


Unveiling the Antiviral Potential of Minocycline: Modulation of Nuclear Export of Viral Ribonuclear Proteins during Influenza Virus Infection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Virus Infection
2.2. IAV Infection In Vivo and Ethics Statement
2.3. Reagents and Antibody
2.4. Quantitative Real-Time Reverse Transcription-PCR (qRT-PCR)
2.5. Western Blot Analysis
2.6. Immunofluorescence Microscopy
2.7. Plasmid Transfection
2.8. Nucleus–Cytosol Extraction
2.9. Cell Viability Assay
2.10. Hemagglutination (HA) Assay
2.11. Apoptosis Assay
2.12. Plaque Assay
2.13. Statistical Analysis
3. Results
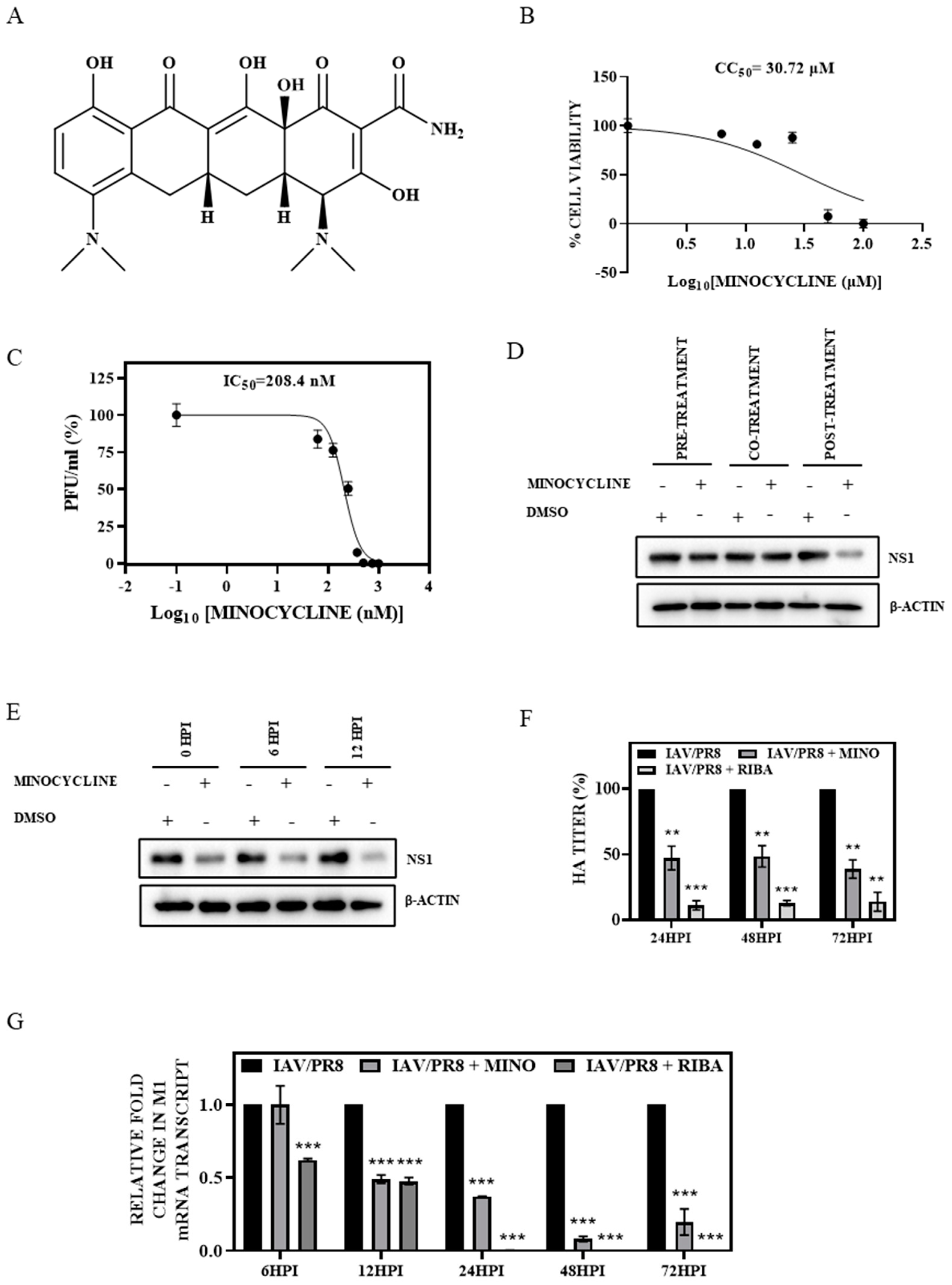
3.1. Minocycline Treatment Showed Potent Anti-IAV Activity In Vitro
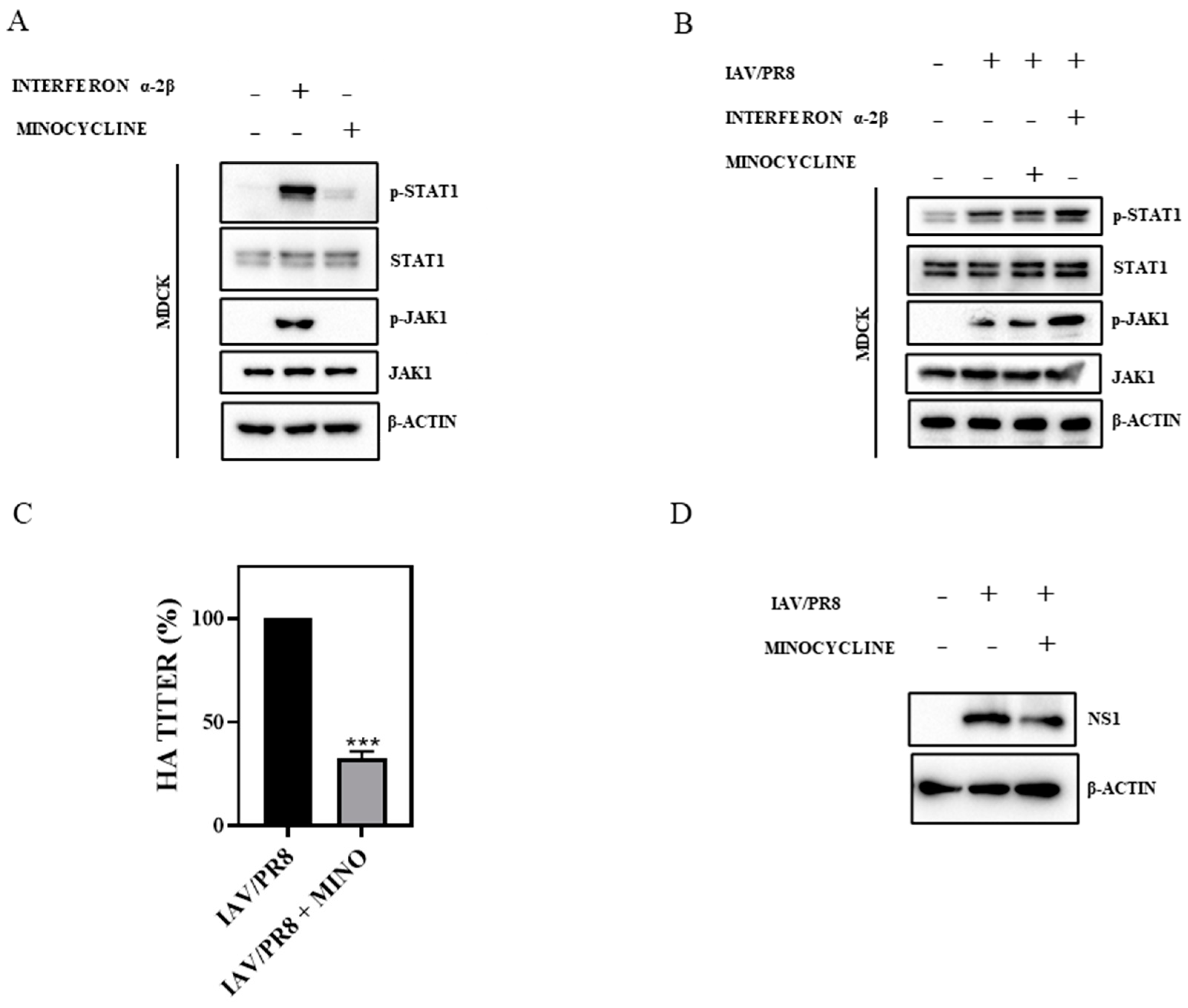
3.2. Minocycline Impedes IAV Infection without Triggering Interferon (IFN) Signaling
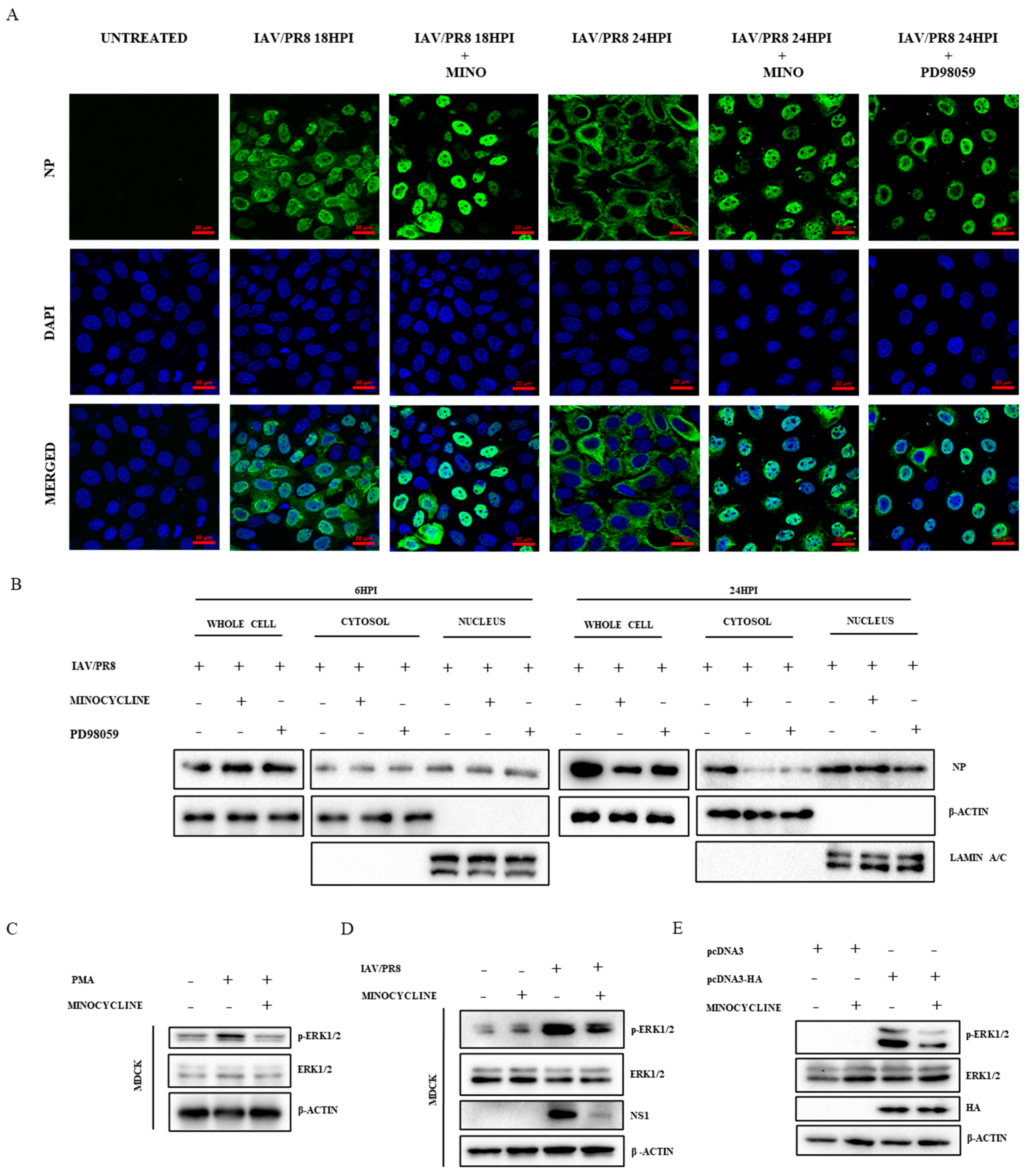
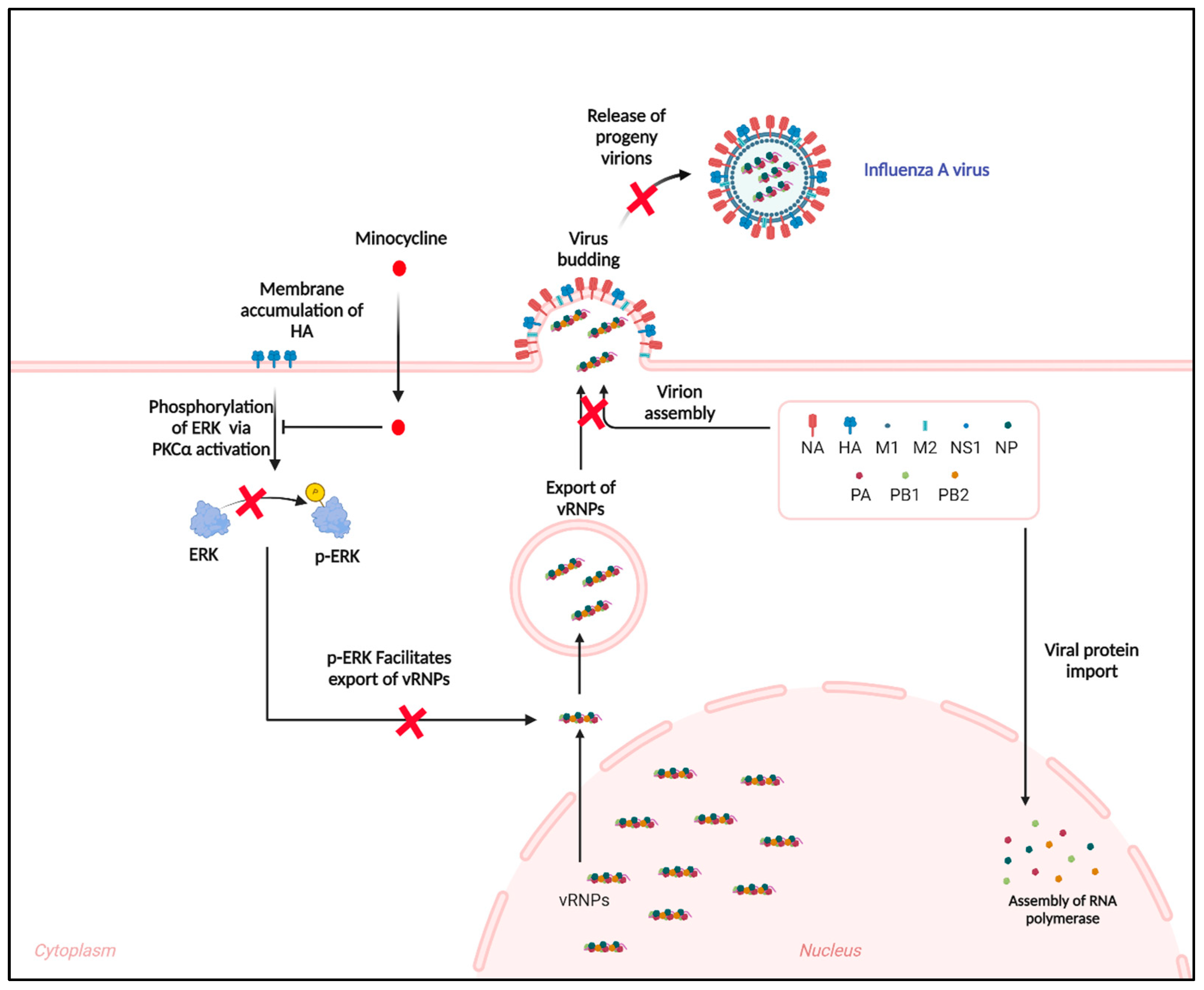
3.3. Minocycline Prevents the Shuttling of vRNPs from the Nucleus to Cytosol by Inhibiting the ERK Pathway
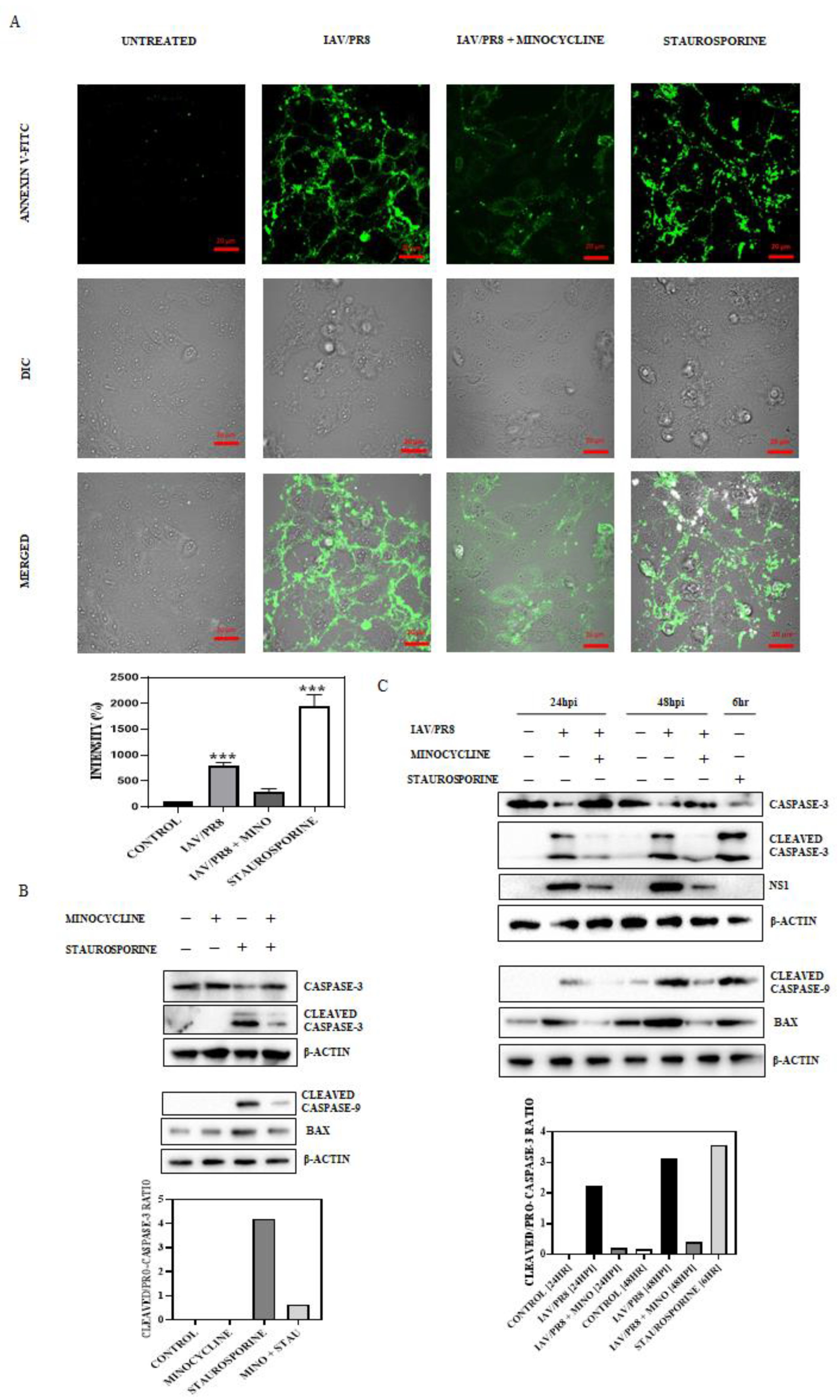
3.4. Minocycline Impedes IAV-Induced Late-Stage Apoptosis
3.5. Minocycline Treatment Attenuates IAV Infection In Vivo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trucchi, C.; Paganino, C.; Orsi, A.; Amicizia, D.; Tisa, V.; Piazza, M.F.; Gallo, D.; Simonetti, S.; Buonopane, B.; Icardi, G. Hospital and economic burden of influenza-like illness and lower respiratory tract infection in adults≥ 50 years-old. BMC Health Serv. Res. 2019, 19, 585. [Google Scholar] [CrossRef] [PubMed]
- Gatherer, D. On the origin of influenza A hemagglutinin. Indian J. Microbiol. 2009, 49, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Cros, J.F.; Palese, P. Trafficking of viral genomic RNA into and out of the nucleus: Influenza, Thogoto and Borna disease viruses. Virus Res. 2003, 95, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.P.; Hui, E.K.-W.; Barman, S. Assembly and budding of influenza virus. Virus Res. 2004, 106, 147–165. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Krug, R.M. Orthomyxoviridae: The viruses and their replication. Fields Virol. 1995, 4, 725–769. [Google Scholar]
- Agrawal, A.S.; Sarkar, M.; Chakrabarti, S.; Rajendran, K.; Kaur, H.; Mishra, A.C.; Chatterjee, M.K.; Naik, T.N.; Chadha, M.S.; Chawla-Sarkar, M. Comparative evaluation of real-time PCR and conventional RT-PCR during a 2 year surveillance for influenza and respiratory syncytial virus among children with acute respiratory infections in Kolkata, India, reveals a distinct seasonality of infection. J. Med. Microbiol. 2009, 58, 1616–1622. [Google Scholar] [CrossRef]
- Agrawal, A.S.; Sarkar, M.; Ghosh, S.; Roy, T.; Chakrabarti, S.; Lal, R.; Mishra, A.C.; Chadha, M.S.; Chawla-Sarkar, M. Genetic characterization of circulating seasonal Influenza A viruses (2005–2009) revealed introduction of oseltamivir resistant H1N1 strains during 2009 in eastern India. Infect. Genet. Evol. 2010, 10, 1188–1198. [Google Scholar] [CrossRef]
- Mukherjee, A.; Roy, T.; Agrawal, A.S.; Sarkar, M.; Lal, R.; Chakrabarti, S.; Chawla-Sarkar, M. Prevalence and epidemiology of pandemic H1N1 strains in hospitals of Eastern India. J. Public Health Epidemiol. 2010, 2, 171–174. [Google Scholar]
- Mukherjee, T.R.; Agrawal, A.S.; Chakrabarti, S.; Chawla-Sarkar, M. Full genomic analysis of an influenza A (H1N2) virus identified during 2009 pandemic in Eastern India: Evidence of reassortment event between co-circulating A (H1N1) pdm09 and A/Brisbane/10/2007-like H3N2 strains. Virol. J. 2012, 9, 233. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Nayak, M.K.; Dutta, S.; Panda, S.; Satpathi, B.R.; Chawla-Sarkar, M. Genetic characterization of circulating 2015 A (H1N1) pdm09 influenza viruses from Eastern India. PLoS ONE 2016, 11, e0168464. [Google Scholar] [CrossRef]
- Mudhigeti, N.; Racherla, R.G.; Mahalakshmi, P.A.; Pamireddy, M.L.; Nallapireddy, U.; Kante, M.; Kalawat, U. A study of influenza 2017–2018 outbreak in Andhra Pradesh, India. Indian J. Med. Microbiol. 2018, 36, 526–531. [Google Scholar] [CrossRef]
- Costantino, C.; Restivo, V.; Amodio, E.; Colomba, G.M.E.; Vitale, F.; Tramuto, F. A mid-term estimate of 2018/2019 vaccine effectiveness to prevent laboratory confirmed A (H1N1) pdm09 and A (H3N2) influenza cases in Sicily (Italy). Vaccine 2019, 37, 5812–5816. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Biswas, M.; Gupta, R.; Majumdar, A.; Mitra, S.; Banerjee, A.; Mukherjee, A.; Dutta, S.; Chawla-Sarkar, M. Molecular characterization of Influenza A pandemic H1N1 viruses circulating in eastern India during 2017–19: Antigenic diversity in comparison to the vaccine strains. Infect. Genet. Evol. 2020, 81, 104270. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Lou, K.; Wang, W. New small-molecule drug design strategies for fighting resistant influenza A. Acta Pharm. Sin. B 2015, 5, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.-L. Current and novel antiviral strategies for influenza infection. Curr. Opin. Virol. 2016, 18, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Peng, C.; Luo, J.; Wang, C.; Han, L.; Wu, B.; Ji, G.; He, H. Adamantane-resistant influenza a viruses in the world (1902–2013): Frequency and distribution of M2 gene mutations. PLoS ONE 2015, 10, e0119115. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, S.; Drozda, M.; Zhang, W.; Stavrovskaya, I.G.; Cattaneo, E.; Ferrante, R.J.; Kristal, B.S.; Friedlander, R.M. Minocycline inhibits caspase-independent and-dependent mitochondrial cell death pathways in models of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 10483–10487. [Google Scholar] [CrossRef] [PubMed]
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [PubMed]
- Garner, S.E.; Eady, A.; Bennett, C.; Newton, J.N.; Thomas, K.; Popescu, C.M. Minocycline for acne vulgaris: Efficacy and safety. Cochrane Database Syst. Rev. 2012, 8, 10. [Google Scholar] [CrossRef]
- Popovic, N.; Schubart, A.; Goetz, B.D.; Zhang, S.C.; Linington, C.; Duncan, I.D. Inhibition of autoimmune encephalomyelitis by a tetracycline. Ann. Neurol. 2002, 51, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, F.; Hader, W.; Yong, V.W. Minocycline attenuates T cell and microglia activity to impair cytokine production in T cell-microglia interaction. J. Leukoc. Biol. 2005, 78, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wei, E.-Q.; Zhang, W.-P.; Zhang, L.; Liu, J.-R.; Chen, Z. Minocycline protects PC12 cells from ischemic-like injury and inhibits 5-lipoxygenase activation. Neuroreport 2004, 15, 2181–2184. [Google Scholar] [CrossRef] [PubMed]
- Darman, J.; Backovic, S.; Dike, S.; Maragakis, N.J.; Krishnan, C.; Rothstein, J.D.; Irani, D.N.; Kerr, D.A. Viral-induced spinal motor neuron death is non-cell-autonomous and involves glutamate excitotoxicity. J. Neurosci. 2004, 24, 7566–7575. [Google Scholar] [CrossRef] [PubMed]
- Richardson-Burns, S.M.; Tyler, K.L. Minocycline delays disease onset and mortality in reovirus encephalitis. Exp. Neurol. 2005, 192, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Irani, D.N.; Prow, N.A. Neuroprotective interventions targeting detrimental host immune responses protect mice from fatal alphavirus encephalitis. J. Neuropathol. Exp. Neurol. 2007, 66, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, M.; Kleinschmidt, M.C.; Doerr, H.W.; Cinatl, J., Jr. Minocycline inhibits West Nile virus replication and apoptosis in human neuronal cells. J. Antimicrob. Chemother. 2007, 60, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.K.; Basu, A. Minocycline neuroprotects, reduces microglial activation, inhibits caspase 3 induction, and viral replication following Japanese encephalitis. J. Neurochem. 2008, 105, 1582–1595. [Google Scholar] [CrossRef]
- Dutta, K.; Kumawat, K.L.; Nazmi, A.; Mishra, M.K.; Basu, A. Minocycline differentially modulates viral infection and persistence in an experimental model of Japanese encephalitis. J. Neuroimmune Pharmacol. 2010, 5, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Szeto, G.L.; Brice, A.K.; Yang, H.-C.; Barber, S.A.; Siliciano, R.F.; Clements, J.E. Minocycline attenuates HIV infection and reactivation by suppressing cellular activation in human CD4+ T cells. J. Infect. Dis. 2010, 201, 1132–1140. [Google Scholar] [CrossRef]
- Enose-Akahata, Y.; Matsuura, E.; Tanaka, Y.; Oh, U.; Jacobson, S. Minocycline modulates antigen-specific CTL activity through inactivation of mononuclear phagocytes in patients with HTLV-I associated neurologic disease. Retrovirology 2012, 9, 16. [Google Scholar] [CrossRef]
- Leela, S.L.; Srisawat, C.; Sreekanth, G.P.; Noisakran, S.; Yenchitsomanus, P.-t.; Limjindaporn, T. Drug repurposing of minocycline against dengue virus infection. Biochem. Biophys. Res. Commun. 2016, 478, 410–416. [Google Scholar] [CrossRef]
- Bawage, S.S.; Tiwari, P.M.; Pillai, S.; Dennis, V.A.; Singh, S.R. Antibiotic minocycline prevents respiratory syncytial virus infection. Viruses 2019, 11, 739. [Google Scholar] [CrossRef]
- Kistner, O.; Barrett, P.; Mundt, W.; Reiter, M.; Schober-Bendixen, S.; Dorner, F. Development of a mammalian cell (Vero) derived candidate influenza virus vaccine. Vaccine 1998, 16, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.J.; Perez, S.; Guo, Z.; Chen, L.-M.; Donis, R.O. Establishment and characterization of a Madin-Darby canine kidney reporter cell line for influenza A virus assays. J. Clin. Microbiol. 2010, 48, 2515–2523. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-C.; Kappes, M.A.; Chen, M.-C.; Lin, C.-C.; Wang, T.T. Distinct susceptibility and applicability of MDCK derivatives for influenza virus research. PLoS ONE 2017, 12, e0172299. [Google Scholar] [CrossRef] [PubMed]
- Courtin, N.; Fotso, A.F.; Fautrad, P.; Mas, F.; Alessi, M.-C.; Riteau, B. Antiviral activity of formyl peptide receptor 2 antagonists against influenza viruses. Antivir. Res. 2017, 143, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Mukherjee, A.; Patra, U.; Chawla-Sarkar, M. Rotavirus disrupts cytoplasmic P bodies during infection. Virus Res. 2015, 210, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, P.; Dutta, D.; Chattopadhyay, S.; Mukherjee, A.; Halder, U.C.; Sarkar, S.; Kobayashi, N.; Komoto, S.; Taniguchi, K.; Chawla-Sarkar, M. Rotavirus nonstructural protein 1 suppresses virus-induced cellular apoptosis to facilitate viral growth by activating the cell survival pathways during early stages of infection. J. Virol. 2010, 84, 6834–6845. [Google Scholar] [CrossRef]
- He, M.-F.; Liang, J.-H.; Shen, Y.-N.; Zhang, C.-W.; Yang, K.-Y.; Liu, L.-C.; Xie, Q.; Hu, C.; Song, X.; Wang, Y. Coptisine Inhibits Influenza Virus Replication by Upregulating p21. Molecules 2023, 28, 5398. [Google Scholar] [CrossRef]
- Fleming, S.B. Viral inhibition of the IFN-induced JAK/STAT signalling pathway: Development of live attenuated vaccines by mutation of viral-encoded IFN-antagonists. Vaccines 2016, 4, 23. [Google Scholar] [CrossRef]
- Bui, M.; Helenius, A. The role of nuclear import and export in influenza virus infection. Trends Cell Biol. 1996, 6, 67–71. [Google Scholar]
- Pleschka, S.; Wolff, T.; Ehrhardt, C.; Hobom, G.; Planz, O.; Rapp, U.R.; Ludwig, S. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell Biol. 2001, 3, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S.; Wolff, T.; Ehrhardt, C.; Wurzer, W.J.; Reinhardt, J.; Planz, O.; Pleschka, S. MEK inhibition impairs influenza B virus propagation without emergence of resistant variants. FEBS Lett. 2004, 561, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Marjuki, H.; Alam, M.I.; Ehrhardt, C.; Wagner, R.; Planz, O.; Klenk, H.-D.; Ludwig, S.; Pleschka, S. Membrane accumulation of influenza A virus hemagglutinin triggers nuclear export of the viral genome via protein kinase Cα-mediated activation of ERK signaling. J. Biol. Chem. 2006, 281, 16707–16715. [Google Scholar] [CrossRef]
- Lowy, R.J. Influenza virus induction of apoptosis by intrinsic and extrinsic mechanisms. Int. Rev. Immunol. 2003, 22, 425–449. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.; Sutton, T.; Weathered, N.; Ray, N.; Caldwell, E.; Plotkin, Z.; Dagher, P. Minocycline inhibits apoptosis and inflammation in a rat model of ischemic renal injury. Am. J. Physiol.-Ren. Physiol. 2004, 287, F760–F766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, P.; Chen, H.; Tan, W.; Lu, J.; Liu, W.; Wang, J.; Zhang, S.; Zhu, W.; Cao, J. The inhibitory effect of minocycline on radiation-induced neuronal apoptosis via AMPKα1 signaling-mediated autophagy. Sci. Rep. 2017, 7, 16373. [Google Scholar]
- He, J.; Mao, J.; Hou, L.; Jin, S.; Wang, X.; Ding, Z.; Jin, Z.; Guo, H.; Dai, R. Minocycline attenuates neuronal apoptosis and improves motor function after traumatic brain injury in rats. Exp. Anim. 2021, 70, 563–569. [Google Scholar] [CrossRef]
- Josset, L.; Zeng, H.; Kelly, S.M.; Tumpey, T.M.; Katze, M.G. Transcriptomic Characterization of the Novel Avian-Origin Influenza A (H7N9) Virus: Specific Host Response and Responses Intermediate between Avian (H5N1 and H7N7) and Human (H3N2) Viruses and Implications for Treatment Options. mBio 2014, 5. [Google Scholar] [CrossRef]
- Fukushi, M.; Ito, T.; Oka, T.; Kitazawa, T.; Miyoshi-Akiyama, T.; Kirikae, T.; Yamashita, M.; Kudo, K. Serial histopathological examination of the lungs of mice infected with influenza A virus PR8 strain. PLoS ONE 2011, 6, e21207. [Google Scholar] [CrossRef]
- Bright, R.A.; Medina, M.-j.; Xu, X.; Perez-Oronoz, G.; Wallis, T.R.; Davis, X.M.; Povinelli, L.; Cox, N.J.; Klimov, A.I. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: A cause for concern. Lancet 2005, 366, 1175–1181. [Google Scholar] [CrossRef]
- Dharan, N.J.; Gubareva, L.V.; Meyer, J.J.; Okomo-Adhiambo, M.; McClinton, R.C.; Marshall, S.A.; George, K.S.; Epperson, S.; Brammer, L.; Klimov, A.I. Infections with oseltamivir-resistant influenza A (H1N1) virus in the United States. Jama 2009, 301, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Krumbholz, A.; Schmidtke, M.; Bergmann, S.; Motzke, S.; Bauer, K.; Stech, J.; Dürrwald, R.; Wutzler, P.; Zell, R. High prevalence of amantadine resistance among circulating European porcine influenza A viruses. J. Gen. Virol. 2009, 90, 900–908. [Google Scholar] [CrossRef]
- Baranovich, T.; Saito, R.; Suzuki, Y.; Zaraket, H.; Dapat, C.; Caperig-Dapat, I.; Oguma, T.; Shabana, I.I.; Saito, T.; Suzuki, H. Emergence of H274Y oseltamivir-resistant A (H1N1) influenza viruses in Japan during the 2008–2009 season. J. Clin. Virol. 2010, 47, 23–28. [Google Scholar] [CrossRef]
- Abdulaziz, L.; Elhadi, E.; Abdallah, E.A.; Alnoor, F.A.; Yousef, B.A. Antiviral activity of approved antibacterial, antifungal, antiprotozoal and anthelmintic drugs: Chances for drug repurposing for antiviral drug discovery. J. Exp. Pharmacol. 2022, 14, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Kaptein, S.J.; De Burghgraeve, T.; Froeyen, M.; Pastorino, B.; Alen, M.M.; Mondotte, J.A.; Herdewijn, P.; Jacobs, M.; de Lamballerie, X.; Schols, D. A derivate of the antibiotic doxorubicin is a selective inhibitor of dengue and yellow fever virus replication in vitro. Antimicrob. Agents Chemother. 2010, 54, 5269–5280. [Google Scholar] [CrossRef]
- Raymonda, M.H.; Ciesla, J.H.; Monaghan, M.; Leach, J.; Asantewaa, G.; Smorodintsev-Schiller, L.A.; Lutz IV, M.M.; Schafer, X.L.; Takimoto, T.; Dewhurst, S. Pharmacologic profiling reveals lapatinib as a novel antiviral against SARS-CoV-2 in vitro. Virology 2022, 566, 60–68. [Google Scholar] [CrossRef] [PubMed]
- TAKATSUKI, A.; TAMURA, G. Tunicamycin, a new antibiotic. II some biological properties of the antiviral activity of tunicamycin. J. Antibiot. 1971, 24, 224–231. [Google Scholar] [CrossRef]
- Shutter, M.; Akhondi, H. Tetracycline; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Tamma, P.D.; Aitken, S.L.; Bonomo, R.A.; Mathers, A.J.; Van Duin, D.; Clancy, C.J. Infectious Diseases Society of America guidance on the treatment of AmpC β-lactamase–producing Enterobacterales, carbapenem-resistant Acinetobacter baumannii, and Stenotrophomonas maltophilia infections. Clin. Infect. Dis. 2022, 74, 2089–2114. [Google Scholar] [CrossRef]
- Inoue, Y.; Hirayama, T.; Kondo, A.; Tomari, S.; Miyazaki, T.; Izumikawa, K.; Kakeya, H.; Yamamoto, Y.; Yanagihara, K.; Tashiro, T. A case of influenza pneumonia following pneumococcal infection in an adult patient with concurrent encephalopathy with a lesion in the splenium of the corpus callosum. Kansenshogaku Zasshi J. Jpn. Assoc. Infect. Dis. 2013, 87, 451–456. [Google Scholar] [CrossRef]
- Barber, S.A.; Bruett, L.; Douglass, B.R.; Herbst, D.S.; Zink, M.C.; Clements, J.E. Visna virus-induced activation of MAPK is required for virus replication and correlates with virus-induced neuropathology. J. Virol. 2002, 76, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Benn, J.; Su, F.; Doria, M.; Schneider, R.J. Hepatitis B virus HBx protein induces transcription factor AP-1 by activation of extracellular signal-regulated and c-Jun N-terminal mitogen-activated protein kinases. J. Virol. 1996, 70, 4978–4985. [Google Scholar] [CrossRef]
- Daum, G.; Eisenmann-Tappe, I.; Fries, H.-W.; Troppmair, J.; Rapp, U.R. The ins and outs of Raf kinases. Trends Biochem. Sci. 1994, 19, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Kujime, K.; Hashimoto, S.; Gon, Y.; Shimizu, K.; Horie, T. p38 mitogen-activated protein kinase and c-jun-NH2-terminal kinase regulate RANTES production by influenza virus-infected human bronchial epithelial cells. J. Immunol. 2000, 164, 3222–3228. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S.; Ehrhardt, C.; Neumeier, E.R.; Kracht, M.; Rapp, U.R.; Pleschka, S. Influenza virus-induced AP-1-dependent gene expression requires activation of the JNK signaling pathway. J. Biol. Chem. 2001, 276, 10990–10998. [Google Scholar] [CrossRef]
- Planz, O.; Pleschka, S.; Ludwig, S. MEK-specific inhibitor U0126 blocks spread of Borna disease virus in cultured cells. J. Virol. 2001, 75, 4871–4877. [Google Scholar] [CrossRef]
- Rodems, S.M.; Spector, D.H. Extracellular signal-regulated kinase activity is sustained early during human cytomegalovirus infection. J. Virol. 1998, 72, 9173–9180. [Google Scholar] [CrossRef] [PubMed]
- Terstegen, L.; Gatsios, P.; Ludwig, S.; Pleschka, S.; Jahnen-Dechent, W.; Heinrich, P.C.; Graeve, L. The vesicular stomatitis virus matrix protein inhibits glycoprotein 130-dependent STAT activation. J. Immunol. 2001, 167, 5209–5216. [Google Scholar] [CrossRef] [PubMed]
- Neumann, G.; Hughes, M.T.; Kawaoka, Y. Influenza A virus NS2 protein mediates vRNP nuclear export through NES-independent interaction with hCRM1. EMBO J. 2000, 19, 6751–6758. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, S.; Pleschka, S.; Planz, O.; Wolff, T. Ringing the alarm bells: Signalling and apoptosis in influenza virus infected cells. Cell. Microbiol. 2006, 8, 375–386. [Google Scholar] [CrossRef]
- Robert, G.; Gastaldi, C.; Puissant, A.; Hamouda, A.; Jacquel, A.; Dufies, M.; Belhacene, N.; Colosetti, P.; Reed, J.C.; Auberger, P. The anti-apoptotic Bcl-B protein inhibits BECN1-dependent autophagic cell death. Autophagy 2012, 8, 637–649. [Google Scholar] [CrossRef]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A. Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef]
- Danthi, P. Viruses and the diversity of cell death. Annu. Rev. Virol. 2016, 3, 533–553. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Brenner, C.; Morselli, E.; Touat, Z.; Kroemer, G. Viral control of mitochondrial apoptosis. PLoS Pathog. 2008, 4, e1000018. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Jiang, W.; Liu, Z.; Liu, S.; Liang, X. Virus infection and death receptor-mediated apoptosis. Viruses 2017, 9, 316. [Google Scholar] [CrossRef]
- Brydon, E.W.; Morris, S.J.; Sweet, C. Role of apoptosis and cytokines in influenza virus morbidity. FEMS Microbiol. Rev. 2005, 29, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Mesa, N.; Zarzuelo, A.; Gálvez, J. Minocycline: Far beyond an antibiotic. Br. J. Pharmacol. 2013, 169, 337–352. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saha, P.; Saha, R.; Datta Chaudhuri, R.; Sarkar, R.; Sarkar, M.; Koley, H.; Chawla-Sarkar, M. Unveiling the Antiviral Potential of Minocycline: Modulation of Nuclear Export of Viral Ribonuclear Proteins during Influenza Virus Infection. Viruses 2024, 16, 1317. https://doi.org/10.3390/v16081317
Saha P, Saha R, Datta Chaudhuri R, Sarkar R, Sarkar M, Koley H, Chawla-Sarkar M. Unveiling the Antiviral Potential of Minocycline: Modulation of Nuclear Export of Viral Ribonuclear Proteins during Influenza Virus Infection. Viruses. 2024; 16(8):1317. https://doi.org/10.3390/v16081317
Chicago/Turabian StyleSaha, Priyanka, Ritubrita Saha, Ratul Datta Chaudhuri, Rakesh Sarkar, Mehuli Sarkar, Hemanta Koley, and Mamta Chawla-Sarkar. 2024. "Unveiling the Antiviral Potential of Minocycline: Modulation of Nuclear Export of Viral Ribonuclear Proteins during Influenza Virus Infection" Viruses 16, no. 8: 1317. https://doi.org/10.3390/v16081317
APA StyleSaha, P., Saha, R., Datta Chaudhuri, R., Sarkar, R., Sarkar, M., Koley, H., & Chawla-Sarkar, M. (2024). Unveiling the Antiviral Potential of Minocycline: Modulation of Nuclear Export of Viral Ribonuclear Proteins during Influenza Virus Infection. Viruses, 16(8), 1317. https://doi.org/10.3390/v16081317