
Experimental Identification of Cross-Reacting IgG Hotspots to Predict Existing Immunity Evasion of SARS-CoV-2 Variants by a New Biotechnological Application of Phage Display

 , , ,
, , ,
Abstract
:1. Introduction
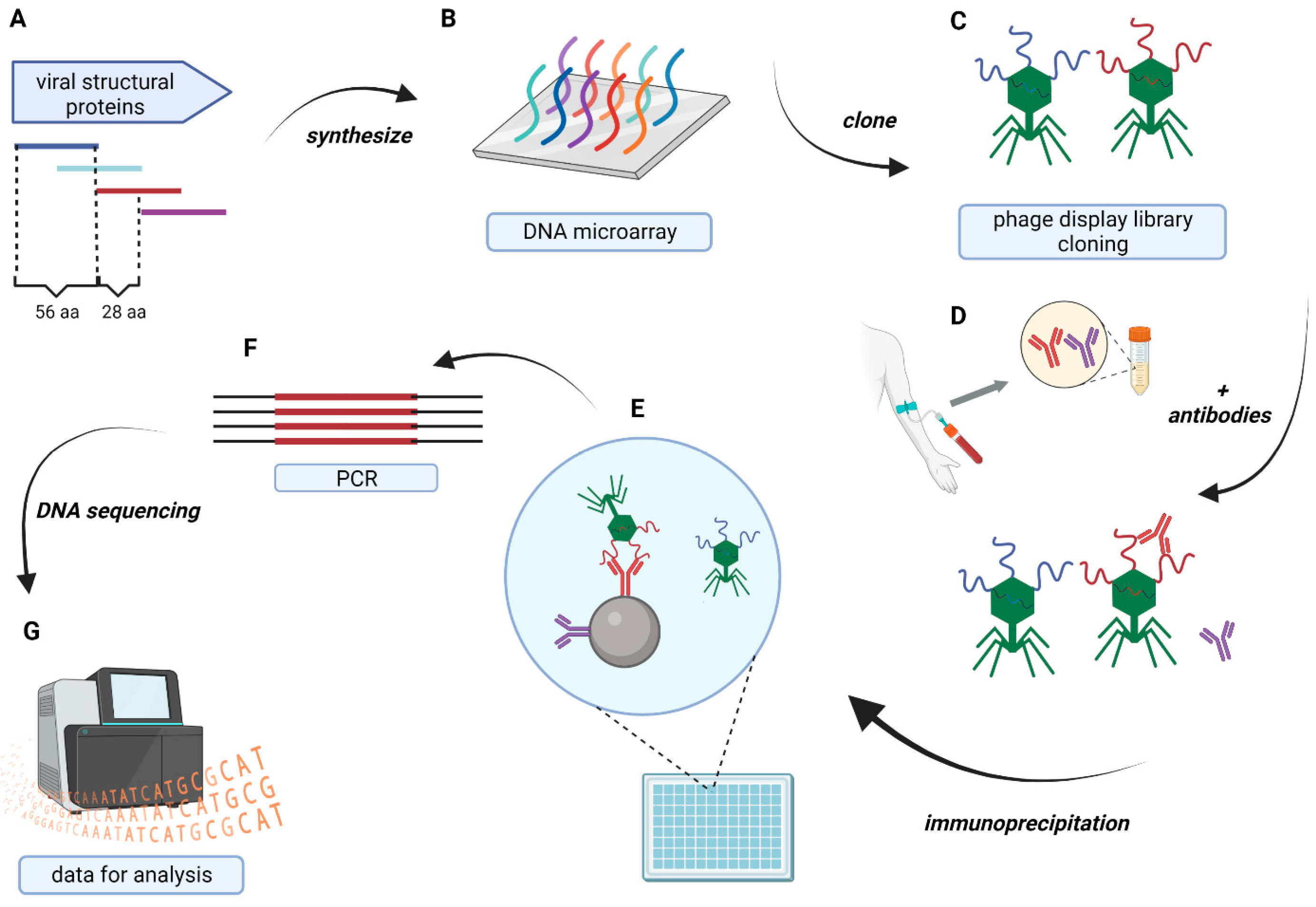
2. Materials and Methods
2.1. Serum Donors
2.2. Bioethics Statements
2.3. Blood Samples
2.4. Identification of SARS-CoV-2 Variants Infecting Investigated Patients
2.5. Epitope Analysis
2.6. Sequencing Data Analysis
2.7. Immunogenicity and Cross-Reacting IgG Hot-Spot Determination—Statistical Model
3. Results
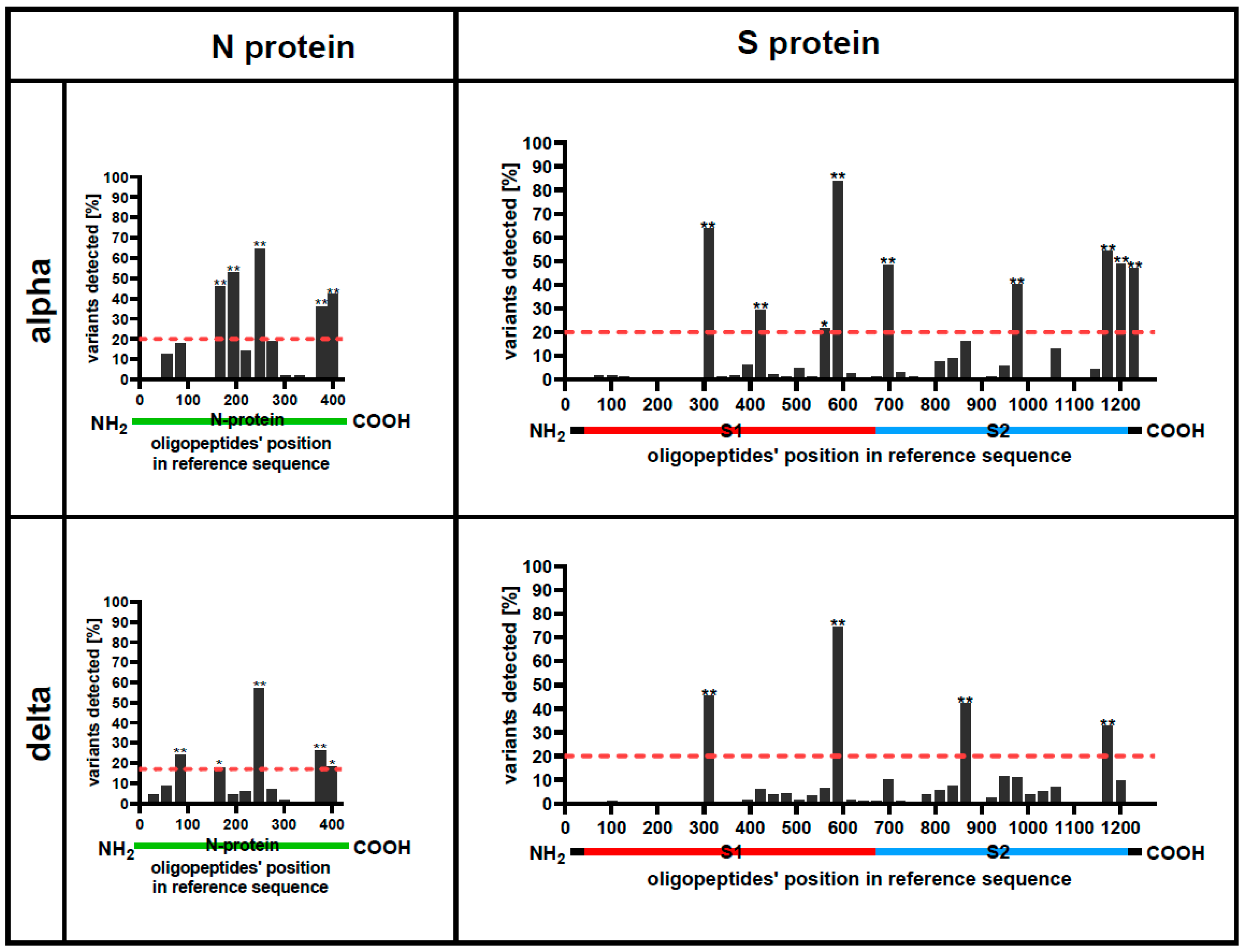
3.1. Some Protein Regions Can Be Altered without Affecting Their Recognition by Patients IgG Antibodies
3.2. Immunogenicity Differs between Protein Regions
3.3. Some Protein Regions Show Both High Immunogenicity and Presence of Cross-Reactive Hotspots
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jacobs, J.L.; Haidar, G.; Mellors, J.W. COVID-19: Challenges of Viral Variants. Annu. Rev. Med. 2023, 74, 31–53. [Google Scholar] [CrossRef] [PubMed]
- Akkız, H. The Biological Functions and Clinical Significance of SARS-CoV-2 Variants of Concern. Front. Med. 2022, 9, 849217. [Google Scholar] [CrossRef] [PubMed]
- Yewdell, J.W. Antigenic drift: Understanding COVID-19. Immunity 2021, 54, 2681–2687. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rao, Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat. Rev. Microbiol. 2021, 19, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Tretyn, A.; Szczepanek, J.; Skorupa, M.; Jarkiewicz-Tretyn, J.; Sandomierz, D.; Dejewska, J.; Ciechanowska, K.; Jarkiewicz-Tretyn, A.; Koper, W.; Pałgan, K. Differences in the Concentration of Anti-SARS-CoV-2 IgG Antibodies Post-COVID-19 Recovery or Post-Vaccination. Cells 2021, 10, 1952. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ong, C.M.; Yun, C.; Mo, W.; Whitman, J.D.; Lynch, K.L.; Wu, A.H.B. Diagnostic Value of Nucleocapsid Protein in Blood for SARS-CoV-2 Infection. Clin. Chem. 2021, 68, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.J.; Kula, T.; Xu, Q.; Li, M.Z.; Vernon, S.D.; Ndung’u, T.; Ruxrungtham, K.; Sanchez, J.; Brander, C.; Chung, R.T.; et al. Viral immunology. Comprehensive serological profiling of human populations using a synthetic human virome. Science 2015, 348, aaa0698. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Goujon, M.; McWilliam, H.; Li, W.; Valentin, F.; Squizzato, S.; Paern, J.; Lopez, R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010, 38, W695–W699. [Google Scholar] [CrossRef] [PubMed]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef]
- Harhala, M.A.; Gembara, K.; Nelson, D.C.; Miernikiewicz, P.; Dąbrowska, K. Immunogenicity of Endolysin PlyC. Antibiotics 2022, 11, 966. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Elshabrawy, H.A.; Coughlin, M.M.; Baker, S.C.; Prabhakar, B.S. Human monoclonal antibodies against highly conserved HR1 and HR2 domains of the SARS-CoV spike protein are more broadly neutralizing. PLoS ONE 2012, 7, e50366. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. Domains and Functions of Spike Protein in Sars-Cov-2 in the Context of Vaccine Design. Viruses 2021, 13, 109. [Google Scholar] [CrossRef] [PubMed]
- Shrock, E.; Fujimura, E.; Kula, T.; Timms, R.T.; Lee, I.H.; Leng, Y.; Robinson, M.L.; Sie, B.M.; Li, M.Z.; Chen, Y.; et al. Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity. Science 2020, 370, eabd4250. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Li, Z.; Guan, Q.; Cui, W.; Zheng, B.; Ding, Q.; Lv, G.; Xu, J.; Zhang, W. Characterization and analysis of linear epitopes corresponding to SARS-CoV-2 outbreak in Jilin Province, China. J. Med. Virol. 2023, 95, e28323. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference Protein Fragment [Amino Acid Position] | Immunogenicity | Cross-Reacting IgG Hot-Spots | ||||
|---|---|---|---|---|---|---|
| N-protein | Start | End | Alpha | Delta | Alpha | Delta |
| 197 | 280 | Very High | High | Strong | Strong | |
| 358 | 419 | Very High | High | Strong | Strong | |
| S-protein | 281 | 337 | Very High | High | Strong | Strong |
| 533 | 617 | High | Medium | Strong | Strong | |
| 925 | 1004 | High | High | Strong | None | |
| 1145 | 1232 | Very High | High | Strong | Strong | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harhala, M.A.; Gembara, K.; Baniecki, K.; Pikies, A.; Nahorecki, A.; Jędruchniewicz, N.; Kaźmierczak, Z.; Rybicka, I.; Klimek, T.; Witkiewicz, W.; et al. Experimental Identification of Cross-Reacting IgG Hotspots to Predict Existing Immunity Evasion of SARS-CoV-2 Variants by a New Biotechnological Application of Phage Display. Viruses 2024, 16, 58. https://doi.org/10.3390/v16010058
Harhala MA, Gembara K, Baniecki K, Pikies A, Nahorecki A, Jędruchniewicz N, Kaźmierczak Z, Rybicka I, Klimek T, Witkiewicz W, et al. Experimental Identification of Cross-Reacting IgG Hotspots to Predict Existing Immunity Evasion of SARS-CoV-2 Variants by a New Biotechnological Application of Phage Display. Viruses. 2024; 16(1):58. https://doi.org/10.3390/v16010058
Chicago/Turabian StyleHarhala, Marek Adam, Katarzyna Gembara, Krzysztof Baniecki, Aleksandra Pikies, Artur Nahorecki, Natalia Jędruchniewicz, Zuzanna Kaźmierczak, Izabela Rybicka, Tomasz Klimek, Wojciech Witkiewicz, and et al. 2024. "Experimental Identification of Cross-Reacting IgG Hotspots to Predict Existing Immunity Evasion of SARS-CoV-2 Variants by a New Biotechnological Application of Phage Display" Viruses 16, no. 1: 58. https://doi.org/10.3390/v16010058
APA StyleHarhala, M. A., Gembara, K., Baniecki, K., Pikies, A., Nahorecki, A., Jędruchniewicz, N., Kaźmierczak, Z., Rybicka, I., Klimek, T., Witkiewicz, W., Barczyk, K., Kłak, M., & Dąbrowska, K. (2024). Experimental Identification of Cross-Reacting IgG Hotspots to Predict Existing Immunity Evasion of SARS-CoV-2 Variants by a New Biotechnological Application of Phage Display. Viruses, 16(1), 58. https://doi.org/10.3390/v16010058