
Limited Short-Term Evolution of SARS-CoV-2 RNA-Dependent RNA Polymerase under Remdesivir Exposure in Upper Respiratory Compartments

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants and Samples
2.2. Viral Load Testing
2.3. SARS-CoV-2 Genotyping
2.4. SARS-CoV-2 Lineages and Variants
2.5. Definitions of RdRp Mutations
2.6. Analyses of RdRp Mutations
2.7. Statistical Analysis
3. Results
3.1. Participants and Sampling
3.2. SARS-CoV-2 Genotyping
3.3. SARS-CoV-2 Lineages and Variants
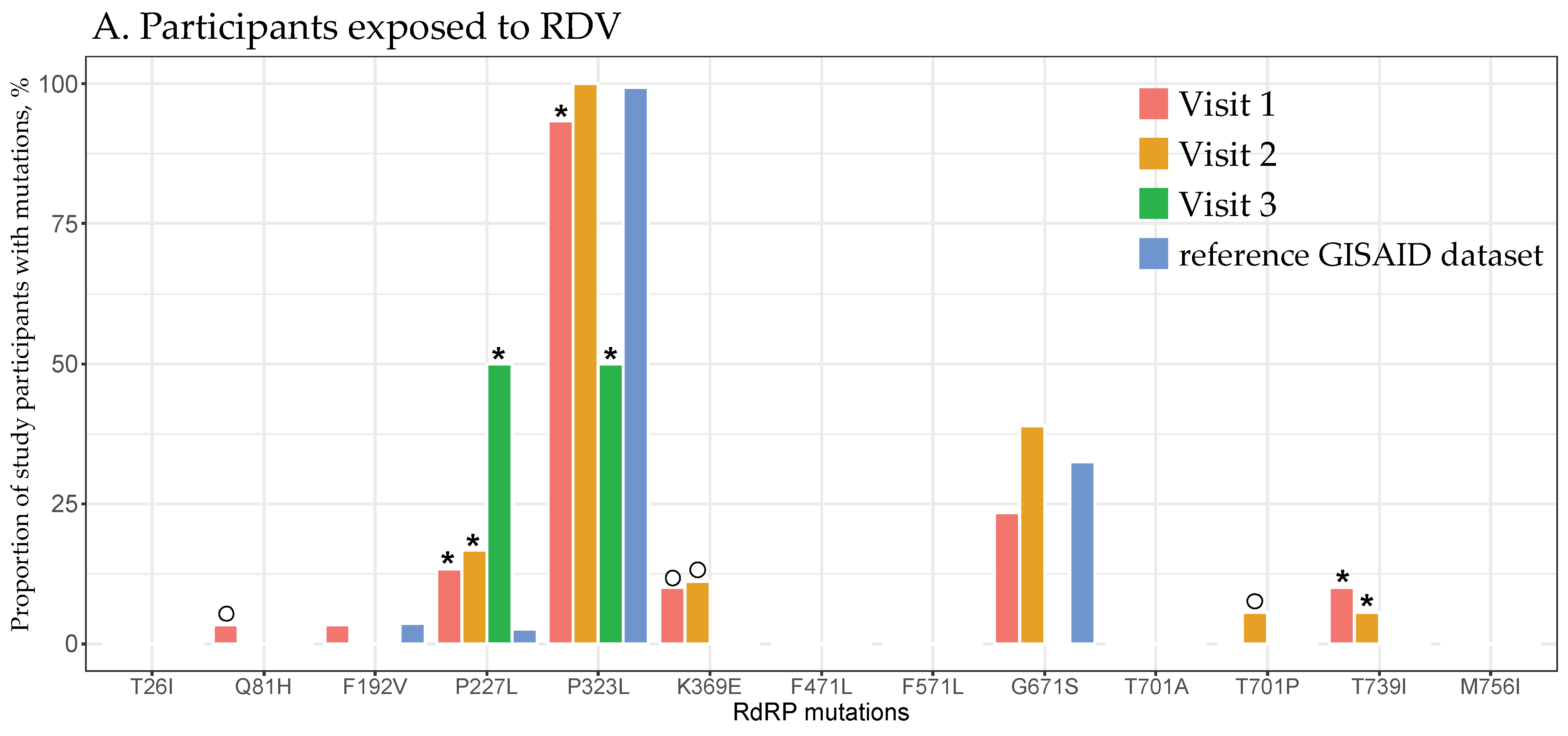
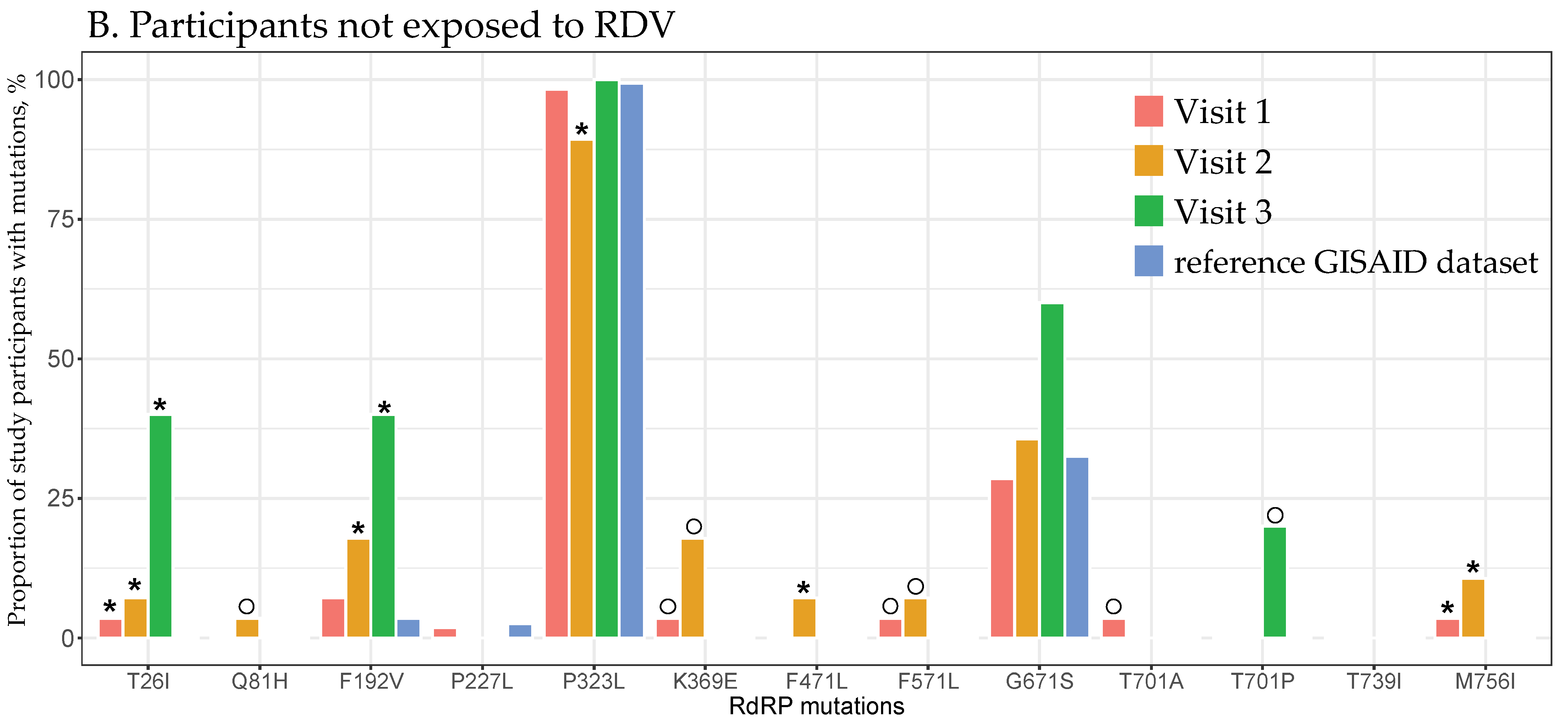
3.4. RdRp Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. WHO Coronovirus (COVID-19) Dashboard. United States of America Situation. Available online: https://covid19.who.int/region/amro/country/us (accessed on 14 June 2024).
- WHO. WHO Coronovirus (COVID-19) Dashboard. Global Situation. Available online: https://covid19.who.int/ (accessed on 14 June 2024).
- CDC CDC Public Health Science Agenda for COVID-19. Available online: https://www.cdc.gov/coronavirus/2019-ncov/science/science-agenda-covid19.html (accessed on 8 June 2024).
- Snijder, E.J.; Decroly, E.; Ziebuhr, J. The Nonstructural Proteins Directing Coronavirus RNA Synthesis and Processing. Adv. Virus Res. 2016, 96, 59–126. [Google Scholar] [PubMed]
- Shannon, A.; Le, N.T.; Selisko, B.; Eydoux, C.; Alvarez, K.; Guillemot, J.C.; Decroly, E.; Peersen, O.; Ferron, F.; Canard, B. Remdesivir and SARS-CoV-2: Structural requirements at both nsp12 RdRp and nsp14 Exonuclease active-sites. Antivir. Res. 2020, 178, 104793. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Subissi, L.; Imbert, I.; Ferron, F.; Collet, A.; Coutard, B.; Decroly, E.; Canard, B. SARS-CoV ORF1b-encoded nonstructural proteins 12-16: Replicative enzymes as antiviral targets. Antivir. Res. 2014, 101, 122–130. [Google Scholar] [CrossRef]
- Jordan, P.C.; Stevens, S.K.; Deval, J. Nucleosides for the treatment of respiratory RNA virus infections. Antivir. Chem. Chemother. 2018, 26, 2040206618764483. [Google Scholar] [CrossRef]
- Focosi, D.; Franchini, M.; Maggi, F.; Shoham, S. COVID-19 therapeutics. Clin. Microbiol. Rev. 2024, 37, e0011923. [Google Scholar] [CrossRef]
- Singh, A.K.; Singh, A.; Singh, R.; Misra, A. Remdesivir in COVID-19: A critical review of pharmacology, pre-clinical and clinical studies. Diabetes Metab. Syndr. 2020, 14, 641–648. [Google Scholar] [CrossRef]
- Lamb, Y.N. Remdesivir: First Approval. Drugs 2020, 80, 1355–1363. [Google Scholar] [CrossRef]
- COVID-19 Treatment Guidelines Panel Coronavirus Disease 2019 (COVID-19) Treatment Guidelines. National Institutes of Health. Available online: https://files.covid19treatmentguidelines.nih.gov/guidelines/covid19treatmentguidelines.pdf (accessed on 8 June 2024).
- NIH. COVID-19 Treatment Guidlines. Remdesivir. Available online: https://www.covid19treatmentguidelines.nih.gov/therapies/antivirals-including-antibody-products/remdesivir/ (accessed on 8 June 2024).
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.X.; et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020, 382, 2327–2336. [Google Scholar] [CrossRef] [PubMed]
- Spinner, C.D.; Gottlieb, R.L.; Criner, G.J.; Arribas Lopez, J.R.; Cattelan, A.M.; Soriano Viladomiu, A.; Ogbuagu, O.; Malhotra, P.; Mullane, K.M.; Castagna, A.; et al. Effect of Remdesivir vs. Standard Care on Clinical Status at 11 Days in Patients With Moderate COVID-19: A Randomized Clinical Trial. JAMA 2020, 324, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.D.; Lye, D.C.B.; Hui, D.S.; Marks, K.M.; Bruno, R.; Montejano, R.; Spinner, C.D.; Galli, M.; Ahn, M.Y.; Nahass, R.G.; et al. Remdesivir for 5 or 10 Days in Patients with Severe COVID-19. N. Engl. J. Med. 2020, 383, 1827–1837. [Google Scholar] [CrossRef] [PubMed]
- Olender, S.A.; Perez, K.K.; Go, A.S.; Balani, B.; Price-Haywood, E.G.; Shah, N.S.; Wang, S.; Walunas, T.L.; Swaminathan, S.; Slim, J.; et al. Remdesivir for Severe COVID-19 versus a Cohort Receiving Standard of Care. Clin. Infect. Dis. 2020, 73, e4166–e4174. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19-Preliminary Report. N. Engl. J. Med. 2020, 383, 1813–1836. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Frediansyah, A.; Nainu, F.; Dhama, K.; Mudatsir, M.; Harapan, H. Remdesivir and its antiviral activity against COVID-19: A systematic review. Clin. Epidemiol. Glob. Health 2020, 9, 123–127. [Google Scholar] [CrossRef]
- Li, Z.; Wang, X.; Cao, D.; Sun, R.; Li, C.; Li, G. Rapid review for the anti-coronavirus effect of remdesivir. Drug Discov. Ther. 2020, 14, 73–76. [Google Scholar] [CrossRef]
- Gottlieb, R.L.; Vaca, C.E.; Paredes, R.; Mera, J.; Webb, B.J.; Perez, G.; Oguchi, G.; Ryan, P.; Nielsen, B.U.; Brown, M.; et al. Early Remdesivir to Prevent Progression to Severe COVID-19 in Outpatients. N. Engl. J. Med. 2022, 386, 305–315. [Google Scholar] [CrossRef]
- Stevens, L.J.; Pruijssers, A.J.; Lee, H.W.; Gordon, C.J.; Tchesnokov, E.P.; Gribble, J.; George, A.S.; Hughes, T.M.; Lu, X.; Li, J.; et al. Mutations in the SARS-CoV-2 RNA-dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci. Transl. Med. 2022, 14, eabo0718. [Google Scholar] [CrossRef]
- Szemiel, A.M.; Merits, A.; Orton, R.J.; MacLean, O.A.; Pinto, R.M.; Wickenhagen, A.; Lieber, G.; Turnbull, M.L.; Wang, S.; Furnon, W.; et al. In vitro selection of Remdesivir resistance suggests evolutionary predictability of SARS-CoV-2. PLoS Pathog. 2021, 17, e1009929. [Google Scholar] [CrossRef] [PubMed]
- Checkmahomed, L.; Carbonneau, J.; Du Pont, V.; Riola, N.C.; Perry, J.K.; Li, J.; Pare, B.; Simpson, S.M.; Smith, M.A.; Porter, D.P.; et al. In Vitro Selection of Remdesivir-Resistant SARS-CoV-2 Demonstrates High Barrier to Resistance. Antimicrob. Agents Chemother. 2022, 66, e0019822. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease. mBio 2018, 9, e00221-18. [Google Scholar] [CrossRef] [PubMed]
- Focosi, D.; Maggi, F.; McConnell, S.; Casadevall, A. Very low levels of remdesivir resistance in SARS-COV-2 genomes after 18 months of massive usage during the COVID19 pandemic: A GISAID exploratory analysis. Antivir. Res. 2022, 198, 105247. [Google Scholar] [CrossRef] [PubMed]
- Mari, A.; Roloff, T.; Stange, M.; Sogaard, K.K.; Asllanaj, E.; Tauriello, G.; Alexander, L.T.; Schweitzer, M.; Leuzinger, K.; Gensch, A.; et al. Global Genomic Analysis of SARS-CoV-2 RNA Dependent RNA Polymerase Evolution and Antiviral Drug Resistance. Microorganisms 2021, 9, 1094. [Google Scholar] [CrossRef]
- Martinot, M.; Jary, A.; Fafi-Kremer, S.; Leducq, V.; Delagreverie, H.; Garnier, M.; Pacanowski, J.; Mekinian, A.; Pirenne, F.; Tiberghien, P.; et al. Emerging RNA-Dependent RNA Polymerase Mutation in a Remdesivir-Treated B-cell Immunodeficient Patient With Protracted Coronavirus Disease 2019. Clin. Infect. Dis. 2021, 73, e1762–e1765. [Google Scholar] [CrossRef]
- Gandhi, S.; Klein, J.; Robertson, A.J.; Pena-Hernandez, M.A.; Lin, M.J.; Roychoudhury, P.; Lu, P.; Fournier, J.; Ferguson, D.; Mohamed Bakhash, S.A.K.; et al. De novo emergence of a remdesivir resistance mutation during treatment of persistent SARS-CoV-2 infection in an immunocompromised patient: A case report. Nat. Commun. 2022, 13, 1547. [Google Scholar] [CrossRef]
- Yang, S.; Multani, A.; Garrigues, J.M.; Oh, M.S.; Hemarajata, P.; Burleson, T.; Green, N.M.; Oliai, C.; Gaynor, P.T.; Beaird, O.E.; et al. Transient SARS-CoV-2 RNA-Dependent RNA Polymerase Mutations after Remdesivir Treatment for Chronic COVID-19 in Two Transplant Recipients: Case Report and Intra-Host Viral Genomic Investigation. Microorganisms 2023, 11, 2096. [Google Scholar] [CrossRef]
- Hogan, J.I.; Duerr, R.; Dimartino, D.; Marier, C.; Hochman, S.E.; Mehta, S.; Wang, G.; Heguy, A. Remdesivir Resistance in Transplant Recipients With Persistent Coronavirus Disease 2019. Clin. Infect. Dis. 2023, 76, 342–345. [Google Scholar] [CrossRef]
- Ahmadi, A.S.; Zadheidar, S.; Sadeghi, K.; Nejati, A.; Salimi, V.; Hajiabdolbaghi, M.; Mokhtari-Azad, T.; Yavarian, J. SARS-CoV-2 intrahost evolution in immunocompromised patients in comparison with immunocompetent populations after treatment. J. Med. Virol. 2023, 95, e28877. [Google Scholar] [CrossRef]
- Heyer, A.; Gunther, T.; Robitaille, A.; Lutgehetmann, M.; Addo, M.M.; Jarczak, D.; Kluge, S.; Aepfelbacher, M.; Schulze Zur Wiesch, J.; Fischer, N.; et al. Remdesivir-induced emergence of SARS-CoV2 variants in patients with prolonged infection. Cell Rep. Med. 2022, 3, 100735. [Google Scholar] [CrossRef] [PubMed]
- Nirmalarajah, K.; Yim, W.; Aftanas, P.; Li, A.X.; Shigayeva, A.; Yip, L.; Zhong, Z.; McGeer, A.J.; Maguire, F.; Mubareka, S.; et al. Use of whole genome sequencing to identify low-frequency mutations in SARS-CoV-2 patients treated with remdesivir. Influenza Other Respir. Viruses 2023, 17, e13179. [Google Scholar] [CrossRef] [PubMed]
- Hedskog, C.; Rodriguez, L.; Roychoudhury, P.; Huang, M.L.; Jerome, K.R.; Hao, L.; Ireton, R.C.; Li, J.; Perry, J.K.; Han, D.; et al. Viral Resistance Analyses From the Remdesivir Phase 3 Adaptive COVID-19 Treatment Trial-1 (ACTT-1). J. Infect. Dis. 2023, 228, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- DNA Pipelines R&D; Farr, B.; Rajan, D.; Dawson, E.; Shirley, L.; Quail, M.; Park, N.; Redshaw, N.; Bronner, I.F.; Aigrain, L.; et al. COVID-19 ARTIC v3 Illumina Library Construction and Sequencing Protocol V.5. Available online: https://www.protocols.io/view/covid-19-artic-v3-illumina-library-construction-an-j8nlke665l5r/v5. (accessed on 5 February 2021).
- Centre for Genomic Pathogen Surveillance Pangolin: Phylogenetic Assignment of Named Global Outbreak LINeages. Available online: https://github.com/cov-lineages/pangolin (accessed on 6 June 2023).
- Nextstrain Project Nextclade: Clade Assignment, Mutation Calling, and Sequence Quality Checks. Available online: https://clades.nextstrain.org/ (accessed on 6 June 2023).
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- A Stanford HIVDB Team Website Stanford University Coronovirus Aniviral & Resistance Database. Available online: https://covdb.stanford.edu/drms/rdrp/ (accessed on 6 June 2023).
- Hirotsu, Y.; Kobayashi, H.; Kakizaki, Y.; Saito, A.; Tsutsui, T.; Kawaguchi, M.; Shimamura, S.; Hata, K.; Hanawa, S.; Toyama, J.; et al. Multidrug-resistant mutations to antiviral and antibody therapy in an immunocompromised patient infected with SARS-CoV-2. Med 2023, 4, 813–824.e4. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- GISAID. Global Initiative on Sharing All Influenza Data (GISAID) Repository. Available online: https://www.epicov.org (accessed on 24 May 2024).
- Carstensen, B.; Plummer, M.; Laara, E.; Hills, M. Epi: Statistical Analysis in Epidemiology. Available online: https://cran.r-project.org/web/packages/Epi/index.html (accessed on 10 June 2024).
- R Core Team. The R Project for Statistical Computing. Available online: http://www.R-project.org/ (accessed on 19 May 2023).
- Troseid, M.; Hentzien, M.; Ader, F.; Cardoso, S.W.; Arribas, J.R.; Molina, J.M.; Mueller, N.; Hites, M.; Bonnet, F.; Manuel, O.; et al. Combine, Immunocompromised patients have been neglected in COVID-19 trials: A call for action. Clin. Microbiol. Infect. 2022, 28, 1182–1183. [Google Scholar] [CrossRef]
- Majchrzak, M.; Madej, L.; Lysek-Gladysinska, M.; Zarebska-Michaluk, D.; Zegadlo, K.; Dziuba, A.; Nogal-Nowak, K.; Kondziolka, W.; Sufin, I.; Myszona-Tarnowska, M.; et al. The RdRp genotyping of SARS-CoV-2 isolated from patients with different clinical spectrum of COVID-19. BMC Infect. Dis. 2024, 24, 281. [Google Scholar] [CrossRef]
- Maurya, R.; Mishra, P.; Swaminathan, A.; Ravi, V.; Saifi, S.; Kanakan, A.; Mehta, P.; Devi, P.; Praveen, S.; Budhiraja, S.; et al. SARS-CoV-2 Mutations and COVID-19 Clinical Outcome: Mutation Global Frequency Dynamics and Structural Modulation Hold the Key. Front. Cell Infect. Microbiol. 2022, 12, 868414. [Google Scholar] [CrossRef]
- Saifi, S.; Ravi, V.; Sharma, S.; Swaminathan, A.; Chauhan, N.S.; Pandey, R. SARS-CoV-2 VOCs, Mutational diversity and clinical outcome: Are they modulating drug efficacy by altered binding strength? Genomics 2022, 114, 110466. [Google Scholar] [CrossRef]
- Sala, E.; Shah, I.S.; Manissero, D.; Juanola-Falgarona, M.; Quirke, A.M.; Rao, S.N. Systematic Review on the Correlation Between SARS-CoV-2 Real-Time PCR Cycle Threshold Values and Epidemiological Trends. Infect. Dis. Ther. 2023, 12, 749–775. [Google Scholar] [CrossRef] [PubMed]
- Hagman, K.; Hedenstierna, M.; Widaeus, J.; Arvidsson, E.; Hammas, B.; Grillner, L.; Jakobsson, J.; Gille-Johnson, P.; Ursing, J. Correlation of SARS-CoV-2 Nasopharyngeal CT Values With Viremia and Mortality in Adults Hospitalized With COVID-19. Open Forum Infect. Dis. 2022, 9, ofac463. [Google Scholar] [CrossRef] [PubMed]
- Juanola-Falgarona, M.; Penarrubia, L.; Jimenez-Guzman, S.; Porco, R.; Congost-Teixidor, C.; Varo-Velazquez, M.; Rao, S.N.; Pueyo, G.; Manissero, D.; Pareja, J. Ct values as a diagnostic tool for monitoring SARS-CoV-2 viral load using the QIAstat-Dx(R) Respiratory SARS-CoV-2 Panel. Int. J. Infect. Dis. 2022, 122, 930–935. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | All Study Participants (n = 44) | Participants Exposed to RDV (n = 18) | Participants Not Exposed to RDV (n = 26) | |||||
|---|---|---|---|---|---|---|---|---|
| Total, with and without Sequences | Genotyped | Not Genotyped | Genotyped | Not Genotyped | Genotyped | Not Genotyped | ||
| Gender | Males, n (%) | 26 (59%) | 23 (62%) | 3 (43%) | 13 (87%) | 2 (67%) | 10 (45%) | 1 (25%) |
| Females, n (%) | 18 (41%) | 14 (38%) | 4 (57%) | 2 (13%) | 1 (33%) | 12 (55%) | 3 (75%) | |
| Median (IQR) age, years | 59 (52–73) | 67 (53–73) | 54 (45–57) | 67 (59–77) | 54 (50–54) | 57 (49–73) | 52 (39–63) | |
| Race | White or Caucasian, n (%) | 33 (75%) | 29 (78%) | 4 (57%) | 15 (100%) | 2 (67%) | 14 (64%) | 2 (50%) |
| Black or African Americans, n (%) | 6 (14%) | 5 (14%) | 1 (14%) | 0 (0%) | 0 (0%) | 5 (23%) | 1 (25%) | |
| Other, n (%) | 5 (11%) | 3 (8%) | 2 (29%) | 0 (0%) | 1 (33%) | 3 (14%) | 1 (25%) | |
| Ethnicity | Not Hispanic or Latino, n (%) | 38 (86%) | 33 (89%) | 5 (71%) | 15 (100%) | 2 (67%) | 18 (82%) | 3 (75%) |
| Hispanic or Latino, n (%) | 6 (14%) | 4 (11%) | 2 (29%) | 0 (0%) | 1 (33%) | 4 (18%) | 1 (25%) | |
| Prior vaccine | 20 (45%) | 19 (51%) | 1 (14%) | 6 (40%) | 0 (0%) | 13 (59%) | 1 (25%) | |
| Median (IQR) days since disease onset | 6 (4–9) | 6 (4–9) | 6 (6–15) | 9 (5–11) | 15 (11–15) | 6 (4–8) | 6 (5–10) | |
| Median (IQR) number of compartments | 3 (2–4) | 4 (3–4) | 3 (2–3) | 3 (2–4) | 3 (3–3) | 4 (3–4) | 3 (2–3) | |
| Median (IQR) number of visits | 2 (1–2) | 2 (1–2) | 2 (1.5–3) | 2 (2–3) | 2 (2–2.5) | 2 (1–2) | 2 (1–3) | |
| Median (IQR) viral load at visit 1, log10 copies/mL | 6.14 (5.12–6.76) | 6.26 (5.55–6.76) | 4.46 (4.32–4.72) | 5.94 (5.12–6.45) | 4.32 (4.32–4.39) | 6.55 (5.98–7.15) | 4.72 (4.32–5.38) | |
| Variable | Compartment | |||
|---|---|---|---|---|
| Nasal Swab | Nasopharyngeal Swab | Oropharyngeal Swab | Saliva | |
| Total number of collected samples per compartment | 71 | 42 | 50 | 79 |
| Genotyping sequences *, n (%) | 40 (56%) | 28 (67%) | 21 (42%) | 50 (63%) |
| Study visit 1 total sequences, n | 25 | 18 | 13 | 30 |
| Study visit 2 total sequences, n | 13 | 9 | 8 | 16 |
| Study visit 3 total sequences, n | 2 | 1 | 0 | 4 |
| Combined visits: | ||||
| Single visit only, n sequences | 12 | 12 | 13 | 17 |
| Study visit 1 | 12 | 11 | 9 | 15 |
| Study visit 2 | 0 | 1 | 4 | 1 |
| Study visit 3 | 0 | 0 | 0 | 1 |
| Two-visit sequences (participants) | 22 (11) | 16 (8) | 8 (4) | 24 (12) |
| Study visits 1 + 2 | 22 | 14 | 8 | 24 |
| Study visits 1 + 3 | 0 | 0 | 0 | 0 |
| Study visits 2 + 3 | 0 | 2 | 0 | 0 |
| Three-visit sequences (participants) | 6 (2) | 0 | 0 | 9 (3) |
| Spike Variant | PANGO Lineage | Study Participants, n | Proportion, % |
|---|---|---|---|
| Alpha | B.1.1.7 | 4 | 10.8 |
| Delta | AY.103 | 2 | 5.4 |
| Delta | AY.119 | 1 | 2.7 |
| Delta | AY.122 | 1 | 2.7 |
| Delta | AY.25 | 1 | 2.7 |
| Delta | AY.3 | 3 | 8.1 |
| Delta | AY.44 | 3 | 8.1 |
| Delta | AY.54 | 1 | 2.7 |
| Epsilon | B.1.427 | 2 | 5.4 |
| Epsilon | B.1.429 | 1 | 2.7 |
| Iota | B.1.526 | 4 | 10.8 |
| Omicron | BA.1.1 | 3 | 8.1 |
| Omicron | BA.1.18 | 1 | 2.7 |
| Omicron | BA.2 | 2 | 5.4 |
| Omicron | BA.2.12.1 | 4 | 10.8 |
| Wild type | B.1.2 | 1 | 2.7 |
| Wild type | B.1.517 | 1 | 2.7 |
| Wild type | B.1.637 | 2 | 5.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novitsky, V.; Beckwith, C.G.; Carpenter-Azevedo, K.; Shin, J.; Hague, J.; Sam, S.; Steingrimsson, J.; Huard, R.C.; Lethbridge, K.; Sahu, S.; et al. Limited Short-Term Evolution of SARS-CoV-2 RNA-Dependent RNA Polymerase under Remdesivir Exposure in Upper Respiratory Compartments. Viruses 2024, 16, 1511. https://doi.org/10.3390/v16101511
Novitsky V, Beckwith CG, Carpenter-Azevedo K, Shin J, Hague J, Sam S, Steingrimsson J, Huard RC, Lethbridge K, Sahu S, et al. Limited Short-Term Evolution of SARS-CoV-2 RNA-Dependent RNA Polymerase under Remdesivir Exposure in Upper Respiratory Compartments. Viruses. 2024; 16(10):1511. https://doi.org/10.3390/v16101511
Chicago/Turabian StyleNovitsky, Vladimir, Curt G. Beckwith, Kristin Carpenter-Azevedo, Jimin Shin, Joel Hague, Soya Sam, Jon Steingrimsson, Richard C. Huard, Kevin Lethbridge, Sujata Sahu, and et al. 2024. "Limited Short-Term Evolution of SARS-CoV-2 RNA-Dependent RNA Polymerase under Remdesivir Exposure in Upper Respiratory Compartments" Viruses 16, no. 10: 1511. https://doi.org/10.3390/v16101511
APA StyleNovitsky, V., Beckwith, C. G., Carpenter-Azevedo, K., Shin, J., Hague, J., Sam, S., Steingrimsson, J., Huard, R. C., Lethbridge, K., Sahu, S., Rapoza, K., Chandran, K., Bazerman, L., Hipolito, E., Diaz, I., Carnevale, D., Guang, A., Gillani, F., Caliendo, A. M., & Kantor, R. (2024). Limited Short-Term Evolution of SARS-CoV-2 RNA-Dependent RNA Polymerase under Remdesivir Exposure in Upper Respiratory Compartments. Viruses, 16(10), 1511. https://doi.org/10.3390/v16101511