
Dimerization of Rabies Virus Phosphoprotein and Phosphorylation of Its Nucleoprotein Enhance Their Binding Affinity



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
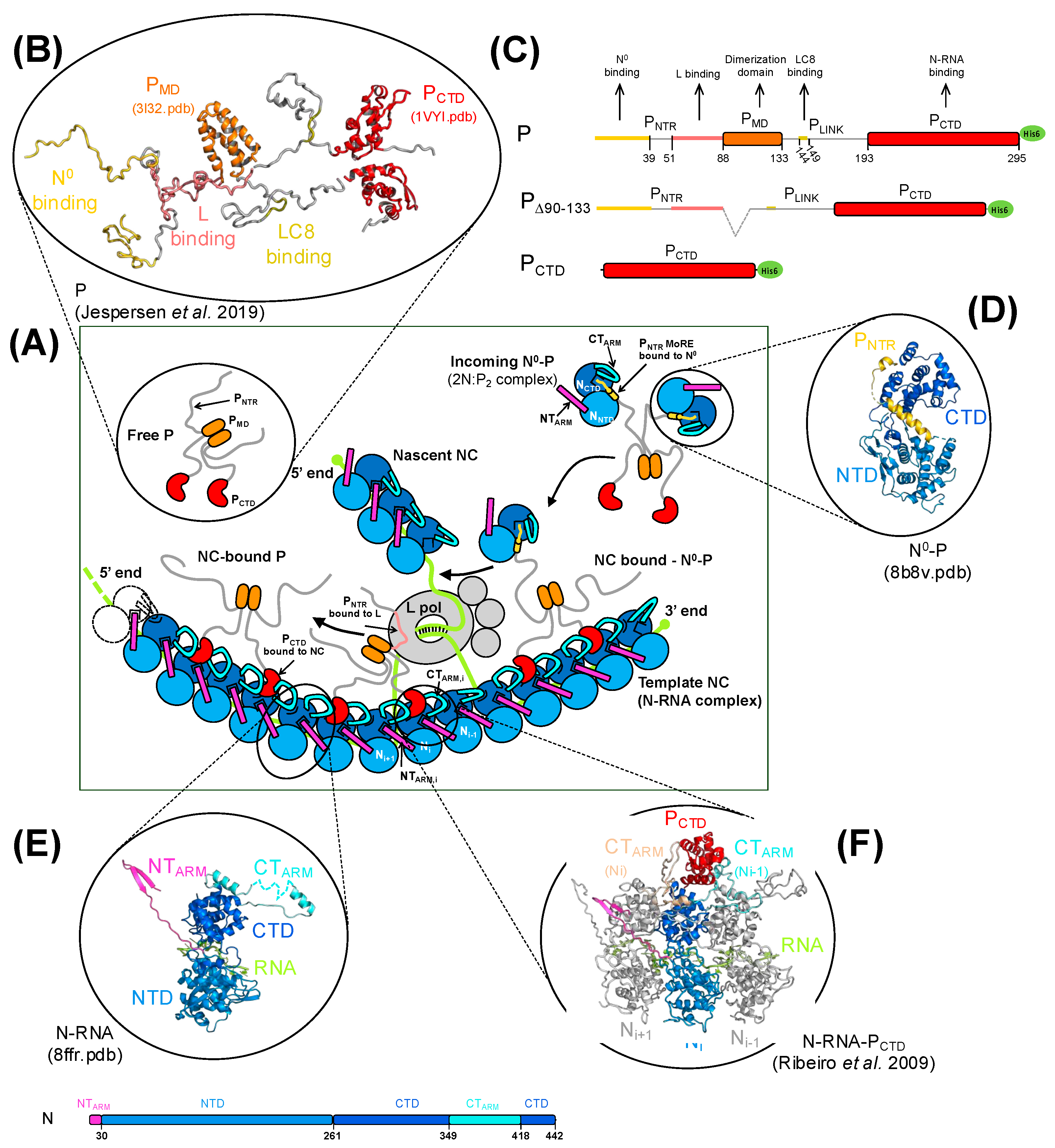
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Native Gel Electrophoresis
2.3. Western Blotting
2.4. Surface Plasmon Resonance
2.5. Modeling Equilibrium Binding Isotherms
2.5.1. Binding of Monomeric Ligand L to Independent Sites (Binding Model 1)
2.5.2. Binding of One Dimeric Ligand L to a Homo-Polymeric Complex (Binding Model 2)
2.5.3. Binding of Two or More of Dimeric Ligand L to a Homo-Polymeric Complex (Binding Model 3)
2.5.4. Calculation of SPR Signal
3. Results
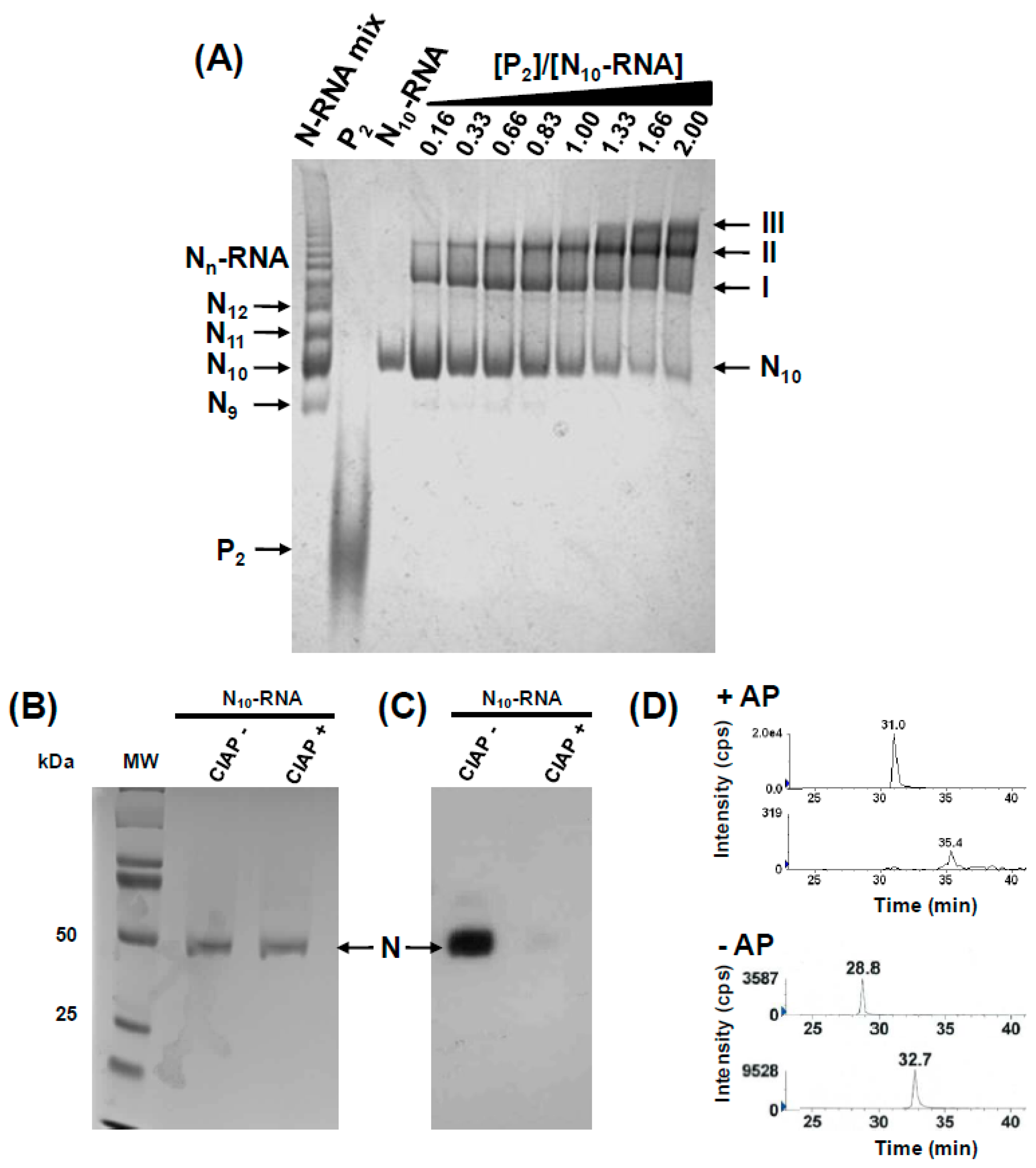
3.1. Binding of P Dimers to Phosphorylated Circular N10-RNA Complex
3.2. Phosphorylation of N Creates High-Affinity Binding Sites for P
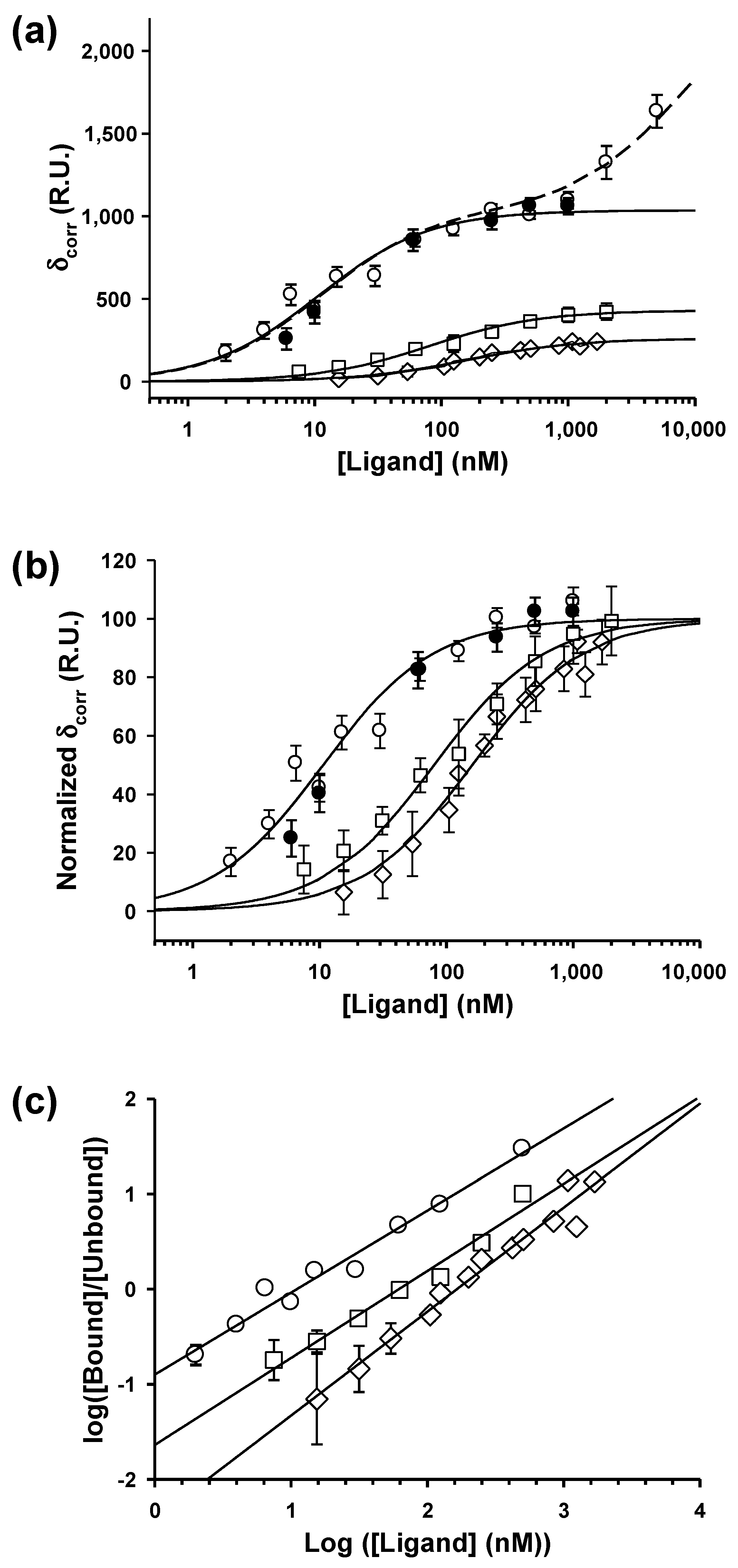
3.3. Equilibrium SPR Analysis
3.4. Dimerization Increases the Affinity of P for the N-RNA Template
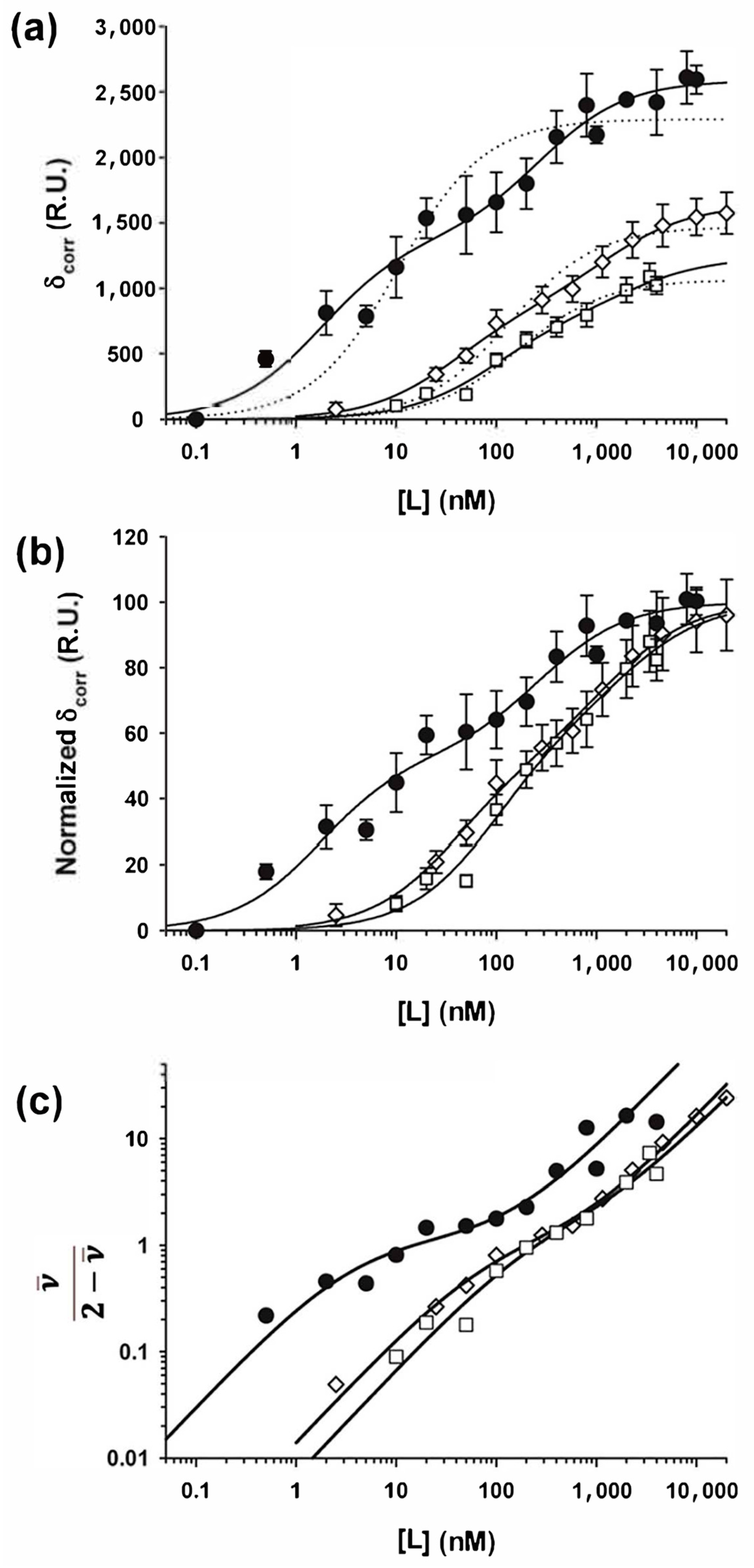
3.5. Interactions with Linear NCs
3.6. Phosphorylation Strongly Enhances the Binding Affinity of P Dimers
4. Discussion
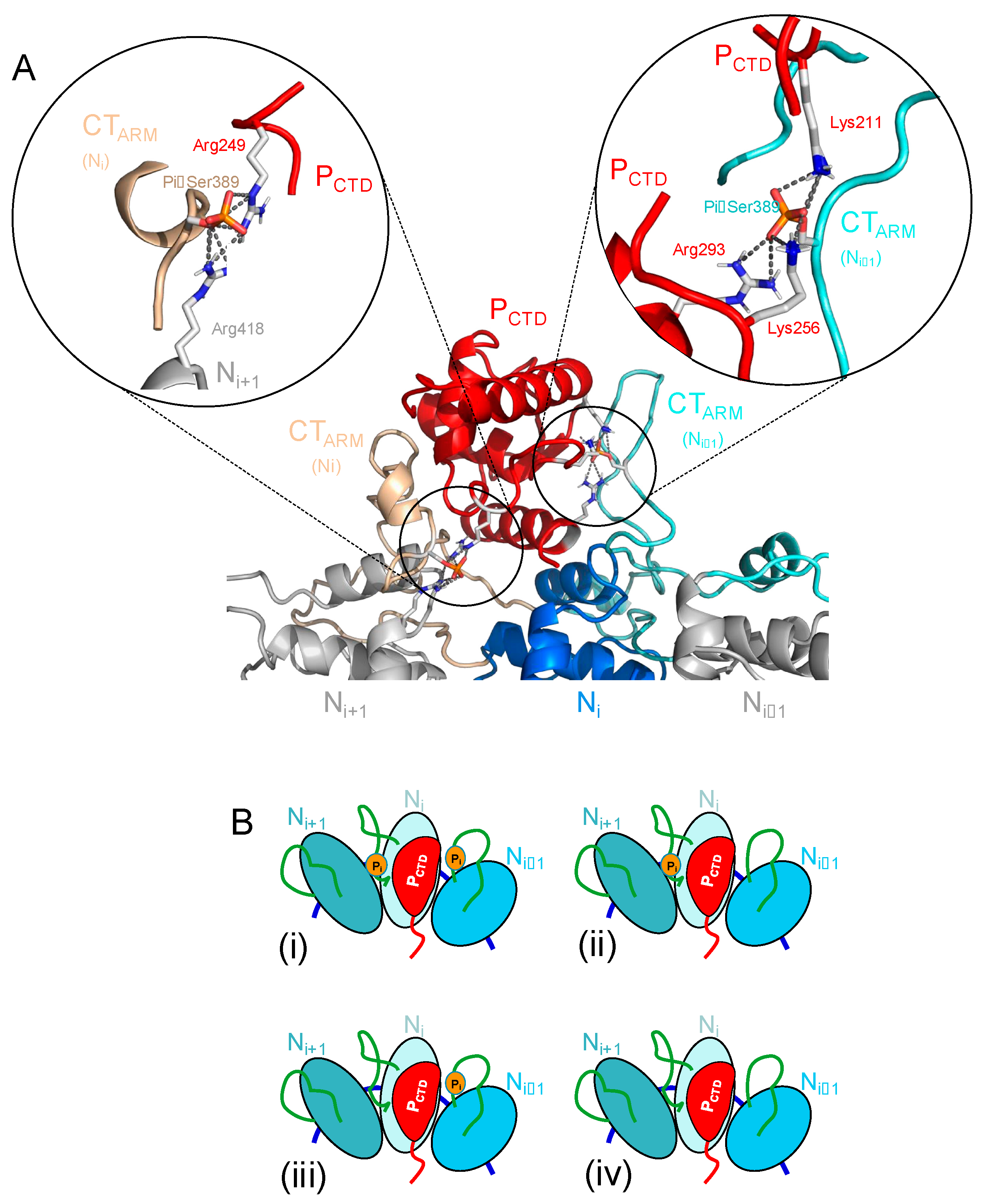
4.1. Phosphorylation of Ser389 in RABV N Enhances Binding Affinity for P CTD
4.2. RABV P Dimerization Moderately Enhances the Affinity for Multimeric N-RNA Complexes
4.3. Interactions Between RABV P and Polymeric N-RNA Complexes
4.4. Relevance of RABV P-N Interaction Dynamics for Polymerase Motion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tarantola, A. Four Thousand Years of Concepts Relating to Rabies in Animals and Humans, Its Prevention and Its Cure. Trop. Med. Infect. Dis. 2017, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.T.; Freuling, C.M. Rabies control in Europe: An overview of past, current and future strategies. Rev. Sci. Tech. 2018, 37, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Fehlner-Gardiner, C. Rabies control in North America—Past, present and future. Rev. Sci. Tech. 2018, 37, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Hampson, K.; Coudeville, L.; Lembo, T.; Sambo, M.; Kieffer, A.; Attlan, M.; Barrat, J.; Blanton, J.D.; Briggs, D.J.; Cleaveland, S.; et al. Estimating the global burden of endemic canine rabies. PLoS Negl. Trop. Dis. 2015, 9, e0003709. [Google Scholar]
- WHO Rabies—Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/rabies (accessed on 15 July 2024).
- Knobel, D.L.; Cleaveland, S.; Coleman, P.G.; Fèvre, E.M.; Meltzer, M.I.; Miranda, M.E.; Shaw, A.; Zinnstag, J.; Meslin, F.X. Re-evaluating the burden of rabies in Africa and Asia. Bull. World Health Organ. 2005, 83, 360–368. [Google Scholar]
- Dietzschold, B.; Li, J.; Faber, M.; Schnell, M. Concepts in the pathogenesis of rabies. Future Virol. 2008, 3, 481–490. [Google Scholar] [CrossRef]
- Schnell, M.J.; McGettigan, J.P.; Wirblich, C.; Papaneri, A. The cell biology of rabies virus: Using stealth to reach the brain. Nat. Rev. Microbiol. 2010, 8, 51–61. [Google Scholar] [CrossRef]
- Fooks, A.R.; Cliquet, F.; Finke, S.; Freuling, C.; Hemachudha, T.; Mani, R.S.; Muller, T.; Nadin-Davis, S.; Picard-Meyer, E.; Wilde, H.; et al. Rabies. Nat. Rev. Dis. Primers 2017, 3, 17091. [Google Scholar] [CrossRef]
- Fisher, C.R.; Streicker, D.G.; Schnell, M.J. The spread and evolution of rabies virus: Conquering new frontiers. Nat. Rev. Microbiol. 2018, 16, 241–255. [Google Scholar] [CrossRef]
- Litchy, B.D.; Power, A.T.; Stojdl, D.F.; Bell, J.C. Vesicular stomatitis virus: Re-inventing the bullet. Trends Mol. Med. 2004, 10, 210–216. [Google Scholar]
- Basak, A.; Mondal, A.; Polley, S.; Mukhopadhyay, S.; Chattopadhyay, D. Reviewing Chandipura: A Vesiculovirus in Human Epidemics. Biosci. Rep. 2007, 27, 275–298. [Google Scholar] [CrossRef] [PubMed]
- Hogenhout, S.A.; Redinbaugh, M.G.; Ammar, E.-D. Plant and animal rhabdovirus host range: A bug’s view. Trends Microbiol. 2003, 11, 264–271. [Google Scholar] [CrossRef]
- Pringle, C.R. The order Mononegavirales. Arch. Virol. 1997, 142, 2321–2326. [Google Scholar]
- Kuhn, J.H.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Amarasinghe, G.K.; Anthony, S.J.; Avsic-Zupanc, T.; Ayllon, M.A.; Bahl, J.; Balkema-Buschmann, A.; et al. 2020 taxonomic update for phylum Negarnaviricota (Riboviria: Orthornavirae), including the large orders Bunyavirales and Mononegavirales. Arch. Virol. 2020, 165, 3023–3072. [Google Scholar] [CrossRef]
- Albertini, A.A.; Wernimont, A.K.; Muziol, T.; Ravelli, R.B.; Clapier, C.R.; Schoehn, G.; Weissenhorn, W.; Ruigrok, R.W. Crystal structure of the rabies virus nucleoprotein-RNA complex. Science 2006, 313, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Morin, B.; Liang, B.; Gardner, E.; Ross, R.A.; Whelan, S.P.J. An In Vitro RNA Synthesis Assay for Rabies Virus Defines Ribonucleoprotein Interactions Critical for Polymerase Activity. J. Virol. 2017, 91, e01508-16. [Google Scholar] [CrossRef]
- Gérard, F.C.A.; Ribeiro, E.A.J.; Leyrat, C.; Ivanov, I.; Blondel, D.; Longhi, S.; Ruigrok, R.W.; Jamin, M. Modular organization of rabies virus phosphoprotein. J. Mol. Biol. 2009, 388, 978–996. [Google Scholar] [CrossRef] [PubMed]
- Chenik, M.; Chebli, K.; Blondel, D. Translation initiation at alternate in-frame AUG codons in the rabies virus phosphoprotein mRNA Is mediated by a ribosomal leaky scanning mechanism. J. Virol. 1995, 69, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, N.E.; Leyrat, C.; Gerard, F.C.; Bourhis, J.M.; Blondel, D.; Jamin, M.; Barbar, E. The LC8-RavP ensemble Structure Evinces A Role for LC8 in Regulating Lyssavirus Polymerase Functionality. J. Mol. Biol. 2019, 431, 4959–4977. [Google Scholar] [CrossRef]
- Scrima, N.; Le Bars, R.; Nevers, Q.; Glon, D.; Chevreux, G.; Civas, A.; Blondel, D.; Lagaudriere-Gesbert, C.; Gaudin, Y. Rabies virus P protein binds to TBK1 and interferes with the formation of innate immunity-related liquid condensates. Cell Rep. 2023, 42, 111949. [Google Scholar] [CrossRef]
- Ito, N.; Moseley, G.W.; Sugiyama, M. The importance of immune evasion in the pathogenesis of rabies virus. J. Vet. Med. Sci. 2016, 78, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Vidy, A.; Chelbi-Alix, M.; Blondel, D. Rabies virus P protein interacts with STAT1 and inhibits interferon signal transduction pathways. J. Virol. 2005, 79, 14411–14420. [Google Scholar] [CrossRef] [PubMed]
- Gérard, F.C.A.; Ribeiro, E.A.J.; Albertini, A.A.; Gutsche, I.; Zaccai, G.; Ruigrok, R.W.; Jamin, M. Unphosphorylated Rhabdoviridae phosphoproteins form elongated dimers in solution. Biochemistry 2007, 46, 10328–10338. [Google Scholar] [CrossRef]
- Ivanov, I.; Crepin, T.; Jamin, M.; Ruigrok, R.W. Structure of the dimerization domain of the rabies virus phosphoprotein. J. Virol. 2010, 84, 3707–3710. [Google Scholar] [CrossRef]
- Mavrakis, M.; McCarthy, A.A.; Roche, S.; Blondel, D.; Ruigrok, R.W. Structure and function of the C-terminal domain of the polymerase cofactor of rabies virus. J. Mol. Biol. 2004, 343, 819–831. [Google Scholar] [CrossRef]
- Gérard, F.C.A.; Bourhis, J.M.; Mas, C.; Branchard, A.; Vu, D.D.; Varhoshkova, S.; Leyrat, C.; Jamin, M. Structure and Dynamics of the Unassembled Nucleoprotein of Rabies Virus in Complex with Its Phosphoprotein Chaperone Module. Viruses 2022, 14, 2813. [Google Scholar] [CrossRef]
- Mavrakis, M.; Iseni, F.; Mazza, C.; Schoehn, G.; Ebel, C.; Gentzel, M.; Franz, T.; Ruigrok, R.W. Isolation and characterisation of the rabies virus N degrees-P complex produced in insect cells. Virology 2003, 305, 406–414. [Google Scholar] [CrossRef]
- Howard, M.; Wertz, G. Vesicular Stomatitis Virus RNA Replication: A Role for the NS Protein. J. Gen. Virol. 1989, 70, 2683–2694. [Google Scholar] [CrossRef] [PubMed]
- Masters, P.S.; Banerjee, A.K. Complex Formation with Vesicular Stomatitis Virus Phosphoprotein NS prevents Binding of Nucleocapsid Protein N to Nonspecific RNA. J. Virol. 1988, 62, 2658–2664. [Google Scholar] [CrossRef]
- Mavrakis, M.; Mehouas, S.; Real, E.; Iseni, F.; Blondel, D.; Tordo, N.; Ruigrok, R.W. Rabies virus chaperone: Identification of the phosphoprotein peptide that keeps nucleoprotein soluble and free from non-specific RNA. Virology 2006, 349, 422–429. [Google Scholar] [CrossRef]
- Horwitz, J.A.; Jenni, S.; Harrison, S.C.; Whelan, S.P.J. Structure of a rabies virus polymerase complex from electron cryo-microscopy. Proc. Natl. Acad. Sci. USA 2020, 117, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, E.A.J.; Leyrat, C.; Gerard, F.C.; Albertini, A.A.; Falk, C.; Ruigrok, R.W.; Jamin, M. Binding of rabies virus polymerase cofactor to recombinant circular nucleoprotein-RNA complexes. J. Mol. Biol. 2009, 394, 558–575. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Watts, E.; Brice, A.M.; Metcalfe, R.D.; Rozario, A.M.; Sethi, A.; Yan, F.; Bell, T.D.M.; Griffin, M.D.W.; Moseley, G.W.; et al. Molecular Basis of Functional Effects of Phosphorylation of the C-Terminal Domain of the Rabies Virus P Protein. J. Virol. 2022, 96, e00111-22. [Google Scholar] [CrossRef]
- Nikolic, J.; Le Bars, R.; Lama, Z.; Scrima, N.; Lagaudriere-Gesbert, C.; Gaudin, Y.; Blondel, D. Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 2017, 8, 58. [Google Scholar] [CrossRef]
- Lahaye, X.; Vidy, A.; Pomier, C.; Obiang, L.; Harper, F.; Gaudin, Y.; Blondel, D. Functional Characterization of Negri Bodies (NBs) in Rabies Virus-Infected Cells: Evidence that NBs Are Sites of Viral Transcription and Replication. J. Virol. 2009, 83, 7948–7958. [Google Scholar] [CrossRef]
- Ding, H.; Green, T.J.; Lu, S.; Luo, M. Crystal structure of the oligomerization domain of the phosphoprotein of vesicular stomatitis virus. J. Virol. 2006, 80, 2808–2814. [Google Scholar] [CrossRef]
- Jensen, M.R.; Yabukarski, F.; Communie, G.; Condamine, E.; Mas, C.; Volchkova, V.; Tarbouriech, N.; Bourhis, J.M.; Volchkov, V.; Blackledge, M.; et al. Structural Description of the Nipah Virus Phosphoprotein and Its Interaction with STAT1. Biophys. J. 2020, 118, 2470–2488. [Google Scholar] [CrossRef] [PubMed]
- Tarbouriech, N.; Curran, J.; Ebel, C.; Ruigrok, R.W.; Burmeister, W.P. On the domain structure and the polymerization state of the sendai virus P protein. Virology 2000, 266, 99–109. [Google Scholar] [CrossRef]
- Bruhn, J.F.; Kirchdoerfer, R.N.; Urata, S.M.; Li, S.; Tickle, I.J.; Bricogne, G.; Saphire, E.O. Crystal Structure of the Marburg Virus VP35 Oligomerization Domain. J. Virol. 2017, 91, e01085-16. [Google Scholar] [CrossRef]
- Leyrat, C.; Renner, M.; Harlos, K.; Grimes, J.M. Solution and crystallographic structures of the central region of the phosphoprotein from human metapneumovirus. PLoS ONE 2013, 8, e80371. [Google Scholar] [CrossRef]
- Curran, J. Reexamination of the Sendai virus P protein domains required for RNA synthesis: A possible supplemental role for the P protein. Virology 1996, 221, 130–140. [Google Scholar] [CrossRef]
- Bourhis, J.M.; Yabukarski, F.; Communie, G.; Schneider, R.; Volchkova, V.A.; Freneat, M.; Gerard, F.C.; Ducournau, C.; Mas, C.; Tarbouriech, N.; et al. Structural Dynamics of the C-terminal X Domain of Nipah and Hendra Viruses Controls the Attachment to the C-terminal Tail of the Nucleocapsid Protein. J. Mol. Biol. 2022, 434, 167551. [Google Scholar] [CrossRef] [PubMed]
- Sokol, F.; Clark, H.F. Phosphoproteins, structural components of rhabdoviruses. Virology 1973, 52, 246–263. [Google Scholar] [CrossRef]
- Yang, J.; Koprowski, H.; Dietzschold, B.; Fu, Z.F. Phosphorylation of rabies virus nucleoprotein regulates viral RNA transcription and replication by modulating leader RNA encapsidation. J. Virol. 1999, 73, 1661–1664. [Google Scholar] [CrossRef]
- Wu, X.; Gong, X.; Foley, H.D.; Schnell, M.J.; Fu, Z.F. Both viral transcription and replication are reduced when the rabies virus nucleoprotein is not phosphorylated. J. Virol. 2002, 76, 4153–4161. [Google Scholar] [CrossRef] [PubMed]
- Préhaud, C.; Harris, R.D.; Fulop, V.; Koh, C.; Wong, J.; Flamand, A.; Bishop, D.H.L. Expression, characterization, and purification of a phosphorylated rabies nucleoprotein synthesized in insect cells by baculovirus vectors. Virology 1990, 178, 486–497. [Google Scholar] [CrossRef]
- Dietzschold, B.; Lafon, M.; Wang, H.; Otvos, L., Jr.; Celis, E.; Wunner, W.H.; Koprowski, H. Localization and immunological characterization of antigenic domains of the rabies virus internal N and NS proteins. Virus Res. 1987, 8, 103–125. [Google Scholar] [CrossRef]
- Wu, X.; Lei, X.; Fu, Z.F. Rabies virus nucleoprotein is phosphorylated by cellular casein kinase II. Biochem. Biophys. Res. Commun. 2003, 304, 333–338. [Google Scholar] [CrossRef]
- Kawai, A.; Toriumi, H.; Tochikura, T.S.; Takahashi, T.; Honda, Y.; Morimoto, K. Nucleocapsid formation and/or subsequent conformational change of rabies virus nucleoprotein (N) is a prerequisite step for acquiring the phosphatase-sensitive epitope of monoclonal antibody 5-2-26. Virology 1999, 263, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Toriumi, H.; Kawai, A. Association of rabies virus nominal phosphoprotein (P) with viral nucleocapsid (NC) is enhanced by phosphorylation of the viral nucleoprotein (N). Microbiol. Immunol. 2004, 48, 399–409. [Google Scholar] [CrossRef]
- Leyrat, C.; Schneider, R.; Ribeiro, E.A., Jr.; Yabukarski, F.; Yao, M.; Gerard, F.C.; Jensen, M.R.; Ruigrok, R.W.; Blackledge, M.; Jamin, M. Ensemble structure of the modular and flexible full-length vesicular stomatitis virus phosphoprotein. J. Mol. Biol. 2012, 423, 182–197. [Google Scholar] [CrossRef]
- Edelhoch, H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 1967, 6, 1948–1954. [Google Scholar] [CrossRef] [PubMed]
- Albertini, A.A.; Clapier, C.R.; Wernimont, A.K.; Schoehn, G.; Weissenhorn, W.; Ruigrok, R.W. Isolation and crystallization of a unique size category of recombinant Rabies virus Nucleoprotein-RNA rings. J. Struct. Biol. 2007, 158, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Jencks, W.P. On the attribution and additivity of binding energies. Proc. Natl. Acad. Sci. USA 1991, 78, 4046–4050. [Google Scholar] [CrossRef]
- McGhee, J.D.; von Hippel, P.H. Theoretical aspects of DNA-Protein Interactions: Co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 1974, 86, 469–489. [Google Scholar] [CrossRef]
- Neri, D.; Momo, M.; Prospero, T.; Winter, G. High-affinity Antigen Binding by Chelating Recombinant Antibodies (CRAbs). J. Mol. Biol. 1995, 246, 367–373. [Google Scholar] [CrossRef]
- Crothers, D.M.; Metzger, H. The influence of polyvalency on the binding properties of antibodies. Immunochemistry 1972, 9, 341–357. [Google Scholar] [CrossRef]
- Klemm, J.D.; Pabo, C.O. Oct-1 POU domain-DNA interactions: Cooperative binding of isolated subdomams and effects of covalent linkage. Genes Dev. 1996, 10, 27–36. [Google Scholar] [CrossRef]
- Zhou, H.-X. Loops in Proteins Can Be Modeled as Worm-Like Chains. J. Phys. Chem. B 2001, 105, 6763–6766. [Google Scholar] [CrossRef]
- Zhou, H.-X. Quantitative Account of the Enhanced Affinity of Two Linked scFvs Specific for Different Epitopes on the Same Antigen. J. Mol. Biol. 2003, 329, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jacob, Y.; Real, E.; Tordo, N. Functional interaction map of lyssavirus phosphoprotein: Identification of the minimal transcription domains. J. Virol. 2001, 75, 9613–9622. [Google Scholar] [CrossRef] [PubMed]
- Gérard, F.C.A.; Jamin, M.; Blackledge, M.; Blondel, D.; Bourhis, J.M. Vesicular Stomatitis Virus Phosphoprotein Dimerization Domain Is Dispensable for Virus Growth. J. Virol. 2020, 94, e01789-19. [Google Scholar] [CrossRef] [PubMed]
- Bloyet, L.M.; Morin, B.; Brusic, V.; Gardner, E.; Ross, R.A.; Vadakkan, T.; Kirchhausen, T.; Whelan, S.P.J. Oligomerization of the Vesicular Stomatitis Virus Phosphoprotein Is Dispensable for mRNA Synthesis but Facilitates RNA Replication. J. Virol. 2020, 94, e00115-20. [Google Scholar] [CrossRef]
- Nevers, Q.; Scrima, N.; Glon, D.; Le Bars, R.; Decombe, A.; Garnier, N.; Ouldali, M.; Lagaudriere-Gesbert, C.; Blondel, D.; Albertini, A.; et al. Properties of rabies virus phosphoprotein and nucleoprotein biocondensates formed in vitro and in cellulo. PLoS Pathog. 2022, 18, e1011022. [Google Scholar] [CrossRef]
- Thomas, D.; Newcomb, W.W.; Brown, J.C.; Wall, J.S.; Hainfeld, J.F.; Trus, B.L.; Steven, A.C. Mass and molecular composition of vesicular stomatitis virus: A scanning transmission electron microscopy analysis. J. Virol. 1985, 54, 598–607. [Google Scholar] [CrossRef]
- Zaccai, N.; Serdyuk, I.N.; Zaccai, G. Methods in Molecular Biophysics—Structure, Dynamics, Function for Biology and Medicine, 2nd ed.; Cambridge University Press: Cambridge, UK, 2017. [Google Scholar]
- Stillman, E.A.; Whitt, M. Transcript initiation and 5’-end modifications are separable events during vesicular stomatitis virus transcription. J. Virol. 1999, 73, 7199–7209. [Google Scholar] [CrossRef]
- Morin, B.; Rahmeh, A.A.; Whelan, S.P. Mechanism of RNA synthesis initiation by the vesicular stomatitis virus polymerase. Embo J. 2012, 31, 1320–1329. [Google Scholar] [CrossRef]
- Kolakofsky, D.; Le Mercier, P.; Iseni, F.; Garcin, D. Viral RNA polymerase scanning and the gymnastics of Sendai virus RNA synthesis. Virology 2004, 318, 463–473. [Google Scholar] [CrossRef]
- Barr, J.N.; Whelan, S.P.; Wertz, G.W. Transcriptional control of the RNA-dependent RNA polymerase of vesicular stomatitis virus. Biochim. Biophys. Acta 2002, 1577, 337–353. [Google Scholar] [CrossRef]
- Gubbay, O.; Curran, J.; Kolakofsky, D. Sendai virus genome synthesis and assembly are coupled: A possible mechanism to promote viral RNA polymerase processivity. J. Gen. Virol. 2001, 82 Pt 12, 2895–2903. [Google Scholar] [CrossRef]
- Plumet, S.; Duprex, W.P.; Gerlier, D. Dynamics of viral RNA synthesis during measles virus infection. J. Virol. 2005, 79, 6900–6908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Feng, Y.; Tu, Z.; Lou, Z.; Tu, C. Proteomic Profiling of Purified Rabies Virus Particles. Virol. Sin. 2020, 35, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Richard, C.A.; Rincheval, V.; Lassoued, S.; Fix, J.; Cardone, C.; Esneau, C.; Nekhai, S.; Galloux, M.; Rameix-Welti, M.A.; Sizun, C.; et al. RSV hijacks cellular protein phosphatase 1 to regulate M2-1 phosphorylation and viral transcription. PLoS Pathog. 2018, 14, e1006920. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Sato, H.; Hagiwara, K.; Watanabe, A.; Sugai, A.; Ikeda, F.; Kozuka-Hata, H.; Oyama, M.; Yoneda, M.; Kai, C. Determination of a phosphorylation site in Nipah virus nucleoprotein and its involvement in virus transcription. J. Gen. Virol. 2011, 92 Pt 9, 2133–2141. [Google Scholar] [CrossRef]
- Kruse, T.; Biedenkopf, N.; Thrane Hertz, E.P.; Dietzel, E.; Stalmann, G.; Lopez-Mendez, B.; Davey, N.E.; Nilsson, J.; Becker, S. The Ebola Virus Nucleoprotein Recruits the Host PP2A-B56 Phosphatase to Activate Transcriptional Support Activity of VP30. Mol. Cell 2018, 69, 136–145. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, E.d.A., Jr.; Leyrat, C.; Gérard, F.C.A.; Jamin, M. Dimerization of Rabies Virus Phosphoprotein and Phosphorylation of Its Nucleoprotein Enhance Their Binding Affinity. Viruses 2024, 16, 1735. https://doi.org/10.3390/v16111735
Ribeiro EdA Jr., Leyrat C, Gérard FCA, Jamin M. Dimerization of Rabies Virus Phosphoprotein and Phosphorylation of Its Nucleoprotein Enhance Their Binding Affinity. Viruses. 2024; 16(11):1735. https://doi.org/10.3390/v16111735
Chicago/Turabian StyleRibeiro, Euripedes de Almeida, Jr., Cédric Leyrat, Francine C. A. Gérard, and Marc Jamin. 2024. "Dimerization of Rabies Virus Phosphoprotein and Phosphorylation of Its Nucleoprotein Enhance Their Binding Affinity" Viruses 16, no. 11: 1735. https://doi.org/10.3390/v16111735
APA StyleRibeiro, E. d. A., Jr., Leyrat, C., Gérard, F. C. A., & Jamin, M. (2024). Dimerization of Rabies Virus Phosphoprotein and Phosphorylation of Its Nucleoprotein Enhance Their Binding Affinity. Viruses, 16(11), 1735. https://doi.org/10.3390/v16111735