

In Silico Discovery and Evaluation of Inhibitors of the SARS-CoV-2 Spike Protein–HSPA8 Complex Towards Developing COVID-19 Therapeutic Drugs




Abstract
1. Introduction
2. Materials and Methods
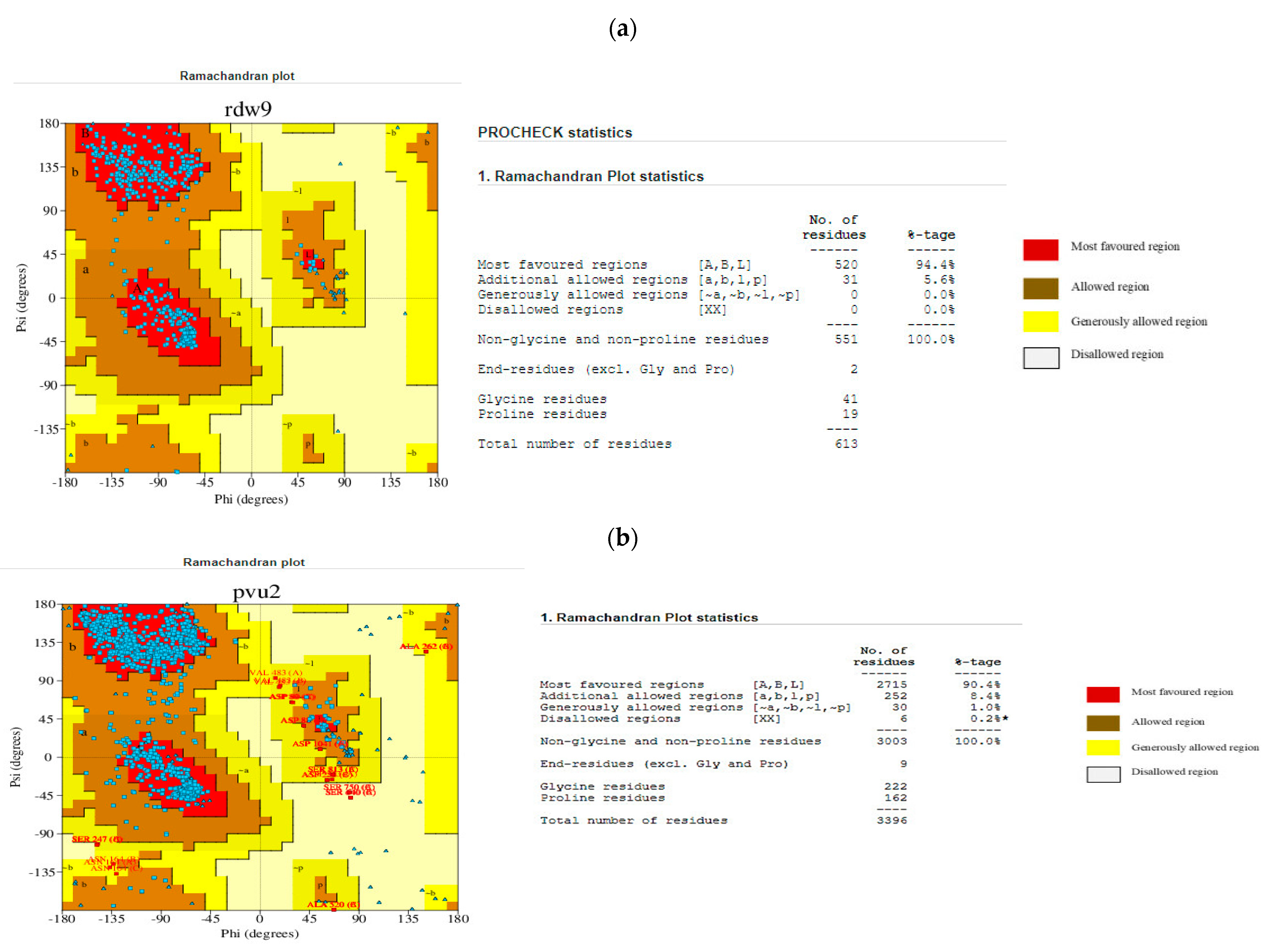
2.1. Homology Modelling and Validation of HSPA8 and SARS-CoV-2 Spike Protein Structures
2.2. Identification and Selection of Potential Small Molecules
2.3. Molecular Docking
2.3.1. Performing Protein–Protein Docking.
2.3.2. Protein–Ligand Docking
2.3.3. Performing Protein Complex–Ligand Docking.
2.4. Prime MM-GBSA
3. Results
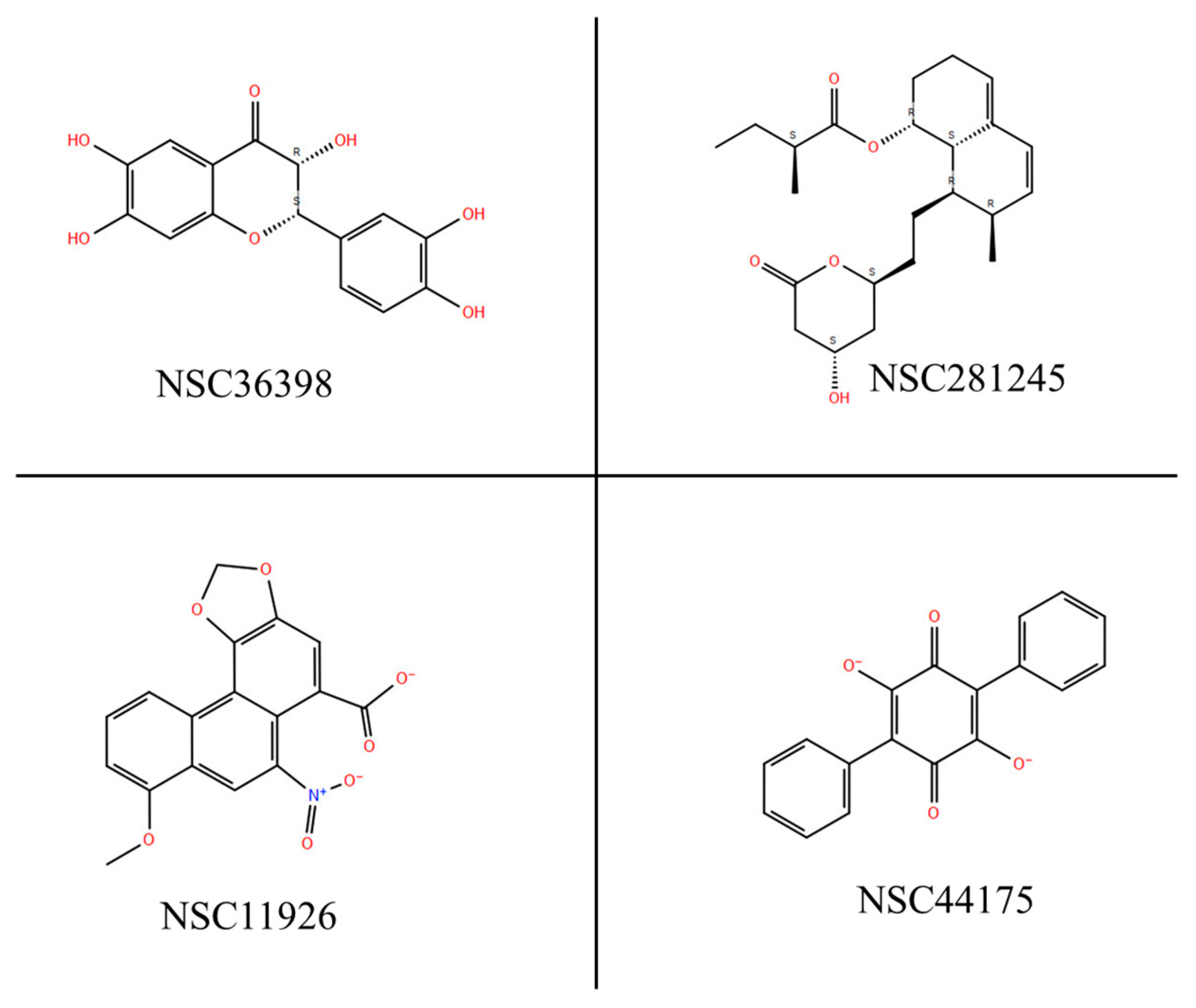
3.1. Screening and Analysis of Drug-Likeness Properties of the Selected Small Molecules
3.2. Three-Dimensional Homology Modelling and Validation
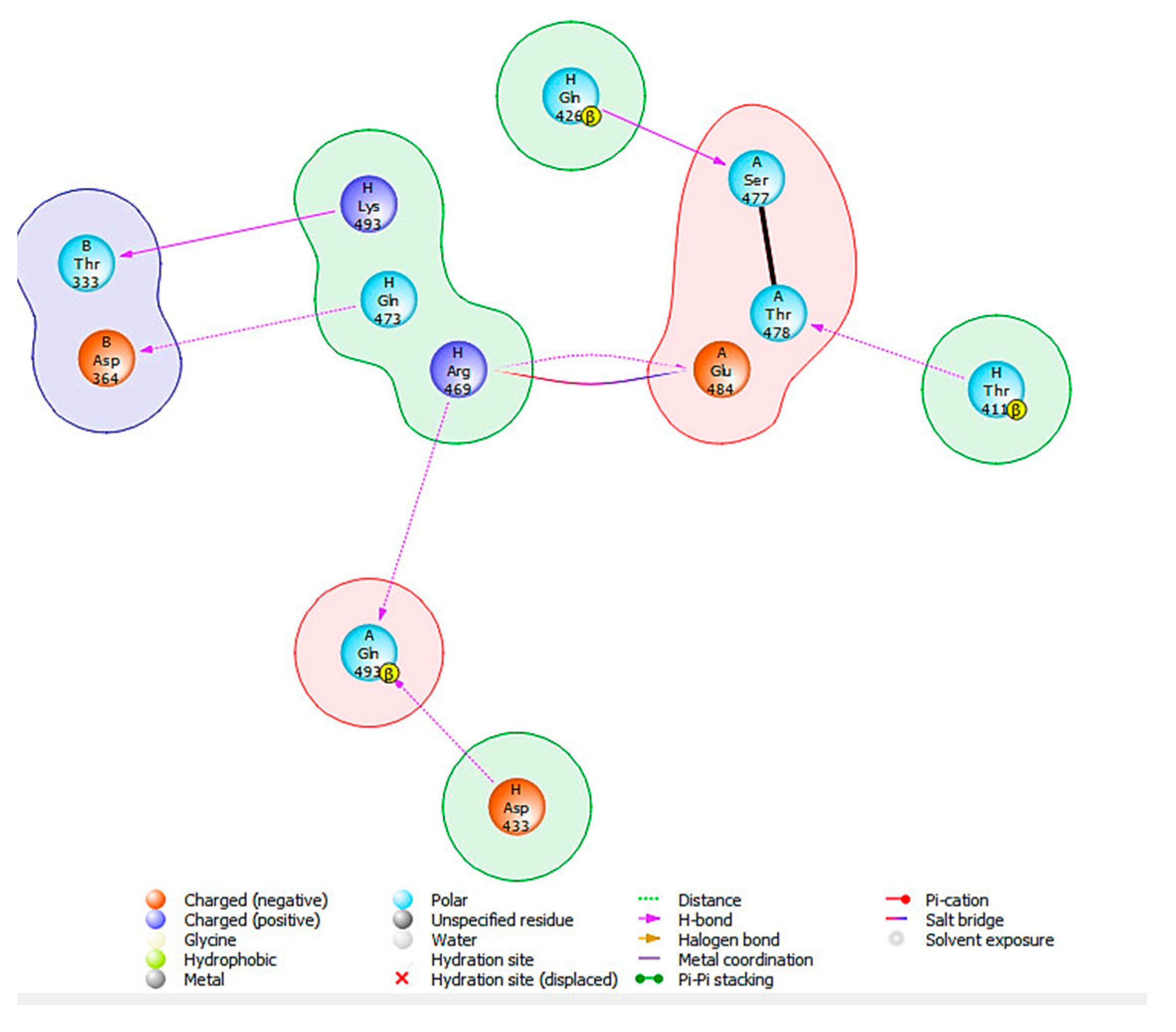
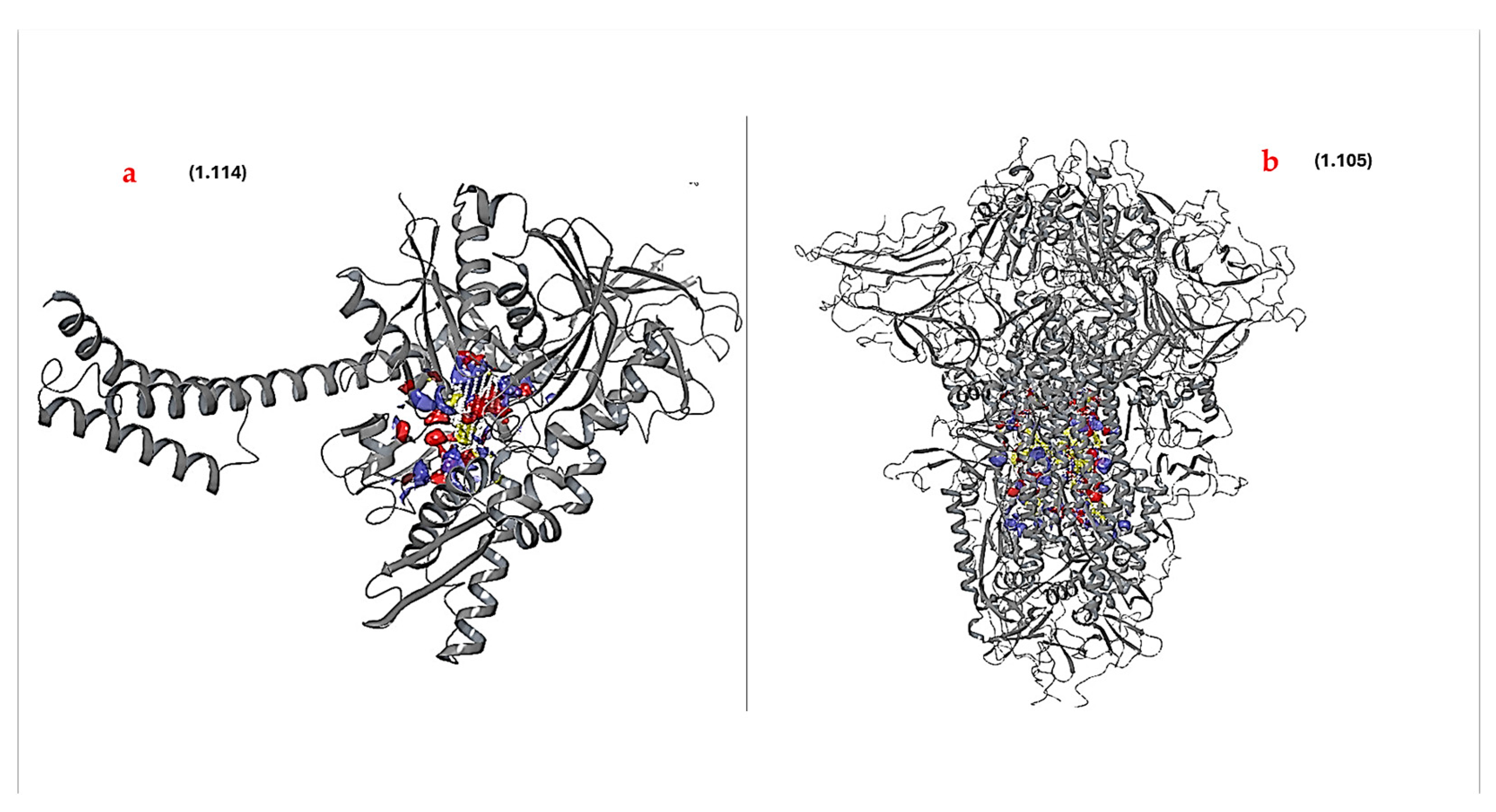
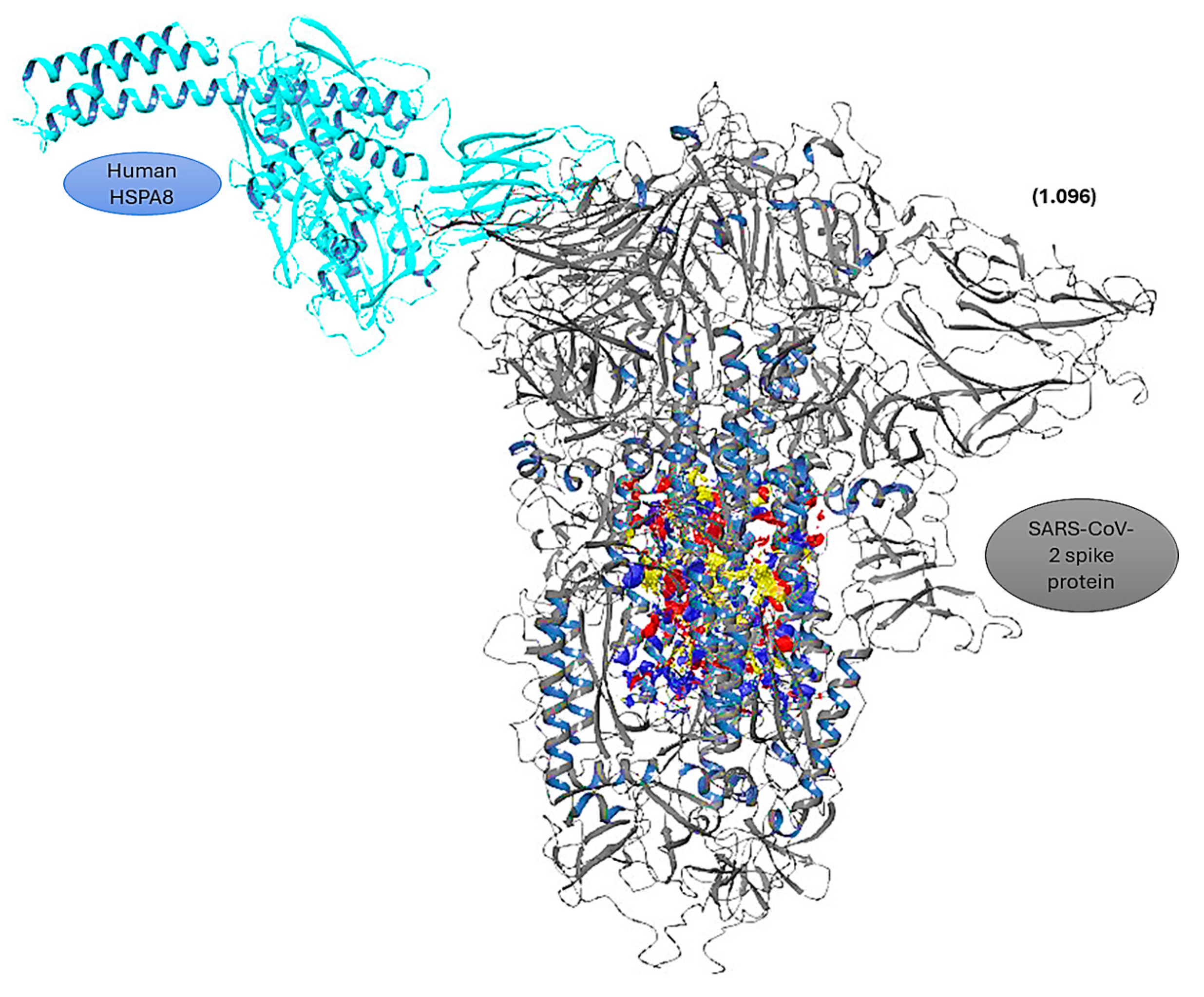
3.3. Protein–Protein Docking
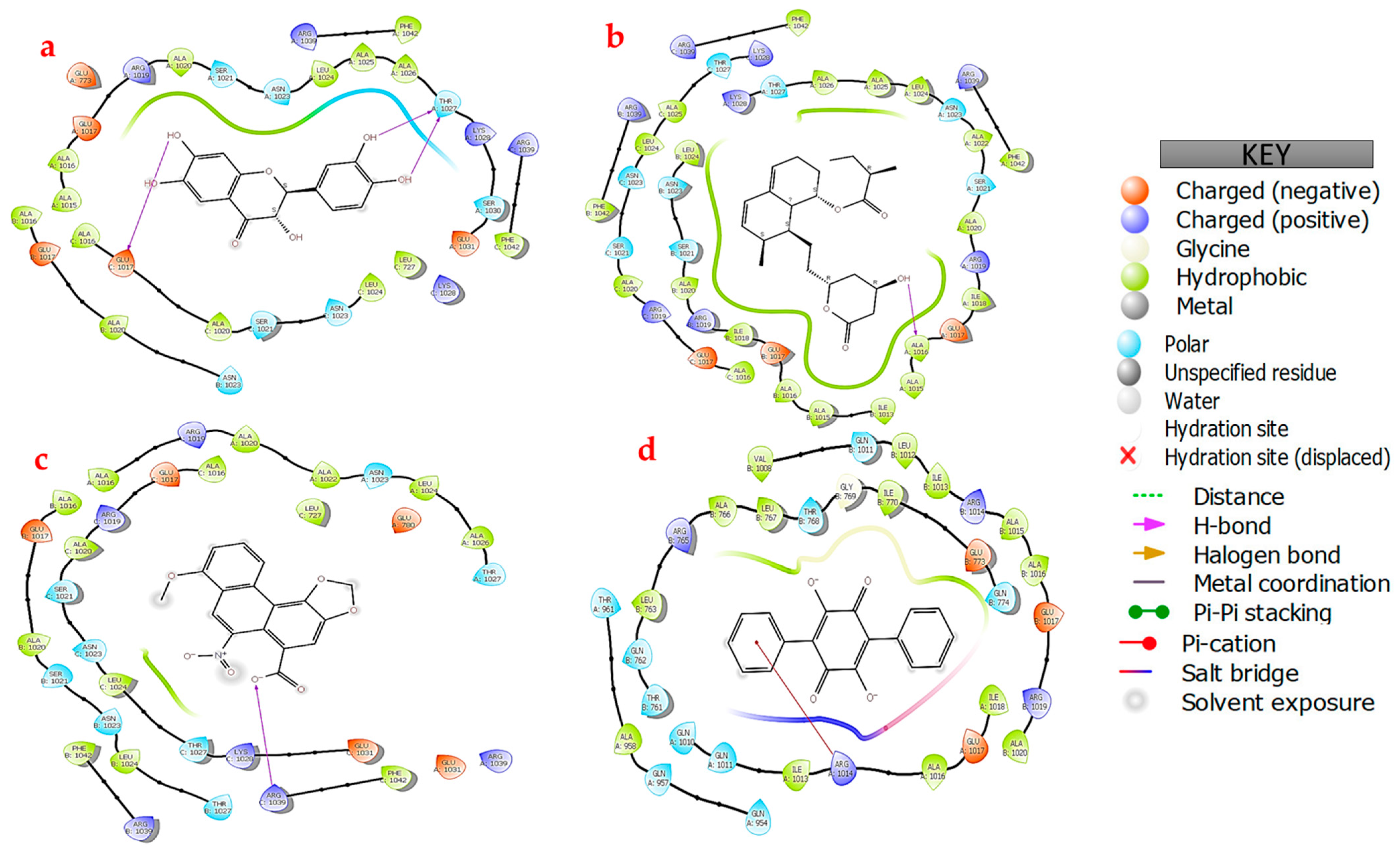
3.4. Protein–Ligand Docking
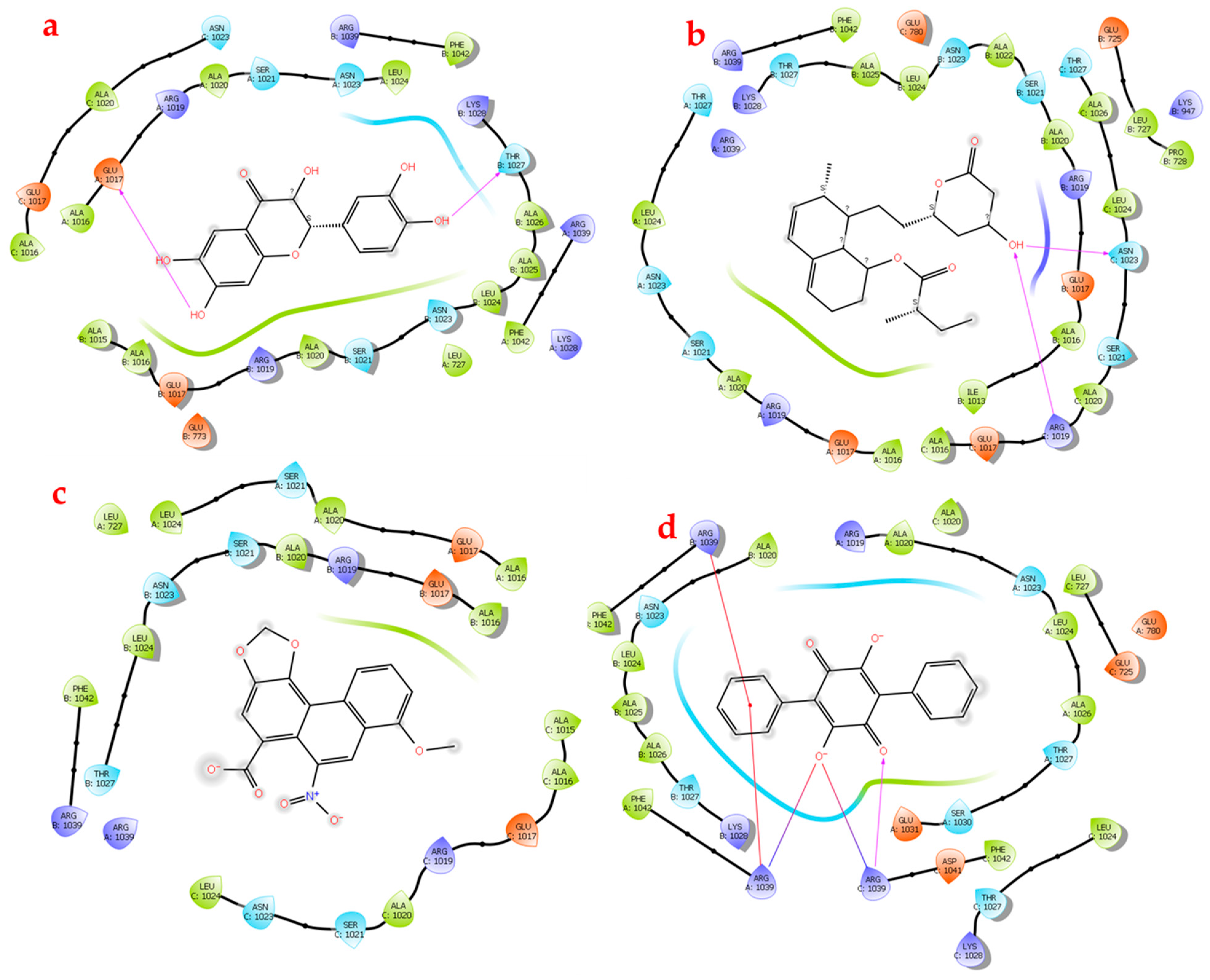
3.5. Protein Complex–Ligand Docking
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Makhoba, X.H.; Makumire, S. The capture of host cell’s resources: The role of heat shock proteins and polyamines in SARS-CoV-2 (COVID-19) pathway to viral infection. Biomol. Concept. 2022, 13, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Mallah, S.I.; Ghorab, O.K.; Al-Salmi, S.; Abdellatif, O.S.; Tharmaratnam, T.; Iskandar, M.A.; Sefen, J.A.N.; Sidhu, P.; Atallah, B.; El-Lababidi, R.; et al. COVID-19: Breaking down a global health crisis. Ann. Clin. Microbiol. Antimicrob. 2021, 20, 35. [Google Scholar] [CrossRef]
- Gil-Manso, S.; Herrero-Quevedo, D.; Carbonell, D.; Martínez-Bonet, M.; Bernaldo-de-Quirós, E.; Kennedy-Batalla, R.; Gallego-Valle, J.; López-Esteban, R.; Blázquez-López, E.; Miguens-Blanco, I.; et al. Multidimensional analysis of immune cells from COVID-19 patients identified cell subsets associated with the severity at hospital admission. PLoS Pathog. 2023, 19, e1011432. [Google Scholar] [CrossRef]
- Islam, M.J.; Islam, N.N.; Alom, M.S.; Kabir, M.; Halim, M.A. A Review on Structural, Non-Structural, and Accessory Proteins of SARS-CoV-2: Highlighting Drug Target Sites. Immunobiology 2023, 228, 152302. [Google Scholar]
- Kakavandi, S.; Zare, I.; VaezJalali, M.; Dadashi, M.; Azarian, M.; Akbari, A.; Ramezani Farani, M.; Zalpoor, H.; Hajikhani, B. Structural and Non-Structural Proteins in SARS-CoV-2: Potential Aspects to COVID-19 Treatment or Prevention of Progression of Related Diseases. Cell Commun. Signal. 2023, 21, 110. [Google Scholar] [CrossRef]
- Aldaais, E.A.; Yegnaswamy, S.; Albahrani, F.; Alsowaiket, F.; Alramadan, S. Sequence and Structural Analysis of COVID-19 E and M Proteins with MERS Virus E and M Proteins—A Comparative Study. Biochem. Biophys. Rep. 2021, 26, 101023. [Google Scholar] [CrossRef]
- Mohammed, M.E. SARS-CoV-2 Proteins: Are They Useful as Targets for COVID-19 Drugs and Vaccines? Curr. Mol. Med. 2022, 22, 50–66. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 Infects Cells After Viral Entry via Clathrin-Mediated Endocytosis. J. Biol. Chem. 2021, 296, 100306. [Google Scholar] [CrossRef]
- Pampel, J. SARS-CoV-2 Life Cycle: Stages and Inhibition Targets. Antibodies-online. Available online: https://www.antibodies-online.com/resources/18/5410/SARS-CoV-2-life-cycle-stages-and-inhibition-targets/#references (accessed on 11 October 2024).
- Alkafaas, S.S.; Abdallah, A.M.; Ghosh, S.; Loutfy, S.A.; Elkafas, S.S.; Abdel Fattah, N.F.; Hessien, M. Insight into the Role of Clathrin-Mediated Endocytosis Inhibitors in SARS-CoV-2 Infection. Rev. Med. Virol. 2023, 33, e2403. [Google Scholar] [CrossRef] [PubMed]
- Paladino, L.; Vitale, A.M.; Caruso Bavisotto, C.; Conway de Macario, E.; Cappello, F.; Macario, A.J.; Marino Gammazza, A. The Role of Molecular Chaperones in Virus Infection and Implications for Understanding and Treating COVID-19. J. Clin. Med. 2020, 9, 3518. [Google Scholar] [CrossRef]
- Neckers, L.; Tatu, U. Molecular Chaperones in Pathogen Virulence: Emerging New Targets for Therapy. Cell Host Microbe 2008, 4, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.K.; Shin, Y.; Ju, S.; Han, S.; Choe, W.; Yoon, K.S.; Kim, S.S.; Kang, I. Heat Shock Response and Heat Shock Proteins: Current Understanding and Future Opportunities in Human Diseases. Int. J. Mol. Sci. 2024, 25, 4209. [Google Scholar] [CrossRef]
- Hu, C.; Yang, J.; Qi, Z.; Wu, H.; Wang, B.; Zou, F.; Mei, H.; Liu, J.; Wang, W.; Liu, Q. Heat Shock Proteins: Biological Functions, Pathological Roles, and Therapeutic Opportunities. MedComm 2022, 3, e161. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, Y.; Bao, C.; Xiang, H.; Gao, Q.; Mao, L. Mechanism and Complex Roles of HSC70/HSPA8 in Viral Entry. Virus Res. 2024, 347, 199433. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Yang, X.; Zhao, J.; Cheng, Y.; Wang, J. Mechanism and Complex Roles of HSC70 in Viral Infections. Front. Microbiol. 2020, 11, 1577. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Lv, C.; Fang, C.; Peng, X.; Sheng, H.; Xiao, P.; Kumar Ojha, N.; Yan, Y.; Liao, M.; Zhou, J. Heat Shock Protein Member 8 Is an Attachment Factor for Infectious Bronchitis Virus. Front. Microbiol. 2020, 11, 1630. [Google Scholar] [CrossRef]
- Navhaya, L.T.; Blessing, D.M.; Yamkela, M.; Godlo, S.; Makhoba, X.H. A comprehensive review of the interaction between COVID-19 spike proteins with mammalian small and major heat shock proteins. Biomol. Concept. 2024, 15, 20220027. [Google Scholar] [CrossRef]
- Lubkowska, A.; Pluta, W.; Strońska, A.; Lalko, A. Role of Heat Shock Proteins (HSP70 and HSP90) in Viral Infection. Int. J. Mol. Sci. 2021, 22, 9366. [Google Scholar] [CrossRef]
- Zhou, Y.W.; Xie, Y.; Tang, L.S.; Pu, D.; Zhu, Y.J.; Liu, J.Y.; Ma, X.L. Therapeutic Targets and Interventional Strategies in COVID-19: Mechanisms and Clinical Studies. Signal Transduct. Target. Ther. 2021, 6, 317. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, M.; Galdiero, M.; Folliero, V.; Zannella, C.; De Filippis, A.; Mali, A.; Rinaldi, L.; Franci, G. SARS-CoV-2 Vaccine Development: Where Are We. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 2752–2784. [Google Scholar] [PubMed]
- Romeo, I.; Mesiti, F.; Lupia, A.; Alcaro, S. Current Updates on Naturally Occurring Compounds Recognizing SARS-CoV-2 Druggable Targets. Molecules 2021, 26, 632. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, A.; Jabeen, T.; Aslam, S.; Ahmad, M.; Ashfaq, U.A.; Mohsin, N.U.A.; Zaki, M.E.; Al-Hussain, S.A. A comprehensive update of various attempts by medicinal chemists to combat COVID-19 through natural products. Molecules 2023, 28, 4860. [Google Scholar] [CrossRef]
- Durgam, L.; Guruprasad, L. Computational Studies on the Design of NCI Natural Products as Inhibitors to SARS-CoV-2 Main Protease. J. Biomol. Struct. Dyn. 2023, 41, 3741–3751. [Google Scholar] [CrossRef]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo—Distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef]
- Laskowski, R.A. PDBsum 1: A standalone program for generating PDBsum analyses. Protein Sci. 2022, 31, e4473. [Google Scholar] [CrossRef]
- Knox, C.; Wilson, M.; Klinger, C.M.; Franklin, M.; Oler, E.; Wilson, A.; Pon, A.; Cox, J.; Chin, N.E.; Strawbridge, S.A.; et al. Drugbank 6.0: The drugbank knowledgebase for 2024. Nucleic Acids Res. 2024, 52, D1265–D1275. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef]
- Jakhmola, V.; Parashar, T.; Ghildiyal, P.; Ansori, A.N.M.; Sharma, R.K.; Rao, N.R.; Kalra, K.; Singh, N.; Nainwal, N.; Singh, R.K.; et al. An In Silico Study to Explore the Role of EGFR in Ovarian Cancer. Pharmacogn. J. 2022, 14, 817–821. [Google Scholar] [CrossRef]
- Bayrak, C.; Taslimi, P.; Karaman, H.S.; Gulcin, I.; Menzek, A. The first synthesis, carbonic anhydrase inhibition and anticholinergic activities of some bromophenol derivatives with S including natural products. Bioorg. Chem. 2019, 85, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Sesethu, G.; Nombalentle, M.; Yamkela, M.; Anelisa, M.; Makumire, S.; Mkwetshana, N.; Govender, K.K.; Makhoba, X.H. In silico evaluation of heat shock proteins reveals an interplay with polyamines as a survival strategy for the Plasmodium falciparum. INNOSC Theranostics Pharmacol. Sci. 2023, 7, 1228. [Google Scholar] [CrossRef]
- Acharya, A.; Pandey, K.; Thurman, M.; Klug, E.; Trivedi, J.; Sharma, K.; Lorson, C.L.; Singh, K.; Byrareddy, S.N. Discovery and evaluation of entry inhibitors for SARS-CoV-2 and its emerging variants. J. Virol. 2021, 95, 10–1128. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Pattar, S.V.; Adhoni, S.A.; Kamanavalli, C.M.; Kumbar, S.S. In silico molecular docking studies and MM/GBSA analysis of coumarin-carbonodithioate hybrid derivatives divulge the anticancer potential against breast cancer. Beni-Suef Univ. J. Basic Appl. Sci. 2020, 9, 36. [Google Scholar] [CrossRef]
- Muddagoni, N.; Bathula, R.; Dasari, M.; Potlapally, S.R. Homology modeling, virtual screening, prime-MMGBSA, AutoDock-identification of inhibitors of FGR protein. Biointerface Res. Appl. Chem. 2021, 11, 11088–11103. [Google Scholar]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high-resolution protein structure modeling. Proteins Struct. Funct. Bioinform. 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Schyman, P.; Liu, R.; Desai, V.; Wallqvist, A. vNN web server for ADMET predictions. Front. Pharmacol. 2017, 8, 889. [Google Scholar] [CrossRef]
- Adebiyi, M.O.; Obagbuwa, I.C. Homology Modeling and Binding Site Analysis of SARS-CoV-2 (COVID-19) Main Protease 3D Structure. In Research Anthology on Bioinformatics, Genomics, and Computational Biology; IGI Global: Hershey, PA, USA, 2024; pp. 933–947. [Google Scholar]
- Yamkela, M.; Sitobo, Z.; Makhoba, X.H. In Silico Analysis of SARS-CoV-2 Non-Structural Proteins Reveals an Interaction with the Host’s Heat Shock Proteins That May Contribute to Viral Replications and Development. Curr. Issues Mol. Biol. 2023, 45, 10225–10247. [Google Scholar] [CrossRef]
- Ncube, N.B.; Govender, K.K.; Tukulula, M. A critical analysis of the binding pocket of Plasmodium falciparum Phosphatidylinositol-4-kinase enzyme. ChemistrySelect 2023, 8, e202302189. [Google Scholar] [CrossRef]
- Harrison, M.C.; Lai, P.K. Investigating the mechanisms of antibody binding to alpha-synuclein for the treatment of Parkinson’s Disease. Mol. Pharm. 2024, 21, 5326–5334. [Google Scholar] [CrossRef] [PubMed]
- Belete, T.M. Recent progress in the development of new antimalarial drugs with novel targets. Drug Des. Devel. Ther. 2020, 14, 3875–3889. [Google Scholar] [CrossRef] [PubMed]
- Venugopala, K.N.; Habeebuddin, M.; Aldhubiab, B.E.; Asif, A.H. Design, Synthesis, and In Vitro Evaluation of Novel Indolyl DiHydropyrazole Derivatives as Potential Anticancer Agents. Molecules 2021, 26, 5235. [Google Scholar] [CrossRef]
- El-Sewedy, A.; El-Bordany, E.A.; Mahmoud, N.F.; Ali, K.A.; Ramadan, S.K. One-pot synthesis, computational chemical study, molecular docking, biological study, and in silico prediction ADME/pharmacokinetics properties of 5-substituted 1 H-tetrazole derivatives. Sci. Rep. 2023, 13, 17869. [Google Scholar] [CrossRef]
- Parveen, D.; Ali, R.; Shaquiquzzaman, M.; Azam, F.; Akhter, M.; Gupta, A.; Kumar, V.; Saifullah, M.K.; Khan, M.A.; Parvez, S.; et al. Design, Molecular Docking and MD Simulation of Novel Estradiol-Pyrimidine Analogues as potential inhibitors of Mpro and ACE2 for COVID-19. Chem. Phys. Impact 2024, 8, 100560. [Google Scholar] [CrossRef]
- Parveen, D.; Das, A.; Amin, S.; Alam, M.M.; Akhter, M.; Ahmed Khan, M.; Ali, R.; Anwer, T.; Sheikh, K.A.; Azam, F.; et al. Effectiveness of estrogen and its derivatives over dexamethasone in the treatment of COVID-19. J. Biomol. Struct. Dyn. 2024, 42, 1858–1874. [Google Scholar] [CrossRef]
- Moschovou, K.; Antoniou, M.; Chontzopoulou, E.; Papavasileiou, K.D.; Melagraki, G.; Afantitis, A.; Mavromoustakos, T. Exploring the binding effects of natural products and antihypertensive drugs on SARS-CoV-2: An in silico investigation of main protease and spike protein. Int. J. Mol. Sci. 2023, 24, 15894. [Google Scholar] [CrossRef]
- Elekofehinti, O.O.; Iwaloye, O.; Josiah, S.S.; Lawal, A.O.; Akinjiyan, M.O.; Ariyo, E.O. Molecular docking studies, molecular dynamics and ADME/tox reveal therapeutic potentials of STOCK1N-69160 against papain-like protease of SARS-CoV-2. Mol. Divers. 2021, 25, 1761–1773. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A.; Garg, N.; Giri, R. An insight into SARS-CoV-2 membrane protein interaction with spike, envelope, and nucleocapsid proteins. J. Biomol. Struct. Dyn. 2023, 41, 1062–1071. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Structure |
|---|---|
| Polyporic acid NSC44175 |  |
| Aristolochic acid NSC11926 | 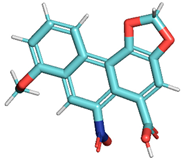 |
| 2-(3,4-Dihydroxyphenyl)-3,6,7-trihydroxy-2,3-dihydro-4H-chromen-4-one NSC36398 |  |
| Mevastatin NSC281245 | 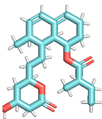 |
| Name of Ligand | Polyporic Acid | Aristolochic Acid | 2-(3,4-Dihydroxyphenyl)-3,6,7-trihydroxy-2,3-dihydro-4H-chromen-4-one | Mevastatin |
|---|---|---|---|---|
| Physiochemical properties | ||||
| Molecular formula | C18H12O4 | C17H11NO7 | C15H12O7 | C23H34O5 |
| Hydrogen Bond Donor Count | 2 | 1 | 5 | 1 |
| Hydrogen Bond Acceptor Count | 4 | 7 | 7 | 5 |
| Topological Polar Surface Area | 74.60 Å2 | 110.81 Å2 | 127.45 Å2 | 72.83 Å2 |
| Fraction CSP3 | 0.00 | 0.12 | 0.13 | 0.74 |
| Water Solubility | ||||
| Log S (SILICOS-IT) | −5.14 | −4.325 | −2.03 | −3.04 |
| Class | Moderately soluble | Moderately soluble | Soluble | Soluble |
| Solubility | 2.11 × 10−3 mg/mL; 7.21 × 10−6 mol/L | 1.52 × 10−2 mg/mL; 4.47 × 10−5 mol/L | 2.87 mg/mL; 9.42 × 10−3 mol/L | 3.55 × 10−1 mg/mL; 9.08 × 10−4 mol/L |
| Drug likeness | ||||
| Lipinski Rule | Yes; 0 violations | Yes; 0 violations | Yes; 0 violations | Yes; 0 violations |
| Veber (GSK) Rule | Yes | Yes | Yes | Yes |
| Egan (phatmacial) Filter | Yes | Yes | Yes | Yes |
| Muegge (Bayer) Filter | Yes | Yes | Yes | Yes |
| Bioavailability (Abbort) Score | 0.85 | 0.56 | 0.55 | 0.55 |
| Medicinal Chemistry | ||||
| Pan Assay Interference Structures | 1 alert: quinone A | 0 alert | 1 alert: catechol A | 0 alert |
| Brenk | 1 alert: chinone A | 3 alerts: nitro group, oxygen-nitrogen single bond, polycyclic_aromatic_hydrocarbon_3 | 1 alert: catechol | 1 alert: more than 2 esters |
| Lead likeness | Yes | No; 1 violation: XLOGP3 > 3.5 | Yes | No; 2 violations: MW > 350, XLOGP3 > 3.5 |
| Synthetic accessibility | 3.00 | 2.77 | 3.52 | 5.56 |
| HSPA8 Residues | SARS-CoV-2 Spike Protein Residues | Distance (Å) | Specific Interactions | No. of Hydrogen Bonds |
|---|---|---|---|---|
| H: THR411 | A: THR478 | 1.6 | 1× hb to A: THR478 | 1 |
| H: GLN426 | A: SER477 | 1.9 | 1× hb to A: SER477 | 1 |
| H: ARG469 | A: GLU487 | 2.0 | 1× hb, 1× salt bridge to A: GLU484 | 1 |
| H: ARG469 | A: GLN493 | 2.1 | 1× hb to A: GLN493 | 1 |
| H: GLN473 | B: ASP364 | 2.2 | 1× hb to B: ASP364 | 1 |
| H:ASP433 | A: GLN493 | 2.4 | 1× hb to A: GLN493 | 1 |
| H: LYS493 | B: THR333 | 2.5 | 1× hb to B: THR333 | 1 |
| Protein | CID | Docking Scores (kcal/mol) | Glide Gscore (kcal/mol) | Prime MM–GBSA Complex Energy (dG bind) (kcal/mol) |
|---|---|---|---|---|
| HSPA8 | NSC36398 | −7.148 | −7.148 | −37.73 |
| HSPA8 | NSC281245 | −5.224 | −5.224 | −13.08 |
| HSPA8 | NSC11926 | −4.141 | −4.141 | −31.30 |
| HSPA8 | NSC44175 | −2.406 | −2.407 | 14.20 |
| Spike protein | NSC36398 | −7.934 | −7.965 | −39.52 |
| Spike protein | NSC281245 | −5.099 | −5.099 | −44.49 |
| Spike protein | NSC11926 | −3.463 | −3.463 | −23.90 |
| Spike protein | NSC44175 | −2.873 | −2.873 | −7.23 |
| HSPA8–spike protein | NSC36398 | −8.029 | −8.029 | −38.61 |
| HSPA8–spike protein | NSC281245 | −5.285 | −5.285 | −36.65 |
| HSPA8–spike protein | NSC11926 | −4.120 | −4.120 | −27.16 |
| HSPA8–spike protein | NSC44175 | −2.796 | −2.798 | 1.61 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navhaya, L.T.; Matsebatlela, T.M.; Monama, M.Z.; Makhoba, X.H. In Silico Discovery and Evaluation of Inhibitors of the SARS-CoV-2 Spike Protein–HSPA8 Complex Towards Developing COVID-19 Therapeutic Drugs. Viruses 2024, 16, 1726. https://doi.org/10.3390/v16111726
Navhaya LT, Matsebatlela TM, Monama MZ, Makhoba XH. In Silico Discovery and Evaluation of Inhibitors of the SARS-CoV-2 Spike Protein–HSPA8 Complex Towards Developing COVID-19 Therapeutic Drugs. Viruses. 2024; 16(11):1726. https://doi.org/10.3390/v16111726
Chicago/Turabian StyleNavhaya, Liberty T., Thabe M. Matsebatlela, Mokgerwa Z. Monama, and Xolani H. Makhoba. 2024. "In Silico Discovery and Evaluation of Inhibitors of the SARS-CoV-2 Spike Protein–HSPA8 Complex Towards Developing COVID-19 Therapeutic Drugs" Viruses 16, no. 11: 1726. https://doi.org/10.3390/v16111726
APA StyleNavhaya, L. T., Matsebatlela, T. M., Monama, M. Z., & Makhoba, X. H. (2024). In Silico Discovery and Evaluation of Inhibitors of the SARS-CoV-2 Spike Protein–HSPA8 Complex Towards Developing COVID-19 Therapeutic Drugs. Viruses, 16(11), 1726. https://doi.org/10.3390/v16111726