|---|---|---|---|---|---|---|---|
| Physicochemical analysis | |||||||||
| Properties | |||||||||
| Molecular weight (g/mol) | 2384.45 | 2393.55 | 2427.61 | 2676.79 | 2641.74 | 2692.92 | 2692.92 | 2684.90 | 2708.88 |
| Theoretical pI | 4.07 | 4.44 | 4.59 | 4.32 | 4.21 | 4.56 | 4.56 | 4.72 | 4.44 |
| Molecular formula | C104H146N26O3 | C106H153N29O35 | C110H155N29O34 | C114H170N32O43 | C115H165N29O43 | C120H178N32O39 | C120H178N32O39 | C121H174N32O38 | C119H174N32O41 |
| Number of atoms | 315 | 323 | 328 | 359 | 352 | 369 | 369 | 365 | 366 |
| Instability Index | 60.48 | 78.59 | 66.99 | 71.73 | 55.44 | 84.27 | 84.27 | 61.30 | 90.94 |
| Half-life (in h) | 1.1 h | 1.1 h | 1.1 h | 1.1 h | 1.0 h | 1.1 h | 1.1 h | 1.1 h | 1.1 h |
| Hydrophobicity (%) | 31.06 | 35.73 | 35.94 | 26.52 | 33.84 | 36.61 | 36.95 | 37.45 | 31.95 |
| GRAVY | −2.133 | −1.733 | −1.611 | −2.585 | −2.290 | −1.905 | −1.905 | −1.820 | −2.270 |
| ADMET analysis | |||||||||
| Absorption | |||||||||
| Water solubility (log mol/L) | −2.892 | −2.892 | −2.892 | −2.892 | −2.892 | −2.892 | −2.892 | −2.892 | −2.892 |
| Skin perm (log Kp) | −2.735 | −2.735 | −2.735 | −2.735 | −2.735 | −2.735 | −2.735 | −2.735 | −2.735 |
| P-glycoprotein substrate | No | No | No | No | No | No | No | No | No |
| P-glycoprotein inhibitors | No | No | No | No | No | No | No | No | No |
| Distribution | |||||||||
| Fraction unbound (human) (Fu) | 0.381 | 0.381 | 0.381 | 0.381 | 0.381 | 0.381 | 0.381 | 0.381 | 0.381 |
| Blood–brain barrier (BBB) permeability (logBB) | −5.846 | −5.865 | −5.665 | −7.073 | −6.408 | −6.495 | −6.497 | −6.213 | −6.767 |
| Central Nervous System (CNS) permeability (logPS) | −10.995 | −10.913 | −10.75 | −12.389 | −11.982 | −11.384 | −11.459 | −11.668 | −11.914 |
| Metabolism | |||||||||
| Cytochrome P 2D6 substrate | No | No | No | No | No | No | No | No | No |
| Cytochrome P 3A4 substrate | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Cyctochrome P inhibitors | No | No | No | No | No | No | No | No | No |
| Excretion | |||||||||
| Total clearance (log mL/min/kg) | −2.773 | −2.863 | −2.824 | −3.332 | −3.24 | −3.527 | −3.383 | −3.023 | −3.386 |
| Toxicity | |||||||||
| AMES toxicity | No | No | No | No | No | No | No | No | No |
| Maximum Recommended Tolerated Dose (MRTD) human (log mg/kg/day) | 0.438 | 0.438 | 0.438 | 0.438 | 0.438 | 0.438 | 0.438 | 0.438 | 0.438 |
| Rat oral LD50 (mol/kg) | 2.482 | 2.482 | 2.482 | 2.482 | 2.482 | 2.482 | 2.482 | 2.482 | 2.482 |
| Hepatotoxicity | No | No | No | No | No | No | No | No | No |
| Skin sensitization | No | No | No | No | No | No | No | No | No |
| No. of Eigenvectors (PC) | Eigenvalues of First Few PCs (%) | Cosine Content | |
|---|---|---|---|
| S protein and BR556 | 54 | PC1: 56.15 | 0.44 |
| PC2: 21.06 | |||
| PC3: 11.20 | |||
| PC4: 4.07 | |||
| % of eigenvalues in 4 PCs = 92.48 | |||
| S protein and BR563 | 54 | PC1: 47.98 | 0.14 |
| PC2: 21.80 | |||
| PC3: 11.43 | |||
| PC4: 7.07 | |||
| PC5: 3.05 | |||
| % of eigenvalues in 5 PCs = 91.33 | |||
| S protein and BR573 | 54 | PC1: 74.41 | 0.34 |
| PC2: 19.28 | |||
| % of eigenvalues in 2 PCs = 93.69 | |||
| S protein and BR582 | 60 | PC1: 76.66 | 0.57 |
| PC2: 14.76 | |||
| % of eigenvalues in 2 PCs = 91.42 | |||
| S protein and BR599 | 60 | PC1: 65.37 | 0.21 |
| PC2: 12.13 | |||
| PC3: 9.37 | |||
| PC4: 4.94 | |||
| % of eigenvalues in 4 PCs = 91.81 | |||
| S protein and BR605 | 60 | PC1: 92.21 | 0.49 |
| % of eigenvalues in 1 PCs = 92.21 | |||
| S protein and BR606 | 60 | PC1: 89.61 | 0.72 |
| PC2: 6.45 | |||
| % of eigenvalues in 2 PCs = 96.06 | |||
| S protein and BR610 | 60 | PC1: 67.47 | 0.53 |
| PC2: 19.50 | |||
| PC3: 6.41 | |||
| % of eigenvalues in 3 PCs = 93.38 | |||
| S protein and BR614 | 60 | PC1: 80.92 | 0.86 |
| PC2: 11.65 | |||
| % of eigenvalues in 2 PCs = 92.57 |
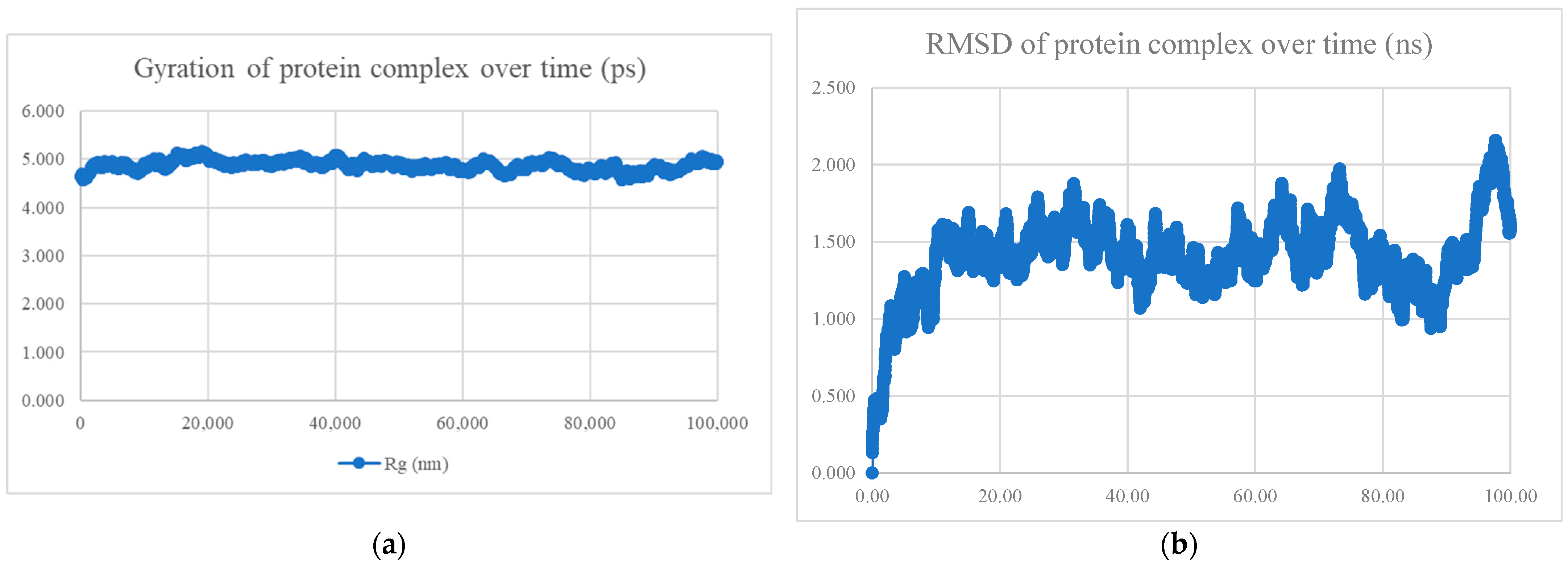
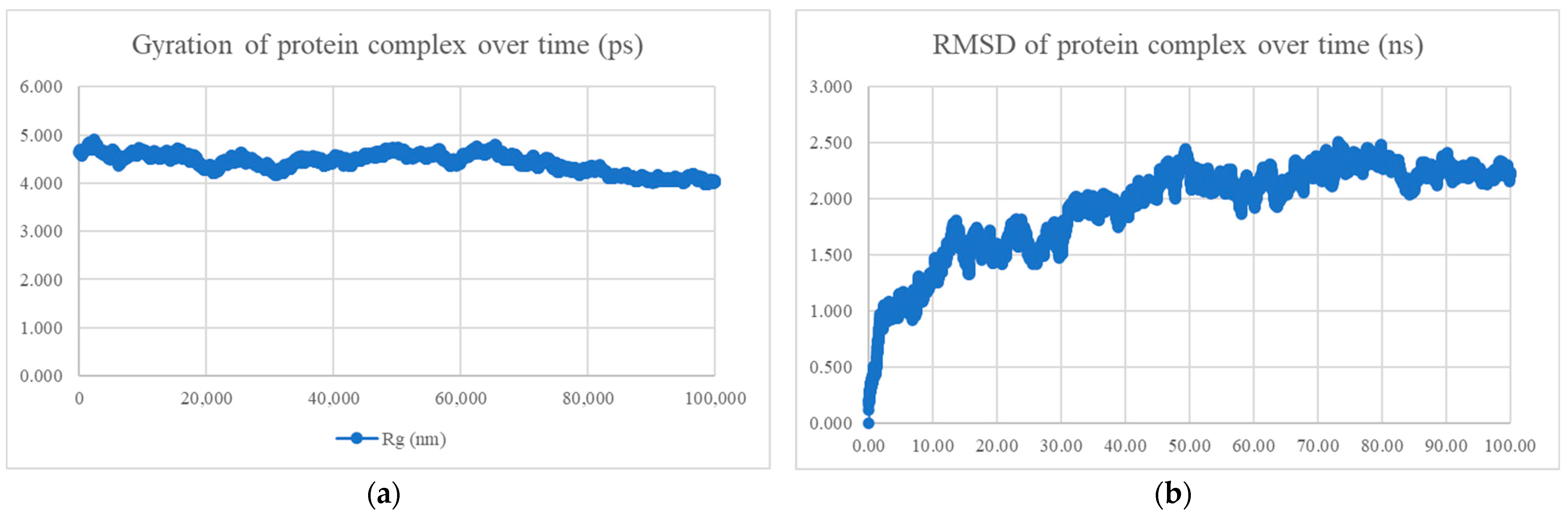
| No. | Peptide Sequence | Amino Acid Length | Models Observed | Average Binding Affinity (kcal/mol) | Radius of Gyration and RMSD | Interaction Energy (kJ/mol) | Mean Number of Hydrogen Bonds |
|---|---|---|---|---|---|---|---|
| BR556 | FDRLYQEFKDDYEDSEER | 18 | 166/200 models are alpha-helix | −10.8 | Compact and stable | −698.794 ± 22.5 | 10.78 |
| BR563 | FDRLRQEFKDDFSEEEFR | 18 | 141/200 models are alpha-helix | −10.1 | Compact and stable | −603.423 ± 44.9 | 7.78 |
| BR573 | FDRLRQEFKDDFYESEFR | 18 | All alpha helix | −10.4 | Compact and stable | −630.395 ± 28.2 | 6.90 |
| BR582 | FDRLRQEFKDDYEDEEERKE | 20 | 110/200 models are alpha-helix | −10.9 | Compact and stable | −1004.769 ± 21.2 | 13.85 |
| BR599 | EKFDRLYQEFKDDYEDSEER | 20 | 156/200 models are alpha-helix | −11.0 | Compact and stable | −1040.334 ± 24.1 | 14.51 |
| BR605 | FDRLRQEFKDDFYEEELREK | 20 | All alpha helix | −10.4 | Not compact, not stable | −809.339 ± 42.3 | 11.53 |
| BR610 | FDRLRQEFKDDFYESEFRKE | 20 | All alpha helix | −10.0 | Compact and stable | −651.934 ± 37.8 | 7.28 |
| BR582-Omicron S Protein | BR5599-Omicron S Protein | ||
|---|---|---|---|
| Docking results | HADDOCK score | −97.4 +/− 4.8 | −100.8 +/− 6.9 |
| Cluster/Position | 3.0/1.0 | 2.0/1.0 | |
| Cluster size | 20 | 17 | |
| RMSD | 0.7 +/− 0.5 | 1.0 +/− 0.6 | |
| Z-score | −2.0 | −2.2 | |
| i-RMSD | 0.0–3.9 | 0.0–3.5 | |
| FCC | 0.5–1.0 | 0.5–1.0 | |
| Average binding affinity ΔG (kcal/mol) | −10.7 | −10.7 | |
| Average Kd (M) at 25.0 °C | 1.58 × 10−8 | 1.65 × 10−8 | |
| PCA and cosine content analysis | No. of Eigenvectors (PC) | 60 | 60 |
| Eigenvalues of first few PCs (%) | PC1: 63.58 PC2: 19.51 PC3: 10.42 % of eigenvalues in 3 PCs = 93.51 | PC1: 77.95 PC2: 16.21 % of eigenvalues in 2 PCs = 94.16 | |
| Cosine content | 0.54 | 0.72 | |
| MDS results | Radius of gyration and RMSD | Compact and stable | - |
| Interaction energy (kJ/mol) | −397.472 ± 20.1 | - | |
| Mean Number of Hydrogen Bonds | 4.90 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, C.; Nazarie, W.F.W.M.; Lee, P.-C. An In Silico Design of Peptides Targeting the S1/S2 Cleavage Site of the SARS-CoV-2 Spike Protein. Viruses 2023, 15, 1930. https://doi.org/10.3390/v15091930
Ho C, Nazarie WFWM, Lee P-C. An In Silico Design of Peptides Targeting the S1/S2 Cleavage Site of the SARS-CoV-2 Spike Protein. Viruses. 2023; 15(9):1930. https://doi.org/10.3390/v15091930
Chicago/Turabian StyleHo, Chian, Wan Fahmi Wan Mohamad Nazarie, and Ping-Chin Lee. 2023. "An In Silico Design of Peptides Targeting the S1/S2 Cleavage Site of the SARS-CoV-2 Spike Protein" Viruses 15, no. 9: 1930. https://doi.org/10.3390/v15091930
APA StyleHo, C., Nazarie, W. F. W. M., & Lee, P.-C. (2023). An In Silico Design of Peptides Targeting the S1/S2 Cleavage Site of the SARS-CoV-2 Spike Protein. Viruses, 15(9), 1930. https://doi.org/10.3390/v15091930