


First Detection and Characterization of Smacovirus in the Human Vagina in Two Sequential Samples over a Twelve-Day Interval

 ,
,  , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Population and Study Design
2.2. Sample Collection and Data
2.3. Extraction, Enrichment of Viral DNA, and Library Preparation
2.4. Database and Viral Sequence Quality
2.5. Alignment and Annotation
2.6. Phylogenetic Reconstruction
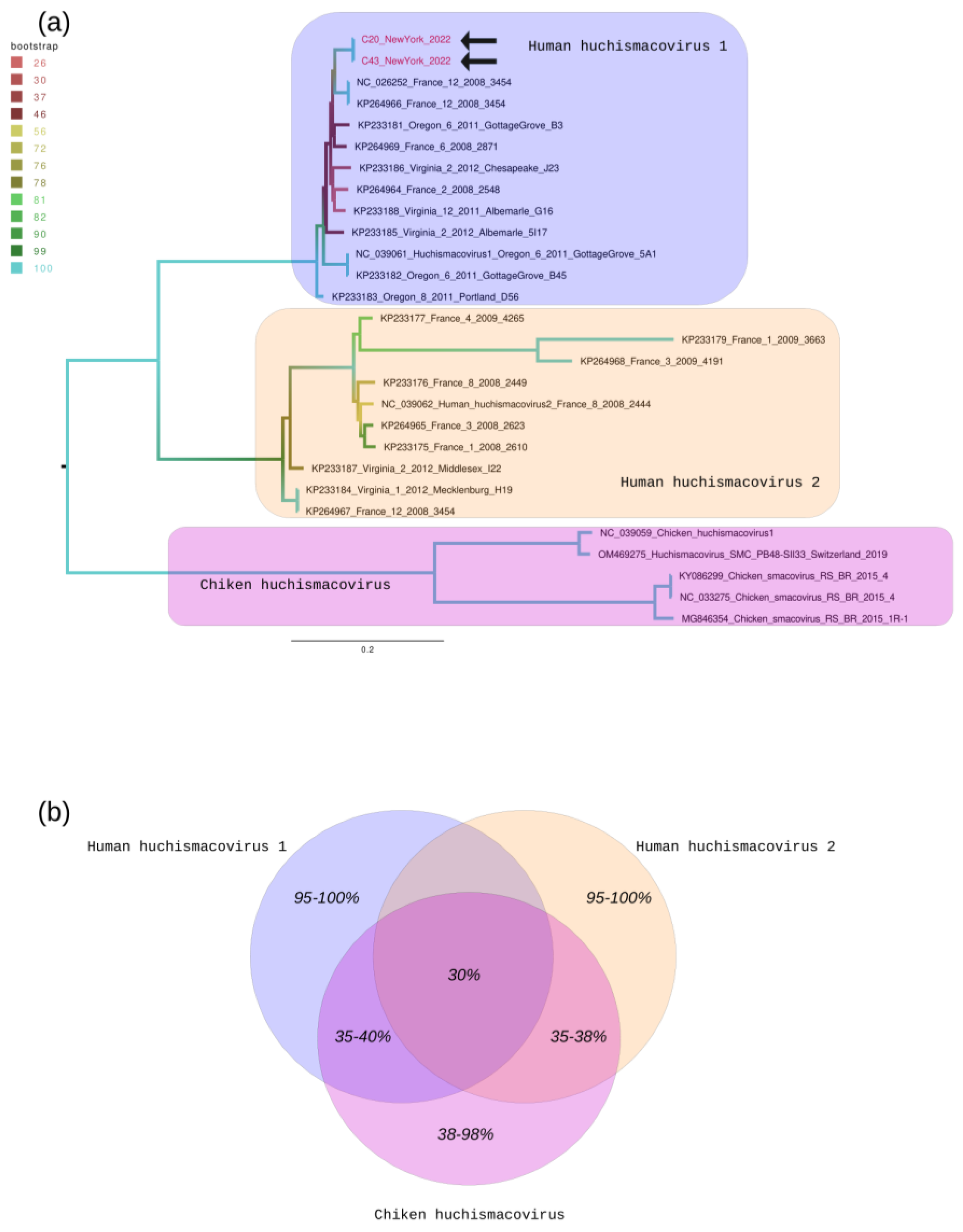
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Varsani, A.; Krupovic, M. Smacoviridae: A new family of animal-associated single-stranded DNA viruses. Arch. Virol. 2018, 163, 2005–2015. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.F.F.; Zhang, W.; Sachsenröder, J.; Kondov, N.O.; da Costa, A.C.; Vega, E.; Holtz, L.R.; Wu, G.; Wang, D.; Stine, C.O.; et al. A diverse group of small circular ssDNA viral genomes in human and non-human primate stools. Virus Evol. 2015, 1, vev017. [Google Scholar] [CrossRef] [PubMed]
- Chibani, C.M.; Mahnert, A.; Borrel, G.; Almeida, A.; Werner, A.; Brugère, J.-F.; Gribaldo, S.; Finn, R.D.; Schmitz, R.A.; Moissl-Eichinger, C. A catalogue of 1167 genomes from the human gut archaeome. Nat. Microbiol. 2021, 7, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Zablocki, O.; Zayed, A.A.; Howell, A.; Bolduc, B.; Sullivan, M.B. The Gut Virome Database Reveals Age-Dependent Patterns of Virome Diversity in the Human Gut. Cell Host Microbe 2020, 28, 724–740.e8. [Google Scholar] [CrossRef] [PubMed]
- Díez-Villaseñor, C.; Rodriguez-Valera, F. CRISPR analysis suggests that small circular single-stranded DNA smacoviruses infect Archaea instead of humans. Nat. Commun. 2019, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.d.S.F.; Tozetto-Mendoza, T.R.; Bortoletto, P.; Ferreira, N.E.; Honorato, L.; Barbosa, E.M.G.; Luchs, A.; Linhares, I.M.; Spandorfer, S.D.; Leal, E.; et al. Characterization of CRESS-DNA viruses in human vaginal secretions: An exploratory metagenomic investigation. J. Med. Virol. 2024, 96, e29750. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, A.C.; Bortoletto, P.; Spandorfer, S.D.; Tozetto-Mendoza, T.R.; Linhares, I.M.; Mendes-Correa, M.C.; Witkin, S.S. Association between torquetenovirus in vaginal secretions and infertility: An exploratory metagenomic analysis. Am. J. Reprod. Immunol. 2023, 90, e13788. [Google Scholar] [CrossRef] [PubMed]
- da Costa, A.C.; Moron, A.F.; Forney, L.J.; Linhares, I.M.; Sabino, E.; Costa, S.F.; Mendes-Correa, M.C.; Witkin, S.S. Identification of bacteriophages in the vagina of pregnant women: A descriptive study. BJOG Int. J. Obstet. Gynaecol. 2021, 128, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.d.S.F.; Rosa, U.A.; Ribeiro, G.d.O.; Villanova, F.; Milagres, F.A.d.P.; Brustulin, R.; Morais, V.d.S.; Araújo, E.L.L.; Pandey, R.P.; Raj, V.S.; et al. Multiple clades of Husavirus in South America revealed by next generation sequencing. PLoS ONE 2021, 16, e0248486. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Naccache, S.N.; Ng, T.; Federman, S.; Li, L.; Chiu, C.Y.; Delwart, E.L. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res. 2015, 43, e46. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; McGinnis, S.; Madden, T.L. BLAST: Improvements for better sequence analysis. Nucleic Acids Res. 2006, 34, W6–W9. [Google Scholar] [CrossRef] [PubMed]
- Altan, E.; Delaney, M.A.; Colegrove, K.M.; Spraker, T.R.; Wheeler, E.A.; Deng, X.; Li, Y.; Gulland, F.M.D.; Delwart, E. Complex Virome in a Mesenteric Lymph Node from a Californian Sea Lion (Zalophus californianus) with Polyserositis and Steatitis. Viruses 2020, 12, 793. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed]
- El Aila, N.A.; Tency, I.; Claeys, G.; Verstraelen, H.; Saerens, B.; Santiago, G.L.d.S.; De Backer, E.; Cools, P.; Temmerman, M.; Verhelst, R.; et al. Identification and genotyping of bacteria from paired vaginal and rectal samples from pregnant women indicates similarity between vaginal and rectal microflora. BMC Infect. Dis. 2009, 9, 167. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Sample 1 | Sample 2 | |
|---|---|---|
| Date of collection | 15 February 2022 | 26 February 2022 |
| Length of fragment (bp) | 2466 | 2466 |
| Coverage | 2350× | 1560× |
| Number of reads | 23,180 | 15,289 |
| a Genbank prototype | KP264966 | KP264966 |
| Country | France | France |
| Percent identity | 96% | 96% |
| Year identified | 2008 | 2008 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Costa, A.C.; Tozetto-Mendoza, T.R.; Foro Ramos, E.d.S.; Bortoletto, P.; Ferreira, N.E.; Honorato, L.; Garcia Barbosa, E.M.; Paião, H.G.O.; de Souza, A.F.; Linhares, I.M.; et al. First Detection and Characterization of Smacovirus in the Human Vagina in Two Sequential Samples over a Twelve-Day Interval. Viruses 2024, 16, 1545. https://doi.org/10.3390/v16101545
da Costa AC, Tozetto-Mendoza TR, Foro Ramos EdS, Bortoletto P, Ferreira NE, Honorato L, Garcia Barbosa EM, Paião HGO, de Souza AF, Linhares IM, et al. First Detection and Characterization of Smacovirus in the Human Vagina in Two Sequential Samples over a Twelve-Day Interval. Viruses. 2024; 16(10):1545. https://doi.org/10.3390/v16101545
Chicago/Turabian Styleda Costa, Antonio Charlys, Tania Regina Tozetto-Mendoza, Endrya do Socorro Foro Ramos, Pietro Bortoletto, Noely Evangelista Ferreira, Layla Honorato, Erick Matheus Garcia Barbosa, Heuder Gustavo Oliveira Paião, Amanda Fernandes de Souza, Iara M. Linhares, and et al. 2024. "First Detection and Characterization of Smacovirus in the Human Vagina in Two Sequential Samples over a Twelve-Day Interval" Viruses 16, no. 10: 1545. https://doi.org/10.3390/v16101545