
Time-Course Transcriptome Analysis Reveals Distinct Phases and Identifies Two Key Genes during Severe Fever with Thrombocytopenia Syndrome Virus Infection in PMA-Induced THP-1 Cells
 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
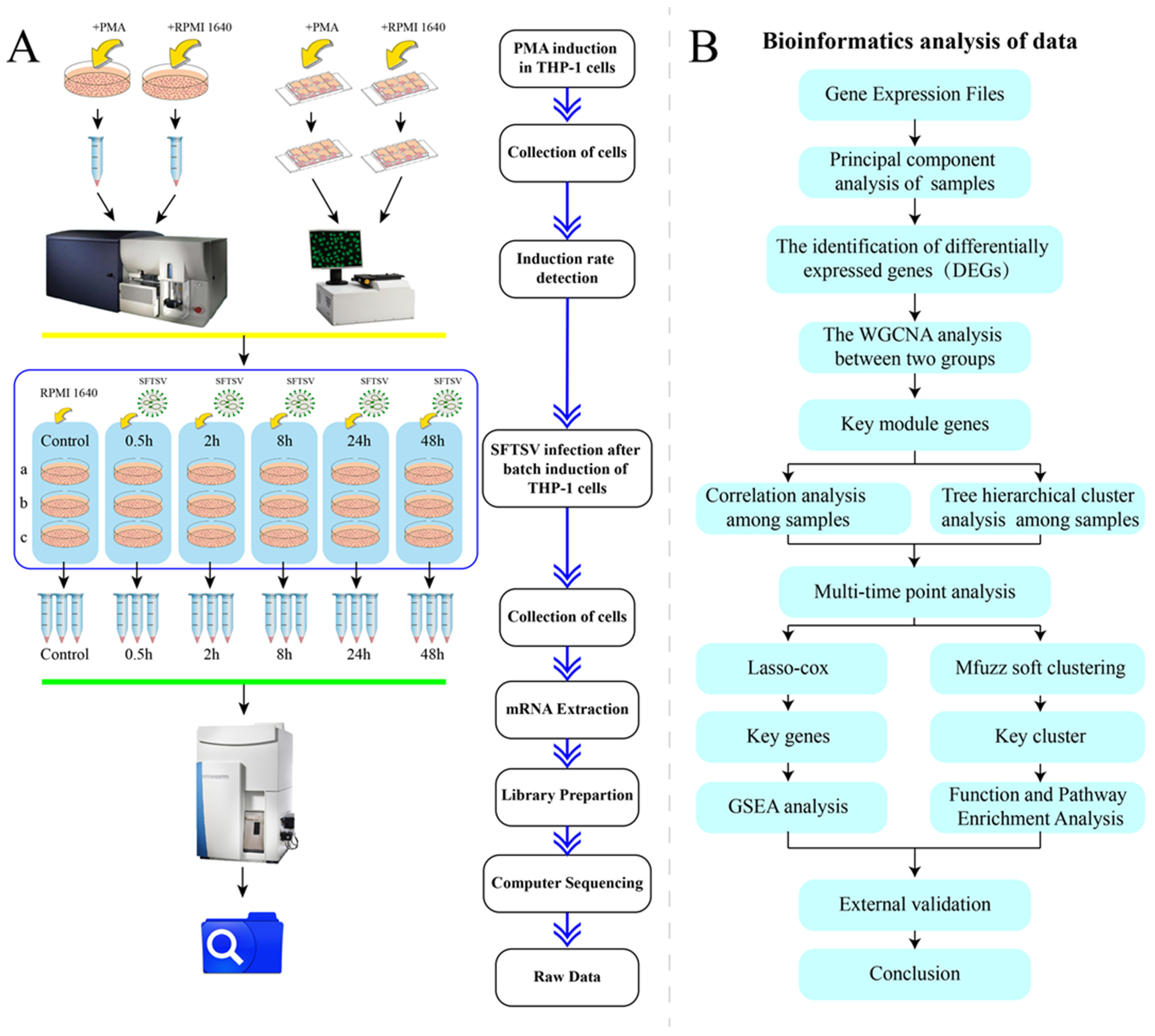
2.1. Cells, Viruses, Antibodies and Other Reagents
2.2. Induction of THP-1
2.3. Cell Samples Preparation for Flow Cytometry
2.4. Cell Samples Preparation for Cellular Immunofluorescence
2.5. SFTSV Infection in dTHP-1 Cells
2.6. Bioinformatics Analysis
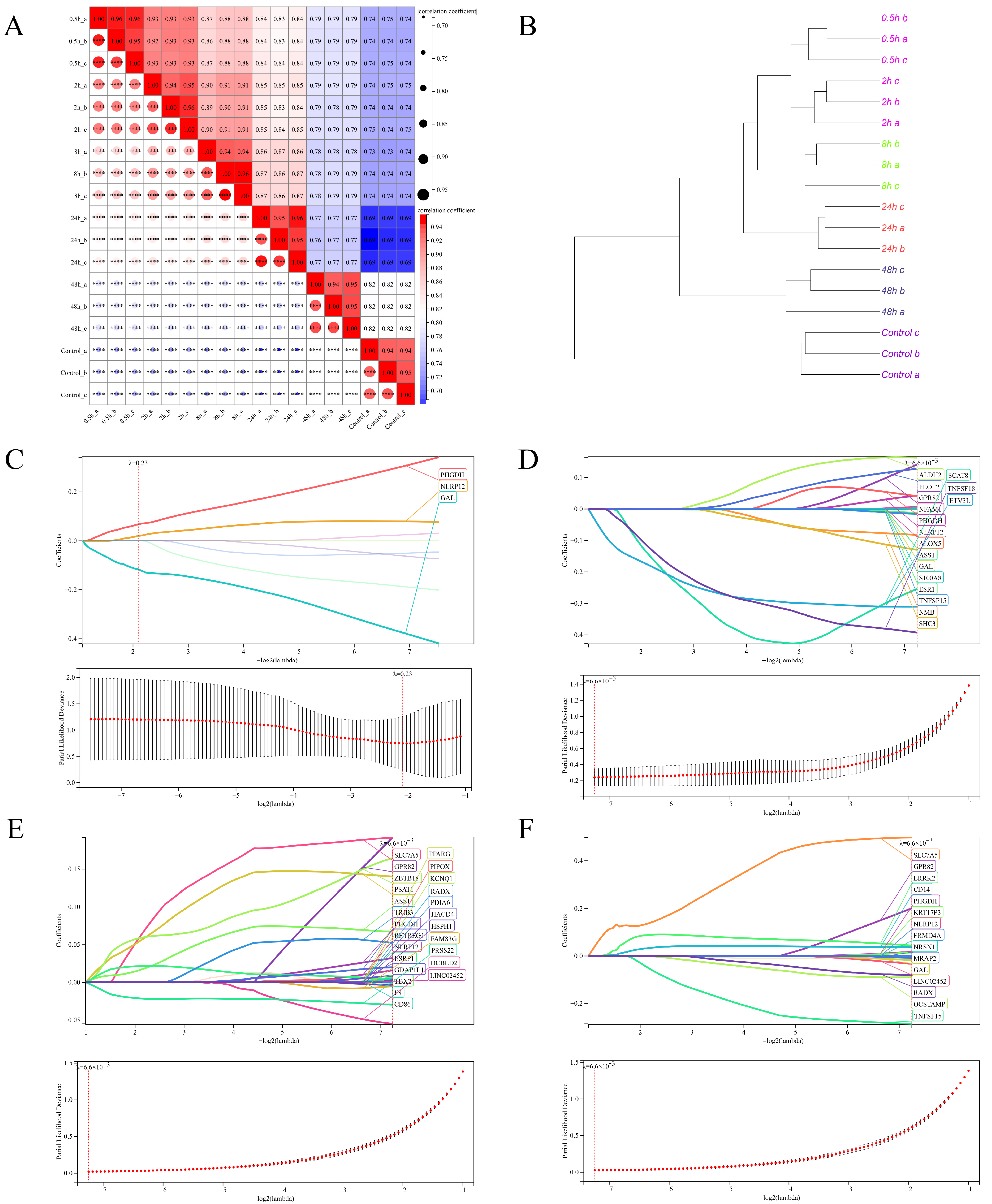
2.6.1. Principal Component Analysis
2.6.2. DEGs Identification
2.6.3. Function and Pathway Enrichment Analysis
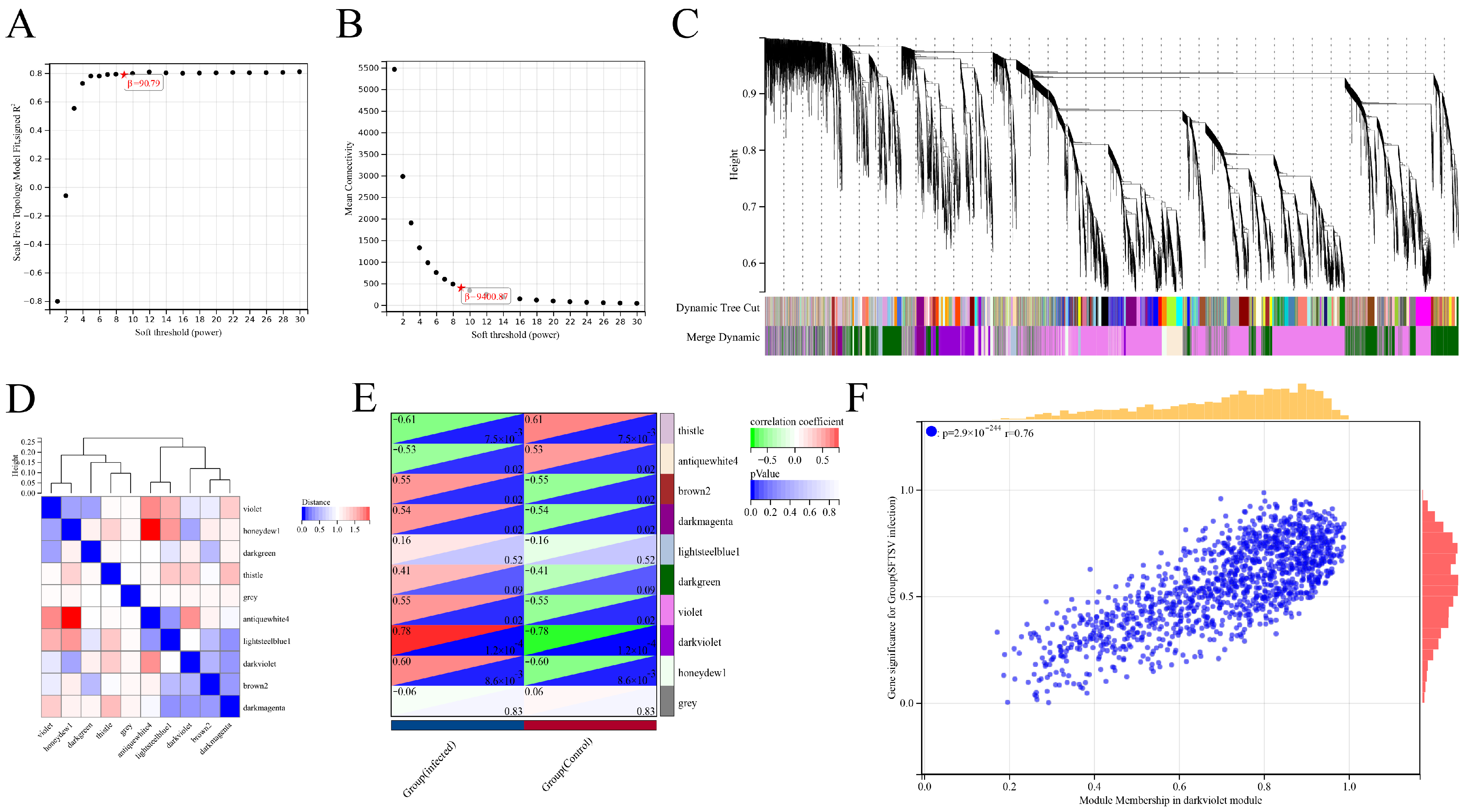
2.6.4. WGCNA Analysis
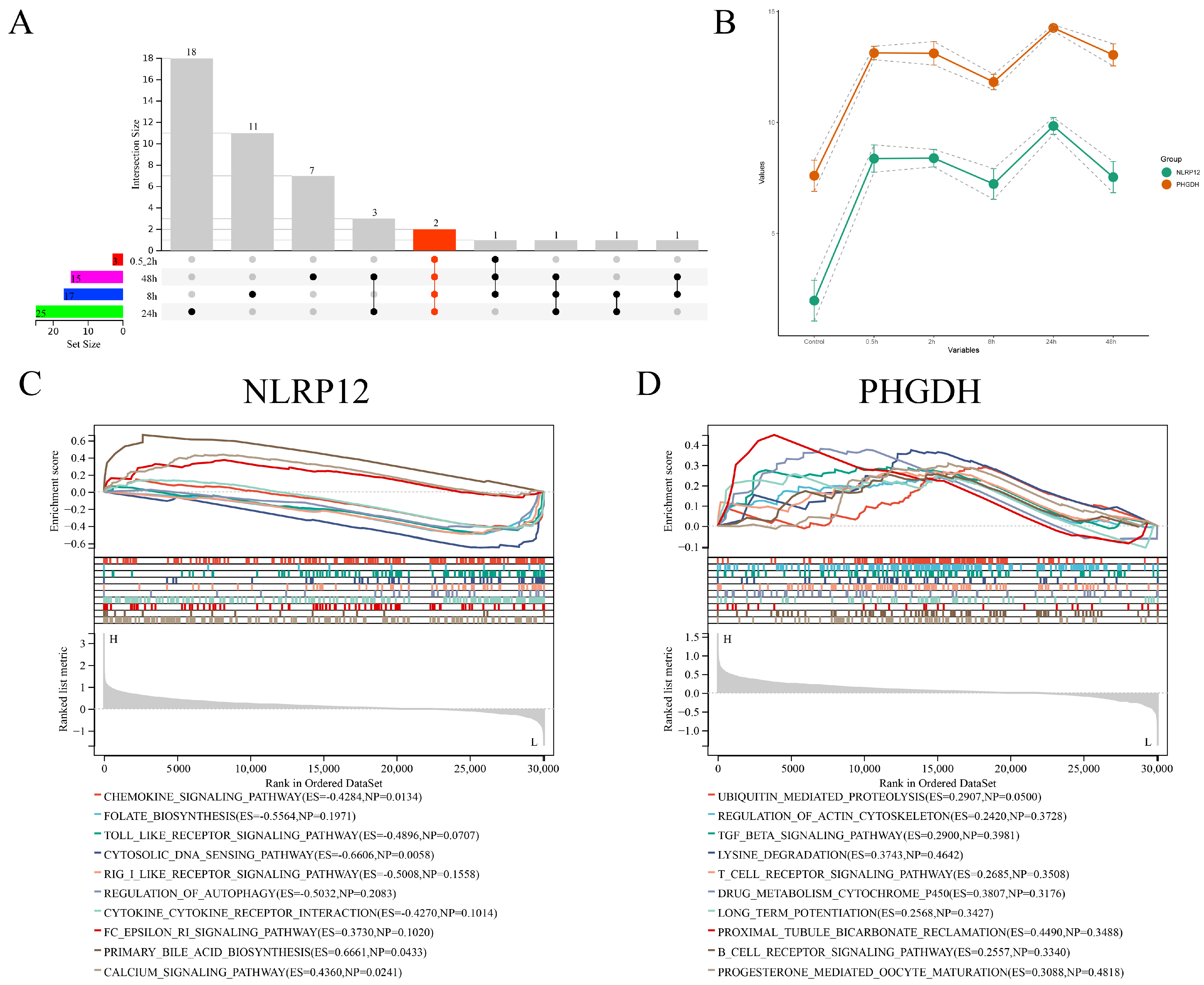
2.6.5. LASSO/Cox Regression Analysis
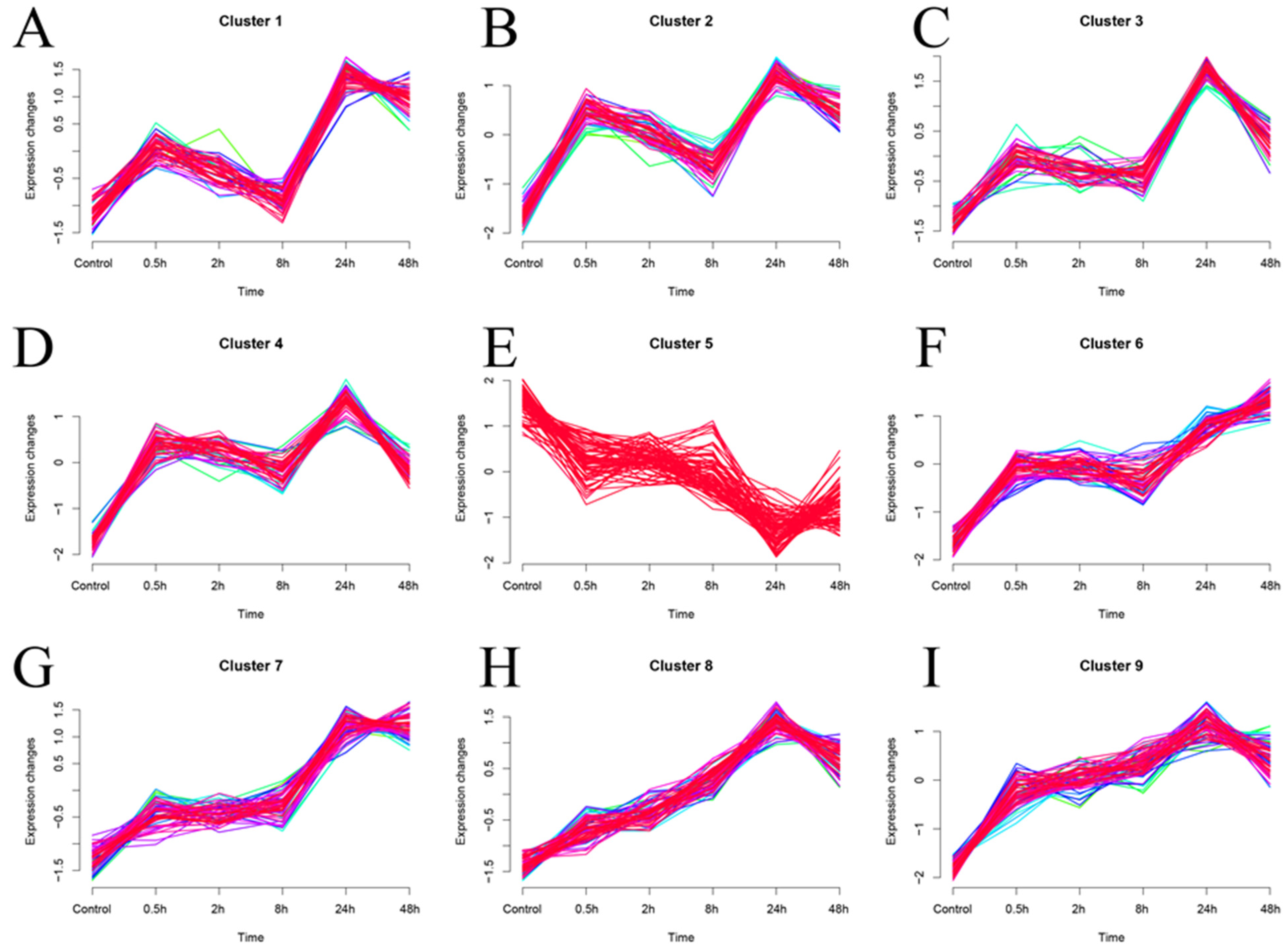
2.6.6. Mfuzz Soft Cluster Analysis
2.6.7. GSEA Results Display
2.6.8. GEO Datasets Involved in This Study
3. Results
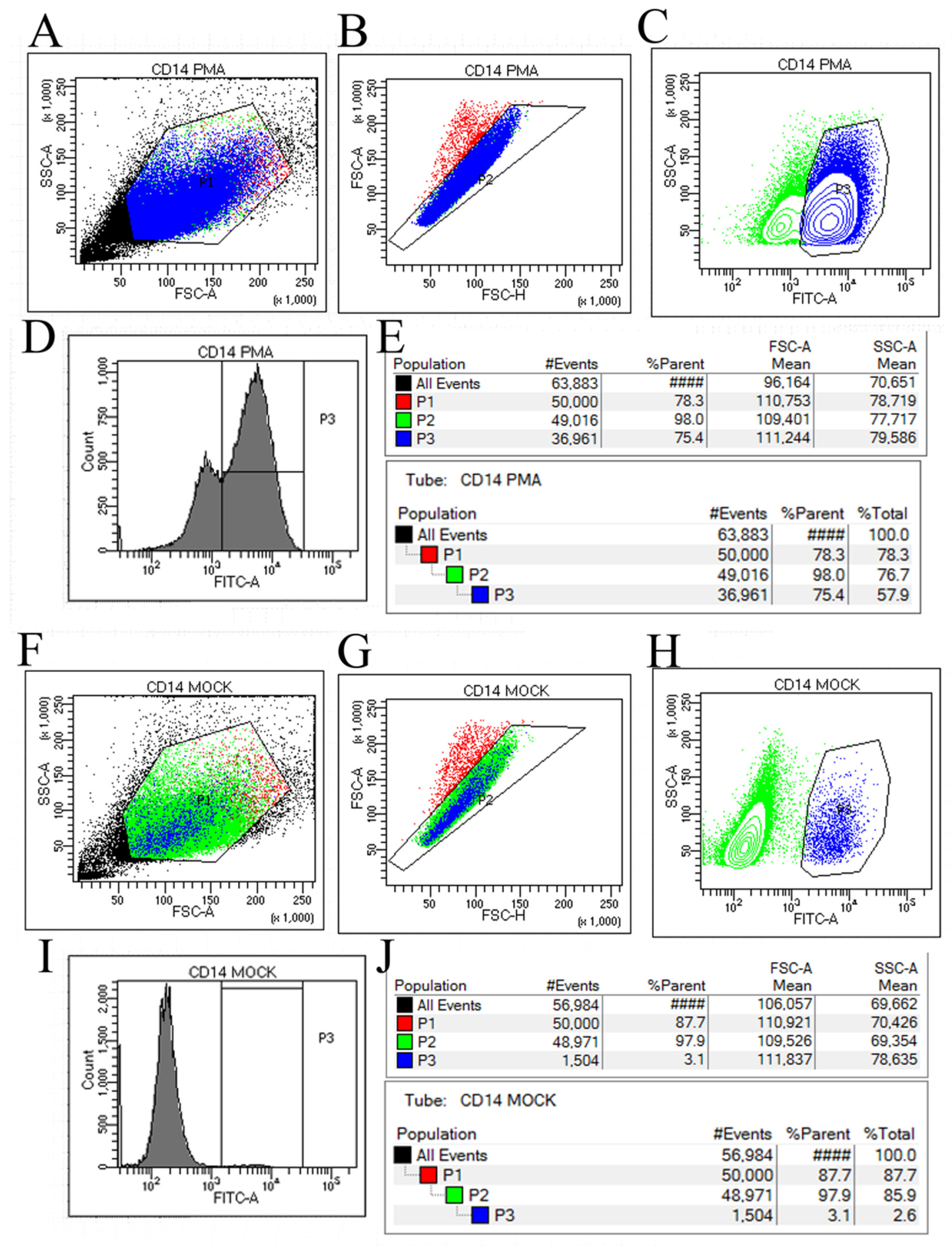
3.1. PMA Induction Results of THP-1 Cells
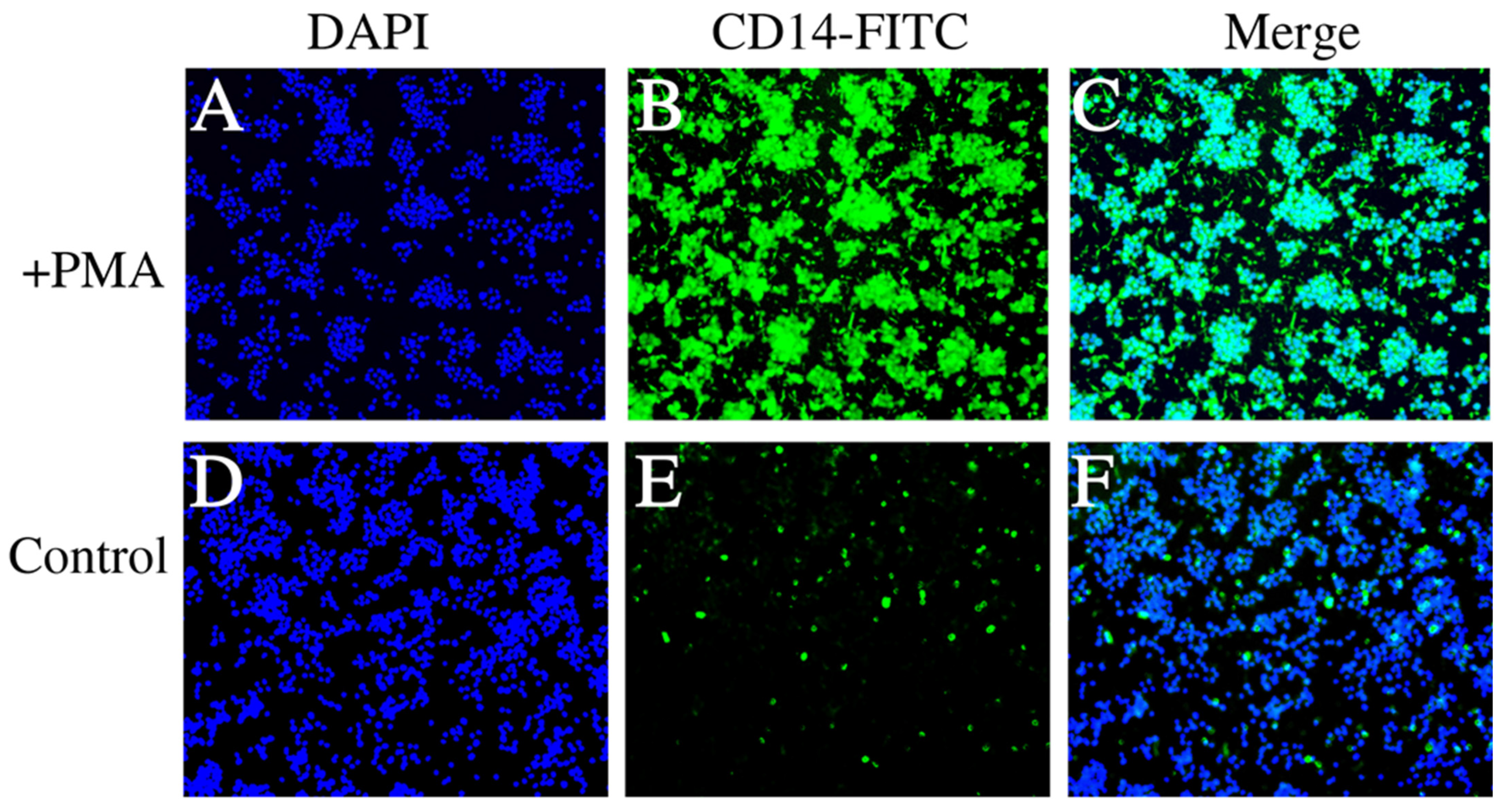
3.2. Fluorescence Microscopy Results of PMA-Induced and Normal Control THP-1 Cells
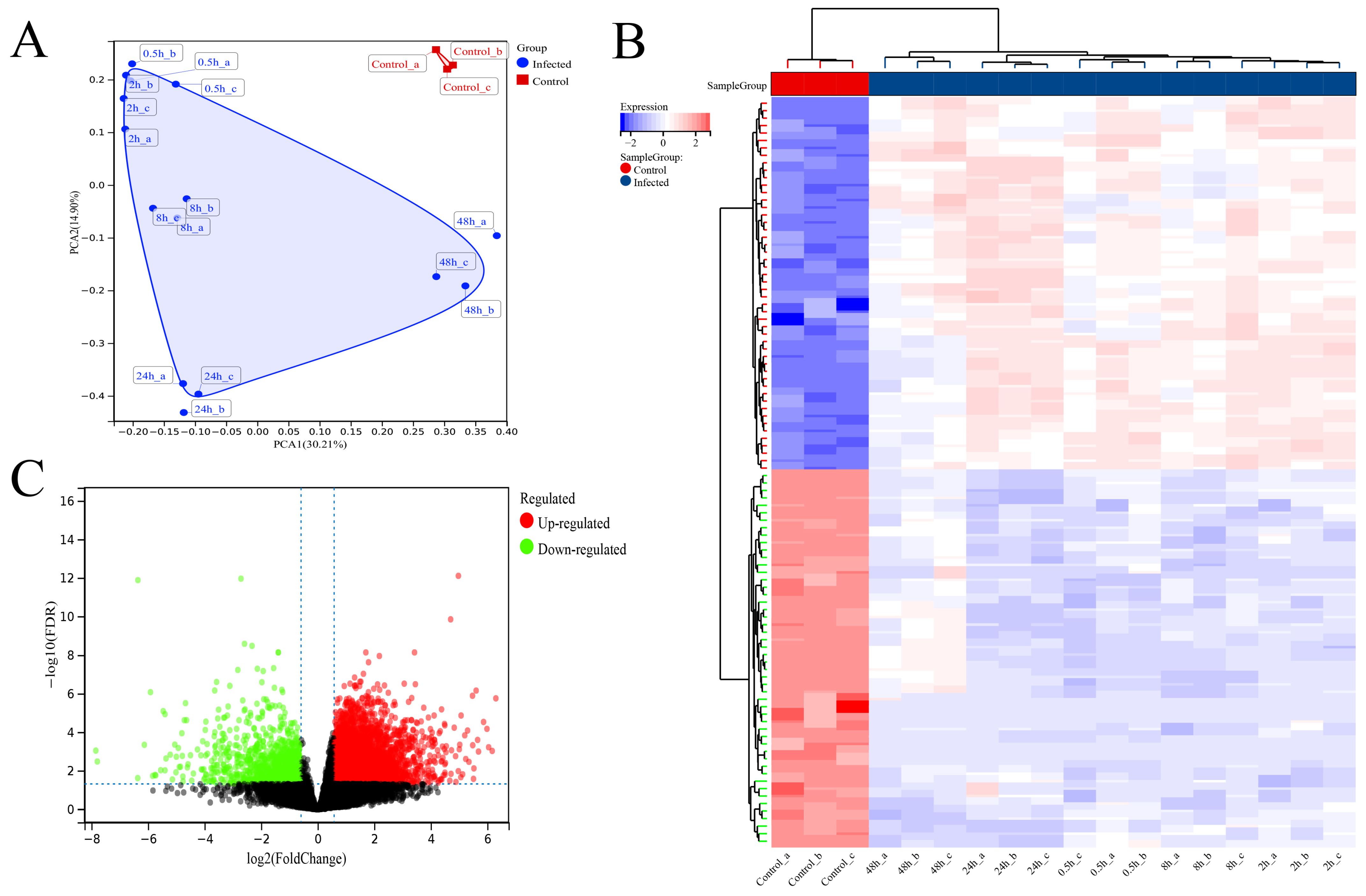
3.3. Transcriptome Sequencing Data Quality Control
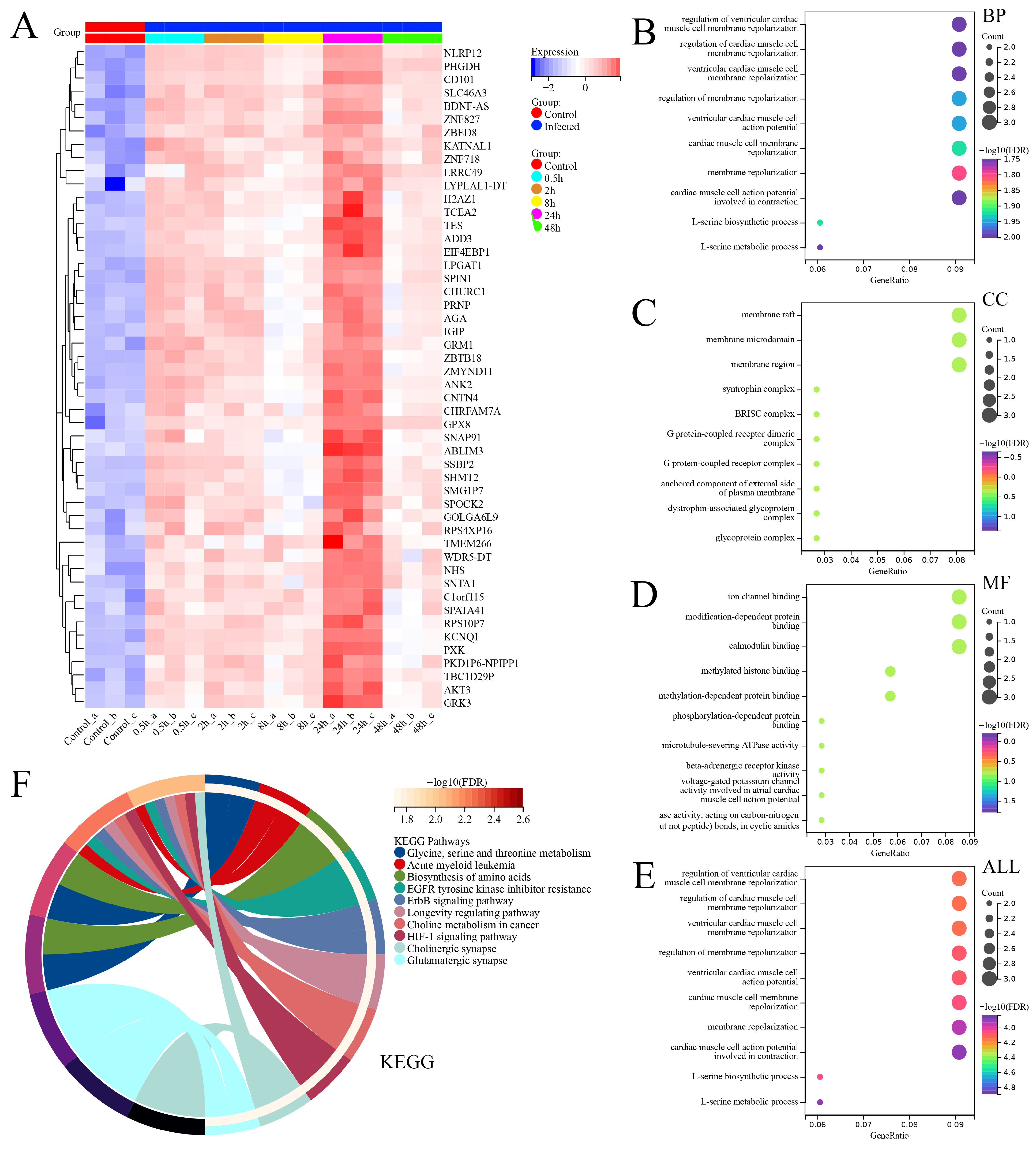
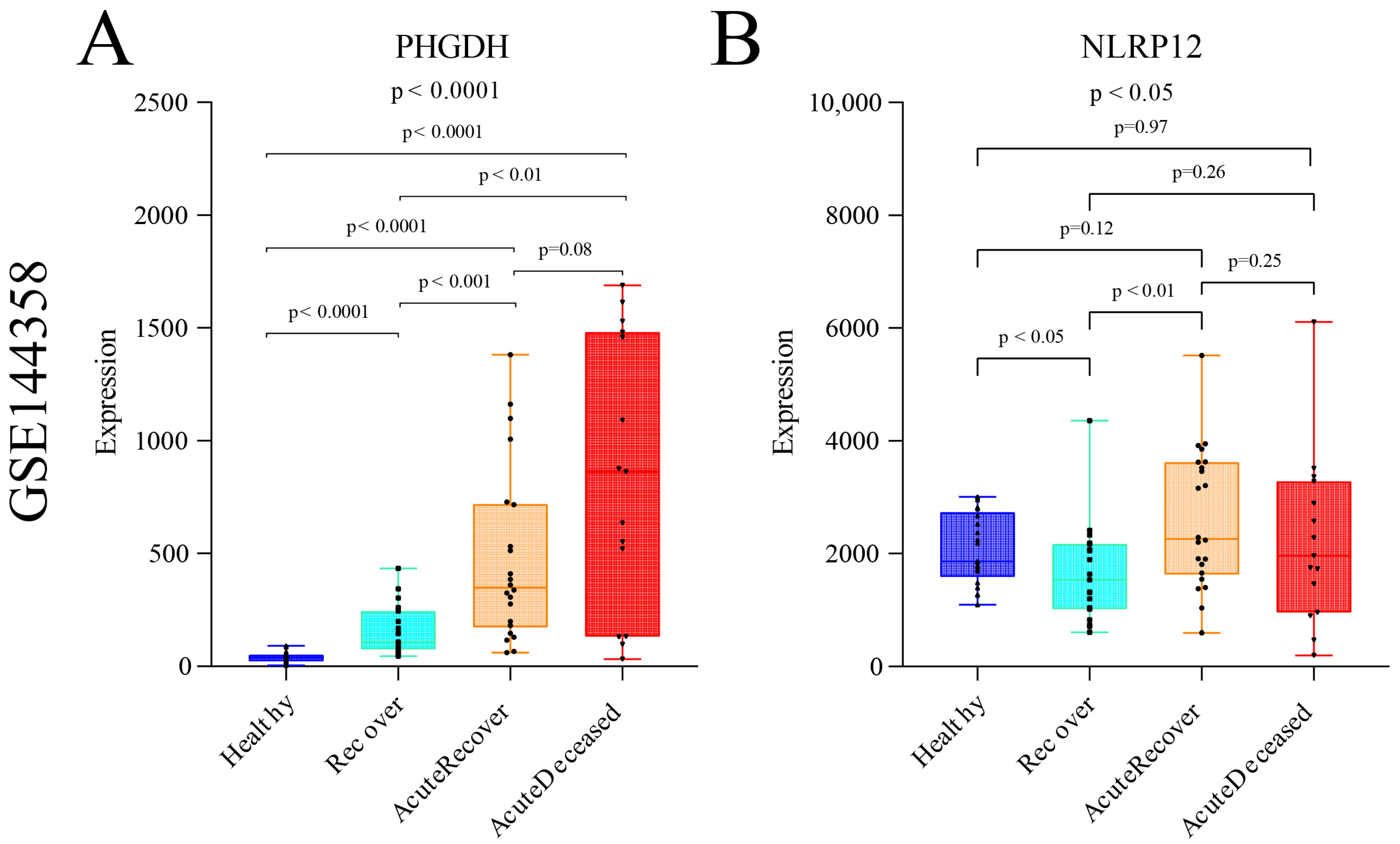
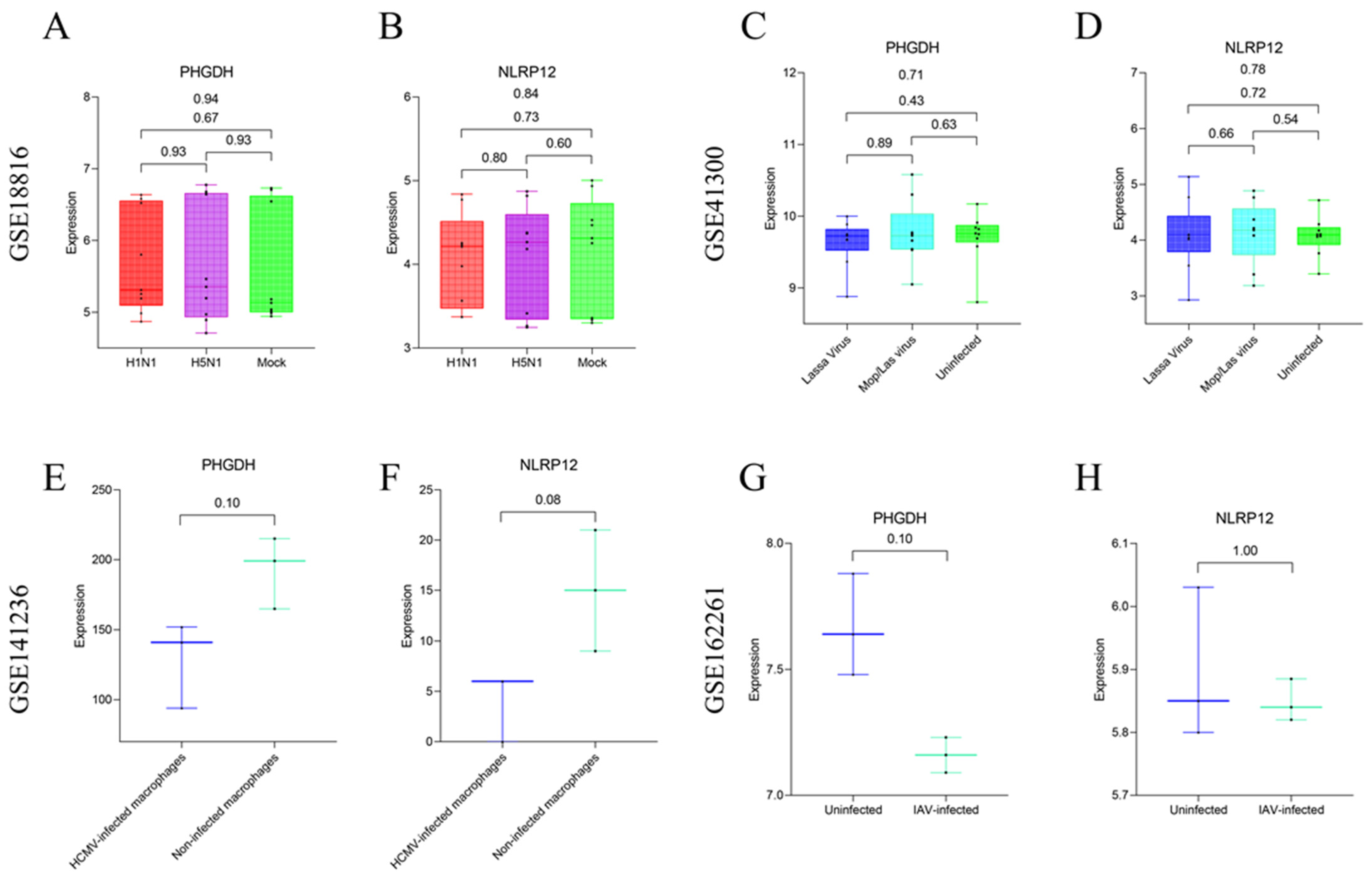
3.4. Differential Analysis between SFTSV-Infected THP-1 Cells and the Control Group
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu, X.J.; Liang, M.F.; Zhang, S.Y.; Liu, Y.; Li, J.D.; Sun, Y.L.; Zhang, L.; Zhang, Q.F.; Popov, V.L.; Li, C.; et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. N. Engl. J. Med. 2011, 364, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.J.; Guo, X.L.; Qi, X.; Hu, J.L.; Zhou, M.H.; Varma, J.K.; Cui, L.B.; Yang, H.T.; Jiao, Y.J.; Klena, J.D.; et al. A family cluster of infections by a newly recognized bunyavirus in eastern China, 2007: Further evidence of person-to-person transmission. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2011, 53, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.Y.; Choi, W.; Kim, J.; Wang, E.; Park, S.W.; Lee, W.J.; Choi, J.; Kim, H.Y.; Uh, Y.; Kim, Y.K. Nosocomial person-to-person transmission of severe fever with thrombocytopenia syndrome. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2019, 25, 633.e1–633.e4. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.L.; Zhang, S.; Jiang, M.; Bi, Z.Q.; Liang, M.F.; Ding, S.J.; Wang, S.W.; Liu, J.Y.; Zhou, S.Q.; Zhang, X.M.; et al. A cluster of person-to-person transmission cases caused by SFTS virus in Penglai, China. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2015, 21, 274–279. [Google Scholar] [CrossRef]
- Tang, X.; Wu, W.; Wang, H.; Du, Y.; Liu, L.; Kang, K.; Huang, X.; Ma, H.; Mu, F.; Zhang, S.; et al. Human-to-human transmission of severe fever with thrombocytopenia syndrome bunyavirus through contact with infectious blood. J. Infect. Dis. 2013, 207, 736–739. [Google Scholar] [CrossRef]
- Gai, Z.; Liang, M.; Zhang, Y.; Zhang, S.; Jin, C.; Wang, S.W.; Sun, L.; Zhou, N.; Zhang, Q.; Sun, Y.; et al. Person-to-person transmission of severe fever with thrombocytopenia syndrome bunyavirus through blood contact. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2012, 54, 249–252. [Google Scholar] [CrossRef]
- Matsuu, A.; Momoi, Y.; Nishiguchi, A.; Noguchi, K.; Yabuki, M.; Hamakubo, E.; Take, M.; Maeda, K. Natural severe fever with thrombocytopenia syndrome virus infection in domestic cats in Japan. Vet. Microbiol. 2019, 236, 108346. [Google Scholar] [CrossRef]
- Bopp, N.E.; Kaiser, J.A.; Strother, A.E.; Barrett, A.D.; Beasley, D.W.; Benassi, V.; Milligan, G.N.; Preziosi, M.P.; Reece, L.M. Baseline mapping of severe fever with thrombocytopenia syndrome virology, epidemiology and vaccine research and development. NPJ Vaccines 2020, 5, 111. [Google Scholar] [CrossRef]
- Chanput, W.; Mes, J.J.; Wichers, H.J. THP-1 cell line: An in vitro cell model for immune modulation approach. Int. Immunopharmacol. 2014, 23, 37–45. [Google Scholar] [CrossRef]
- Aldo, P.B.; Craveiro, V.; Guller, S.; Mor, G. Effect of culture conditions on the phenotype of THP-1 monocyte cell line. Am. J. Reprod. Immunol. 2013, 70, 80–86. [Google Scholar] [CrossRef]
- Fernández-Bravo, A.; Figueras, M.J. Immune Response of the Monocytic Cell Line THP-1 Against Six Aeromonas spp. Front. Immunol. 2022, 13, 875689–875690. [Google Scholar] [CrossRef] [PubMed]
- Maria, H.J.; Nicole, T. The Monocytic Cell Line THP-1 as a Validated and Robust Surrogate Model for Human Dendritic Cells. Int. J. Mol. Sci. 2023, 24, 1452–1454. [Google Scholar]
- Ren, J.; Szombath, G.; Vitale-Cross, L.; Stroncek, D.F.; Robey, P.G.; Hajdara, A.; Szalayova, I.; Mayer, B.; Martin, D.; Mezey, E.; et al. The Potential Use of THP-1, a Monocytic Leukemia Cell Line, to Predict Immune-Suppressive Potency of Human Bone-Marrow Stromal Cells (BMSCs) In Vitro: A Pilot Study. Int. J. Mol. Sci. 2023, 24, 13258–13260. [Google Scholar] [CrossRef] [PubMed]
- Kasoju, N.; Wang, H.; Zhang, B.; George, J.; Gao, S.; Triffitt, J.T.; Cui, Z.; Ye, H. Transcriptomics of human multipotent mesenchymal stromal cells: Retrospective analysis and future prospects. Biotechnol. Adv. 2017, 35, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Huang, T.; Li, J.; Li, A.; Li, C.; Huang, X.; Li, D.; Wang, S.; Liang, M. Optimization of differentiation and transcriptomic profile of THP-1 cells into macrophage by PMA. PLoS ONE 2023, 18, e0286056. [Google Scholar] [CrossRef] [PubMed]
- Daigneault, M.; Preston, J.A.; Marriott, H.M.; Whyte, M.K.; Dockrell, D.H. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS ONE 2010, 5, e8668. [Google Scholar] [CrossRef]
- Goetz, C.; Hammerbeck, C.; Boss, K.; Peterson, R.; Bonnevier, J. Phenotyping of M1 and M2a Macrophages and Differential Expression of ACE-2 on Monocytes by Flow Cytometry: Impact of Cell Culture Conditions and Sample Processing. Methods Mol. Biol. 2023, 2593, 197–212. [Google Scholar]
- Ritchie, M.E.; Phipson, B.; Wu, D.I.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Utazirubanda, J.C.; León, T.M.; Ngom, P. Variable selection with Group LASSO approach: Application to Cox regression with frailty model. Commun. Stat. Simul. Comput. 2021, 50, 881–901. [Google Scholar] [CrossRef] [PubMed]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Mesirov, J.P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Lee, S.M.; Gardy, J.L.; Cheung, C.Y.; Cheung, T.K.; Hui, K.P.; Ip, N.Y.; Peiris, J.M. Systems-level comparison of host-responses elicited by avian H5N1 and seasonal H1N1 influenza viruses in primary human macrophages. PLoS ONE 2009, 4, e8072. [Google Scholar] [CrossRef] [PubMed]
- Zapata, J.C.; Carrion Jr, R.; Patterson, J.L.; Crasta, O.; Zhang, Y.; Mani, S.; Salvato, M.S. Transcriptome analysis of human peripheral blood mononuclear cells exposed to Lassa virus and to the attenuated Mopeia/Lassa reassortant 29 (ML29), a vaccine candidate. PLoS Neglected Trop. Diseases 2013, 7, e2406. [Google Scholar] [CrossRef] [PubMed]
- Quan, H.; Kim, J.; Na, Y.R.; Kim, J.H.; Kim, B.J.; Kim, B.J.; Hong, J.J.; Hwang, E.S.; Seok, S.H. Human Cytomegalovirus-Induced Interleukin-10 Production Promotes the Proliferation of Mycobacterium massiliense in Macrophages. Front. Immunol. 2020, 11, 518605. [Google Scholar] [CrossRef]
- Sohail, A.; Iqbal, A.A.; Sahini, N.; Chen, F.; Tantawy, M.; Waqas, S.F.; Pessler, F. Itaconate and derivatives reduce interferon responses and inflammation in influenza A virus infection. PLoS Pathog. 2022, 18, e1010219. [Google Scholar]
- Tang, J.; Kong, D.; Cui, Q.; Wang, K.; Zhang, D.; Gong, Y.; Wu, G. Prognostic Genes of Breast Cancer Identified by Gene Co-expression Network Analysis. Front. Oncol. 2018, 8, 374. [Google Scholar] [CrossRef]
- Li, S.; Li, H.; Zhang, Y.L.; Xin, Q.L.; Guan, Z.Q.; Chen, X.; Peng, K. SFTSV Infection Induces BAK/BAX-Dependent Mitochondrial DNA Release to Trigger NLRP3 Inflammasome Activation. Cell Rep. 2020, 30, 4370–4385.e7. [Google Scholar] [CrossRef]
- Liu, H.; Lorenzini, P.A.; Zhang, F.; Xu, S.; Wong MS, M.; Zheng, J.; Roca, X. Alternative splicing analysis in human monocytes and macrophages reveals MBNL1 as major regulator. Nucleic Acids Res. 2018, 46, 6069–6086. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Hu, P.; Zhang, Y.; Ji, Z.; Shan, X.; Ni, L.; Yu, Q. Serine metabolism antagonizes antiviral innate immunity by preventing ATP6V0d2-mediated YAP lysosomal degradation. Cell Metab. 2021, 33, 971–987.e6. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, B.; Pandian, N.; Mall, R.; Wang, Y.; Sarkar, R.; Kim, H.J.; Kanneganti, T.D. NLRP12-PANoptosome activates PANoptosis and pathology in response to heme and PAMPs. Cell 2023, 186, 2783–2801.e20. [Google Scholar] [CrossRef] [PubMed]
- Lukens, J.R.; Gurung, P.; Shaw, P.J.; Barr, M.J.; Zaki, M.H.; Brown, S.A.; Kanneganti, T.D. The NLRP12 Sensor Negatively Regulates Autoinflammatory Disease by Modulating Interleukin-4 Production in T Cells. Immunity 2015, 42, 654–664. [Google Scholar] [CrossRef]
- Suzuki, T.; Sato, Y.; Sano, K.; Arashiro, T.; Katano, H.; Nakajima, N.; Hasegawa, H. Severe fever with thrombocytopenia syndrome virus targets B cells in lethal human infections. J. Clin. Investig. 2020, 130, 799–812. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Clean Bases | Error (%) | Q20 (%) | Q30 (%) | GC (%) |
|---|---|---|---|---|---|---|---|
| Con_a | 51351534 | 49380376 | 7.39 G | 0.04 | 97.56 | 93.43 | 47.29 |
| Con_b | 48700228 | 46605374 | 6.97 G | 0.04 | 96.98 | 92.03 | 46.88 |
| Con_c | 48772582 | 46967334 | 7.03 G | 0.04 | 97.15 | 92.17 | 47.08 |
| 0.5h_a | 57442976 | 55542550 | 8.3 G | 0.04 | 97.87 | 94.31 | 46.16 |
| 0.5h_b | 55893148 | 54049126 | 8.08 G | 0.04 | 97.6 | 93.54 | 46.08 |
| 0.5h_c | 48629620 | 47049176 | 7.04 G | 0.04 | 97.39 | 92.87 | 46.2 |
| 2h_a | 56381612 | 54489560 | 8.16 G | 0.04 | 97.56 | 93.4 | 46.79 |
| 2h_b | 55676058 | 53751606 | 8.04 G | 0.04 | 97.51 | 93.26 | 46.71 |
| 2h_c | 55803544 | 53802804 | 8.05 G | 0.04 | 97.46 | 93.18 | 46.67 |
| 8h_a | 50837064 | 49222058 | 7.34 G | 0.04 | 97.89 | 94.25 | 47.43 |
| 8h_b | 47859920 | 46116568 | 6.9 G | 0.04 | 97.46 | 93.2 | 47.03 |
| 8h_c | 54598234 | 52678942 | 7.88 G | 0.04 | 97.4 | 92.93 | 47.07 |
| 24h_a | 50530122 | 48702430 | 7.29 G | 0.04 | 97.17 | 92.27 | 47.38 |
| 24h_b | 50848524 | 48895934 | 7.32 G | 0.04 | 97.59 | 93.54 | 47.09 |
| 24h_c | 47937234 | 46222114 | 6.92 G | 0.04 | 97.35 | 92.78 | 47.15 |
| 48h_a | 47382892 | 45919646 | 6.87 G | 0.04 | 97.85 | 94.12 | 47.41 |
| 48h_b | 53639272 | 51868716 | 7.76 G | 0.04 | 97.65 | 93.62 | 47.16 |
| 48h_c | 58370484 | 56457460 | 8.44 G | 0.04 | 97.64 | 93.49 | 47.51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, T.; Wang, X.; Mi, Y.; Wu, W.; Xu, X.; Li, C.; Wen, Y.; Li, B.; Li, Y.; Sun, L.; et al. Time-Course Transcriptome Analysis Reveals Distinct Phases and Identifies Two Key Genes during Severe Fever with Thrombocytopenia Syndrome Virus Infection in PMA-Induced THP-1 Cells. Viruses 2024, 16, 59. https://doi.org/10.3390/v16010059
Huang T, Wang X, Mi Y, Wu W, Xu X, Li C, Wen Y, Li B, Li Y, Sun L, et al. Time-Course Transcriptome Analysis Reveals Distinct Phases and Identifies Two Key Genes during Severe Fever with Thrombocytopenia Syndrome Virus Infection in PMA-Induced THP-1 Cells. Viruses. 2024; 16(1):59. https://doi.org/10.3390/v16010059
Chicago/Turabian StyleHuang, Tao, Xueqi Wang, Yuqian Mi, Wei Wu, Xiao Xu, Chuan Li, Yanhan Wen, Boyang Li, Yang Li, Lina Sun, and et al. 2024. "Time-Course Transcriptome Analysis Reveals Distinct Phases and Identifies Two Key Genes during Severe Fever with Thrombocytopenia Syndrome Virus Infection in PMA-Induced THP-1 Cells" Viruses 16, no. 1: 59. https://doi.org/10.3390/v16010059