
Epigenetic Restriction Factors (eRFs) in Virus Infection

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Virus Epigenetic Restriction Factors (eRFs)
2.1. eRFs That Target Viral Chromatin
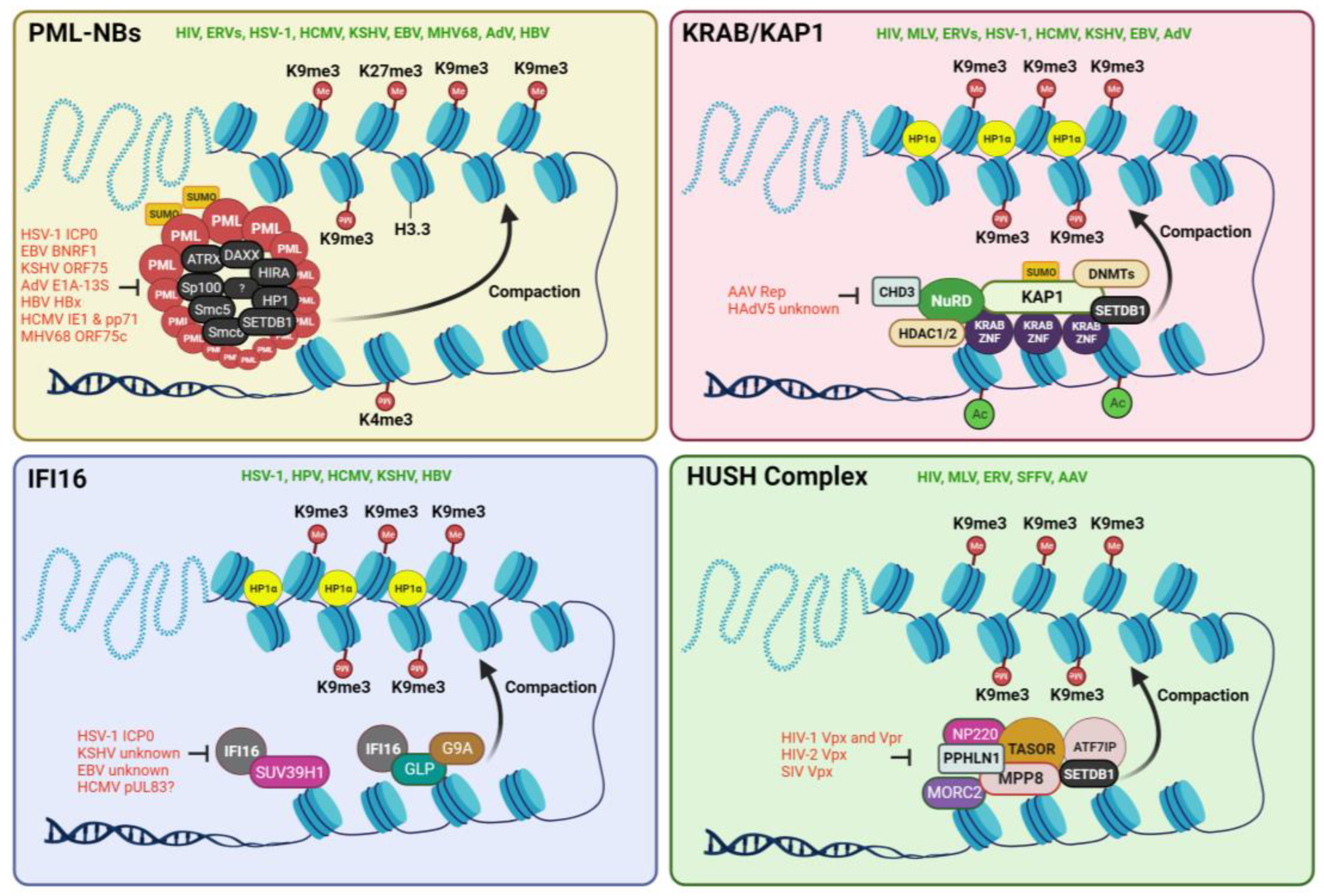
2.1.1. PML-NBs or ND10
2.1.2. KRAB/KAP1 Complex
2.1.3. IFI16
2.1.4. The HUSH Complex
2.2. Epitranscriptomic eRFs
2.2.1. Adenosine Deaminase Acting on RNA (ADAR)
2.2.2. Pseudouridine Synthases (PUS)
2.2.3. N6-Methyladenosine (m6A) Writers, Readers, and Erasers
3. Future Directions
3.1. The Possibility of a Supramolecular ‘Restrictosome’
3.2. Crosstalk between Epitranscriptomics and Epigenetics
3.3. Epidrugs and the Potential of Targeting eRFs
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, L.; Liu, S.; Chen, Z.J. SnapShot: Pathways of antiviral innate immunity. Cell 2010, 140, 436–436.e2. [Google Scholar] [CrossRef]
- Zuniga, E.I.; Macal, M.; Lewis, G.M.; Harker, J.A. Innate and Adaptive Immune Regulation During Chronic Viral Infections. Annu. Rev. Virol. 2015, 2, 573–597. [Google Scholar] [CrossRef] [PubMed]
- Chemudupati, M.; Kenney, A.D.; Bonifati, S.; Zani, A.; McMichael, T.M.; Wu, L.; Yount, J.S. From APOBEC to ZAP: Diverse mechanisms used by cellular restriction factors to inhibit virus infections. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2019, 1866, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Chen, Z.J. Intrinsic antiviral immunity. Nat. Immunol. 2012, 13, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Wang, L.; Ding, H.; Schwamborn, J.C.; Li, S.; Dorf, M.E. TRIM32 Senses and Restricts Influenza A Virus by Ubiquitination of PB1 Polymerase. PLoS Pathog. 2015, 11, e1004960. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Kochs, G. Mx genes: Host determinants controlling influenza virus infection and trans-species transmission. Hum. Genet. 2020, 139, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Stavrou, S.; Ross, S.R. APOBEC3 Proteins in Viral Immunity. J. Immunol. 2015, 195, 4565–4570. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Cullen, B.R. Epigenetic and epitranscriptomic regulation of viral replication. Nat. Rev. Microbiol. 2020, 18, 559–570. [Google Scholar] [CrossRef]
- Gifford, R.; Tristem, M. The evolution, distribution and diversity of endogenous retroviruses. Virus Genes 2003, 26, 291–315. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar]
- Xu, X.; Zhao, H.; Gong, Z.; Han, G.-Z. Endogenous retroviruses of non-avian/mammalian vertebrates illuminate diversity and deep history of retroviruses. PLoS Pathog. 2018, 14, e1007072. [Google Scholar] [CrossRef]
- Mi, S.; Lee, X.; Li, X.-P.; Veldman, G.M.; Finnerty, H.; Racie, L.; LaVallie, E.; Tang, X.-Y.; Edouard, P.; Howes, S.; et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 2000, 403, 785–789. [Google Scholar] [CrossRef]
- Badarinarayan, S.S.; Sauter, D. Switching Sides: How Endogenous Retroviruses Protect Us from Viral Infections. J. Virol. 2021, 95, e02299-20. [Google Scholar] [CrossRef]
- Shimode, S.; Nakagawa, S.; Miyazawa, T. Multiple invasions of an infectious retrovirus in cat genomes. Sci. Rep. 2015, 5, 8164. [Google Scholar] [CrossRef]
- Yuan, P.; Yan, J.; Wang, S.; Guo, Y.; Xi, X.; Han, S.; Yin, J.; Peng, B.; He, X.; Bodem, J.; et al. Trim28 acts as restriction factor of prototype foamy virus replication by modulating H3K9me3 marks and destabilizing the viral transactivator Tas. Retrovirology 2021, 18, 38. [Google Scholar] [CrossRef]
- Wang, X.; Xia, H.; Liu, S.; Cao, L.; You, F. Epigenetic regulation in antiviral innate immunity. Eur. J. Immunol. 2021, 51, 1641–1651. [Google Scholar] [CrossRef] [PubMed]
- Ngo, A.M.; Puschnik, A.S. Genome-Scale Analysis of Cellular Restriction Factors That Inhibit Transgene Expression from Adeno-Associated Virus Vectors. J. Virol. 2023, 97, e0194822. [Google Scholar] [CrossRef] [PubMed]
- Lallemand-Breitenbach, V.; de The, H. PML nuclear bodies: From architecture to function. Curr. Opin. Cell Biol. 2018, 52, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Corpet, A.; Kleijwegt, C.; Roubille, S.; Juillard, F.; Jacquet, K.; Texier, P.; Lomonte, P. PML nuclear bodies and chromatin dynamics: Catch me if you can! Nucleic Acids Res. 2020, 48, 11890–11912. [Google Scholar] [CrossRef] [PubMed]
- Ascoli, C.A.; Maul, G.G. Identification of a novel nuclear domain. J. Cell Biol. 1991, 112, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Maroui, M.A.; El Asmi, F.; Dutrieux, J.; Chelbi-Alix, M.K.; Nisole, S. Implication of PML nuclear bodies in intrinsic and innate immunity. Med. Sci. 2014, 30, 765–771. [Google Scholar]
- Ryabchenko, B.; Šroller, V.; Horníková, L.; Lovtsov, A.; Forstová, J.; Huérfano, S. The interactions between PML nuclear bodies and small and medium size DNA viruses. Virol. J. 2023, 20, 82. [Google Scholar] [CrossRef] [PubMed]
- Scherer, M.; Stamminger, T. Emerging Role of PML Nuclear Bodies in Innate Immune Signaling. J. Virol. 2016, 90, 5850–5854. [Google Scholar] [CrossRef]
- Tsai, K.; Chan, L.; Gibeault, R.; Conn, K.; Dheekollu, J.; Domsic, J.; Marmorstein, R.; Schang, L.M.; Lieberman, P.M. Viral reprogramming of the Daxx histone H3.3 chaperone during early Epstein-Barr virus infection. J. Virol. 2014, 88, 14350–14363. [Google Scholar] [CrossRef]
- Full, F.; Ensser, A. Early Nuclear Events after Herpesviral Infection. J. Clin. Med. 2019, 8, 1408. [Google Scholar] [CrossRef] [PubMed]
- Lusic, M.; Marini, B.; Ali, H.; Lucic, B.; Luzzati, R.; Giacca, M. Proximity to PML Nuclear Bodies Regulates HIV-1 Latency in CD4+ T Cells. Cell Host Microbe 2013, 13, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Dutrieux, J.; Maarifi, G.; Portilho, D.M.; Arhel, N.J.; Chelbi-Alix, M.K.; Nisole, S. PML/TRIM19-Dependent Inhibition of Retroviral Reverse-Transcription by Daxx. PLoS Pathog. 2015, 11, e1005280. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.; Corpet, A.; Roubille, S.; Maroui, M.A.; Poccardi, N.; Rousseau, A.; Kleijwegt, C.; Binda, O.; Texier, P.; Sawtell, N.; et al. Promyelocytic leukemia (PML) nuclear bodies (NBs) induce latent/quiescent HSV-1 genomes chromatinization through a PML NB/Histone H3.3/H3.3 Chaperone Axis. PLoS Pathog. 2018, 14, e1007313. [Google Scholar] [CrossRef] [PubMed]
- Cabral, J.M.; Oh, H.S.; Knipe, D.M. ATRX promotes maintenance of herpes simplex virus heterochromatin during chromatin stress. eLife 2018, 7, e40228. [Google Scholar] [CrossRef]
- Cabral, J.M.; Cushman, C.H.; Sodroski, C.N.; Knipe, D.M. ATRX limits the accessibility of histone H3-occupied HSV genomes during lytic infection. PLoS Pathog. 2021, 17, e1009567. [Google Scholar] [CrossRef]
- Woodhall, D.L.; Groves, I.J.; Reeves, M.B.; Wilkinson, G.; Sinclair, J.H. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 2006, 281, 37652–37660. [Google Scholar] [CrossRef] [PubMed]
- Albright, E.R.; Kalejta, R.F. Canonical and Variant Forms of Histone H3 Are Deposited onto the Human Cytomegalovirus Genome during Lytic and Latent Infections. J. Virol. 2016, 90, 10309–10320. [Google Scholar] [CrossRef] [PubMed]
- Lukashchuk, V.; McFarlane, S.; Everett, R.D.; Preston, C.M. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 2008, 82, 12543–12554. [Google Scholar] [CrossRef] [PubMed]
- Mitra, B.; Guo, H. Hepatitis B virus X protein crosses out Smc5/6 complex to maintain covalently closed circular DNA transcription. Hepatology 2016, 64, 2246–2249. [Google Scholar] [CrossRef] [PubMed]
- Saffert, R.T.; Kalejta, R.F. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 2006, 80, 3863–3871. [Google Scholar] [CrossRef] [PubMed]
- Tavalai, N.; Papior, P.; Rechter, S.; Stamminger, T. Nuclear domain 10 components promyelocytic leukemia protein and hDaxx independently contribute to an intrinsic antiviral defense against human cytomegalovirus infection. J. Virol. 2008, 82, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Wagenknecht, N.; Reuter, N.; Scherer, M.; Reichel, A.; Müller, R.; Stamminger, T. Contribution of the Major ND10 Proteins PML, hDaxx and Sp100 to the Regulation of Human Cytomegalovirus Latency and Lytic Replication in the Monocytic Cell Line THP-1. Viruses 2015, 7, 2884–2907. [Google Scholar] [CrossRef]
- Full, F.; Jungnickl, D.; Reuter, N.; Bogner, E.; Brulois, K.; Scholz, B.; Stürzl, M.; Myoung, J.; Jung, J.U.; Stamminger, T.; et al. Kaposi’s sarcoma associated herpesvirus tegument protein ORF75 is essential for viral lytic replication and plays a critical role in the antagonization of ND10-instituted intrinsic immunity. PLoS Pathog. 2014, 10, e1003863. [Google Scholar] [CrossRef]
- Schreiner, S.; Bürck, C.; Glass, M.; Groitl, P.; Wimmer, P.; Kinkley, S.; Mund, A.; Everett, R.D.; Dobner, T. Control of human adenovirus type 5 gene expression by cellular Daxx/ATRX chromatin-associated complexes. Nucleic Acids Res. 2013, 41, 3532–3550. [Google Scholar] [CrossRef]
- Li, Y.; He, M.; Gong, R.; Wang, Z.; Lu, L.; Peng, S.; Duan, Z.; Feng, Y.; Liu, Y.; Gao, B. Forkhead O Transcription Factor 4 Restricts HBV Covalently Closed Circular DNA Transcription and HBV Replication through Genetic Downregulation of Hepatocyte Nuclear Factor 4 Alpha and Epigenetic Suppression of Covalently Closed Circular DNA via Interacting with Promyelocytic Leukemia Protein. J. Virol. 2022, 96, e0054622. [Google Scholar]
- Decorsière, A.; Mueller, H.; Van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Livingston, C.M.; Li, L.; Beran, R.K.; Daffis, S.; Ramakrishnan, D.; Burdette, D.; Peiser, L.; Salas, E.; Ramos, H.; et al. The Smc5/6 Complex Restricts HBV when Localized to ND10 without Inducing an Innate Immune Response and Is Counteracted by the HBV X Protein Shortly after Infection. PLoS ONE 2017, 12, e0169648. [Google Scholar] [CrossRef] [PubMed]
- Berscheminski, J.; Groitl, P.; Dobner, T.; Wimmer, P.; Schreiner, S. The adenoviral oncogene E1A-13S interacts with a specific isoform of the tumor suppressor PML to enhance viral transcription. J. Virol. 2013, 87, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Fada, B.J.; Guha, U.; Zheng, Y.; Reward, E.; Kaadi, E.; Dourra, A.; Gu, H. A Novel Recognition by the E3 Ubiquitin Ligase of HSV-1 ICP0 Enhances the Degradation of PML Isoform I to Prevent ND10 Reformation in Late Infection. Viruses 2023, 15, 1070. [Google Scholar] [CrossRef]
- Kim, Y.-E.; Lee, J.-H.; Kim, E.T.; Shin, H.J.; Gu, S.Y.; Seol, H.S.; Ling, P.D.; Lee, C.H.; Ahn, J.-H. Human cytomegalovirus infection causes degradation of Sp100 proteins that suppress viral gene expression. J. Virol. 2011, 85, 11928–11937. [Google Scholar] [CrossRef] [PubMed]
- Lanfranca, M.P.; Mostafa, H.H.; Davido, D.J. HSV-1 ICP0: An E3 Ubiquitin Ligase That Counteracts Host Intrinsic and Innate Immunity. Cells 2014, 3, 438–454. [Google Scholar] [CrossRef] [PubMed]
- Maul, G.G.; Everett, R.D. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J. Gen. Virol. 1994, 75, 1223–1233. [Google Scholar] [CrossRef]
- Tsai, K.; Thikmyanova, N.; Wojcechowskyj, J.A.; Delecluse, H.-J.; Lieberman, P.M. EBV tegument protein BNRF1 disrupts DAXX-ATRX to activate viral early gene transcription. PLoS Pathog. 2011, 7, e1002376. [Google Scholar] [CrossRef]
- Lee, H.-R.; Kim, D.-J.; Lee, J.-M.; Choi, C.Y.; Ahn, B.-Y.; Hayward, G.S.; Ahn, J.-H. Ability of the human cytomegalovirus IE1 protein to modulate sumoylation of PML correlates with its functional activities in transcriptional regulation and infectivity in cultured fibroblast cells. J. Virol. 2004, 78, 6527–6542. [Google Scholar] [CrossRef]
- Marcos-Villar, L.; Lopitz-Otsoa, F.; Gallego, P.; Munoz-Fontela, C.; Gonzalez-Santamaria, J.; Campagna, M.; Jiang, G.S.; Rodriguez, M.S.; Rivas, C. Kaposi’s sarcoma-associated herpesvirus protein LANA2 disrupts PML oncogenic domains and inhibits PML-mediated transcriptional repression of the survivin gene. J. Virol. 2009, 83, 8849–8858. [Google Scholar] [CrossRef]
- Dooley, A.L.; O’connor, C.M. Regulation of the MIE Locus During HCMV Latency and Reactivation. Pathogens 2020, 9, 869. [Google Scholar] [CrossRef]
- Catez, F.; Picard, C.; Held, K.; Gross, S.; Rousseau, A.; Theil, D.; Sawtell, N.; Labetoulle, M.; Lomonte, P. HSV-1 genome subnuclear positioning and associations with host-cell PML-NBs and centromeres regulate LAT locus transcription during latency in neurons. PLoS Pathog. 2012, 8, e1002852. [Google Scholar] [CrossRef]
- Bell, P.; Lieberman, P.M.; Maul, G.G. Lytic but not latent replication of epstein-barr virus is associated with PML and induces sequential release of nuclear domain 10 proteins. J. Virol. 2000, 74, 11800–11810. [Google Scholar] [CrossRef]
- Günther, T.; Schreiner, S.; Dobner, T.; Tessmer, U.; Grundhoff, A. Influence of ND10 components on epigenetic determinants of early KSHV latency establishme. PLoS Pathog. 2014, 10, e1004274. [Google Scholar] [CrossRef]
- Hossain, M.G.; Ohsaki, E.; Honda, T.; Ueda, K. Importance of Promyelocytic Leukema Protein (PML) for Kaposi’s Sarcoma-Associated Herpesvirus Lytic Replication. Front. Microbiol. 2018, 9, 2324. [Google Scholar] [CrossRef]
- Guion, L.; Bienkowska-Haba, M.; DiGiuseppe, S.; Florin, L.; Sapp, M. PML nuclear body-residing proteins sequentially associate with HPV genome after infectious nuclear delivery. PLoS Pathog. 2019, 15, e1007590. [Google Scholar] [CrossRef]
- Moosmann, P.; Georgiev, O.; Le Douarin, B.; Bourquin, J.P.; Schaffner, W. Transcriptional repression by RING finger protein TIF1 beta that interacts with the KRAB repressor domain of KOX1. Nucleic Acids Res. 1996, 24, 4859–4867. [Google Scholar] [CrossRef]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J., III. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Ecco, G.; Imbeault, M.; Trono, D. KRAB zinc finger proteins. Development 2017, 144, 2719–2729. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.C.; Friedman, J.R.; Rauscher, F.J., 3rd. Targeting histone deacetylase complexes via KRAB-zinc finger proteins: The PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2α subunit of NuRD. Genes Dev. 2001, 15, 428–443. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, A.L.; Ortiz, J.A.; You, J.; Oulad-Abdelghani, M.; Khechumian, R.; Gansmuller, A.; Chambon, P.; Losson, R. Interaction with members of the heterochromatin protein 1 (HP1) family and histone deacetylation are differentially involved in transcriptional silencing by members of the TIF1 family. EMBO J. 1999, 18, 6385–6395. [Google Scholar] [CrossRef]
- Ryan, R.F.; Schultz, D.C.; Ayyanathan, K.; Singh, P.B.; Friedman, J.R.; Fredericks, W.J.; Rauscher, F.J. KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: A potential role for Kruppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol. Cell. Biol. 1999, 19, 4366–4378. [Google Scholar] [CrossRef]
- Cheng, C.T.; Kuo, C.Y.; Ann, D.K. KAPtain in charge of multiple missions: Emerging roles of KAP1. World J. Biol. Chem. 2014, 5, 308–320. [Google Scholar] [CrossRef]
- Ait-Ammar, A.; Bellefroid, M.; Daouad, F.; Martinelli, V.; Van Assche, J.; Wallet, C.; Rodari, A.; De Rovere, M.; Fahrenkrog, B.; Schwartz, C.; et al. Inhibition of HIV-1 gene transcription by KAP1 in myeloid lineage. Sci. Rep. 2021, 11, 2692. [Google Scholar] [CrossRef]
- Ma, X.; Yang, T.; Luo, Y.; Wu, L.; Jiang, Y.; Song, Z.; Pan, T.; Liu, B.; Liu, G.; Liu, J.; et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. eLife 2019, 8, e42426. [Google Scholar] [CrossRef]
- Wolf, D.; Cammas, F.; Losson, R.; Goff, S.P. Primer binding site-dependent restriction of murine leukemia virus requires HP1 binding by TRIM28. J. Virol. 2008, 82, 4675–4679. [Google Scholar] [CrossRef]
- Wolf, D.; Goff, S.P. TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell 2007, 131, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Rowe, H.M.; Jakobsson, J.; Mesnard, D.; Rougemont, J.; Reynard, S.; Aktas, T.; Maillard, P.V.; Layard-Liesching, H.; Verp, S.; Marquis, J.; et al. KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 2010, 463, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Bürck, C.; Mund, A.; Berscheminski, J.; Kieweg, L.; Müncheberg, S.; Dobner, T.; Schreiner, S. KAP1 Is a Host Restriction Factor That Promotes Human Adenovirus E1B-55K SUMO Modification. J. Virol. 2016, 90, 930–946. [Google Scholar] [CrossRef]
- Reichel, A.; Stilp, A.-C.; Scherer, M.; Reuter, N.; Lukassen, S.; Kasmapour, B.; Schreiner, S.; Cicin-Sain, L.; Winterpacht, A.; Stamminger, T. Chromatin-Remodeling Factor SPOC1 Acts as a Cellular Restriction Factor against Human Cytomegalovirus by Repressing the Major Immediate Early Promoter. J. Virol. 2018, 92, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Bhaduri-McIntosh, S.; McIntosh, M.T. Inflammasome, the Constitutive Heterochromatin Machinery, and Replication of an Oncogenic Herpesvirus. Viruses 2021, 13, 846. [Google Scholar] [CrossRef]
- Chang, P.C.; Fitzgerald, L.D.; Van Geelen, A.; Izumiya, Y.; Ellison, T.J.; Wang, D.H.; Ann, D.K.; Luciw, P.A.; Kung, H.-J. Kruppel-associated box domain-associated protein-1 as a latency regulator for Kaposi’s sarcoma-associated herpesvirus and its modulation by the viral protein kinase. Cancer Res. 2009, 69, 5681–5689. [Google Scholar] [CrossRef]
- Tsai, M.-S.; Chen, S.-H.; Chang, C.-P.; Hsiao, Y.-L.; Wang, L.-C. Integrin-Linked Kinase Reduces H3K9 Trimethylation to Enhance Herpes Simplex Virus 1 Replication. Front. Cell. Infect. Microbiol. 2022, 12, 814307. [Google Scholar] [CrossRef]
- Xu, H.; Li, X.; Rousseau, B.A.; Akinyemi, I.A.; Frey, T.R.; Zhou, K.; Droske, L.E.; Mitchell, J.A.; McIntosh, M.T.; Bhaduri-McIntosh, S. IFI16 Partners with KAP1 to Maintain Epstein-Barr Virus Latency. J. Virol. 2022, 96, e0102822. [Google Scholar] [CrossRef]
- Sun, R.; Liang, D.; Gao, Y.; Lan, K. Kaposi’s sarcoma-associated herpesvirus-encoded LANA interacts with host KAP1 to facilitate establishment of viral latency. J. Virol. 2014, 88, 7331–7344. [Google Scholar] [CrossRef]
- Gjyshi, O.; Roy, A.; Dutta, S.; Veettil, M.V.; Dutta, D.; Chandran, B. Activated Nrf2 Interacts with Kaposi’s Sarcoma-Associated Herpesvirus Latency Protein LANA-1 and Host Protein KAP1 To Mediate Global Lytic Gene Repression. J. Virol. 2015, 89, 7874–7892. [Google Scholar] [CrossRef] [PubMed]
- Smith-Moore, S.; Neil, S.J.D.; Fraefel, C.; Linden, R.M.; Bollen, M.; Rowe, H.M.; Henckaerts, E. Adeno-associated virus Rep proteins antagonize phosphatase PP1 to counteract KAP1 repression of the latent viral genome. Proc. Natl. Acad. Sci. USA 2018, 115, E3529–E3538. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Singh, V.V.; Dutta, S.; Veettil, M.V.; Dutta, D.; Chikoti, L.; Lu, J.; Everly, D.; Chandran, B. Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J. Virol. 2013, 87, 8606–8623. [Google Scholar] [CrossRef]
- Cridland, J.A.; Curley, E.Z.; Wykes, M.N.; Schroder, K.; Sweet, M.J.; Roberts, T.L.; Ragan, M.A.; Kassahn, K.S.; Stacey, K.J. The mammalian PYHIN gene family: Phylogeny, evolution and expression. BMC Evol. Biol. 2012, 12, 140. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.J.; Trapani, J.A. IFI 16 gene encodes a nuclear protein whose expression is induced by interferons in human myeloid leukaemia cell lines. J. Cell. Biochem. 1995, 57, 39–51. [Google Scholar] [CrossRef]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef]
- Mishra, S.; Raj, A.S.; Kumar, A.; Rajeevan, A.; Kumari, P.; Kumar, H. Innate immune sensing of influenza A viral RNA through IFI16 promotes pyroptotic cell death. iScience 2022, 25, 103714. [Google Scholar] [CrossRef] [PubMed]
- Ouchi, M.; Ouchi, T. Role of IFI16 in DNA damage and checkpoint. Front. Biosci. 2008, 13, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.R.; Sharma, S.; Atianand, M. Interferon gamma-inducible protein (IFI) 16 transcriptionally regulates type i interferons and other interferon-stimulated genes and controls the interferon response to both DNA and RNA viruses. J. Biol. Chem. 2014, 289, 23568–23581. [Google Scholar] [CrossRef] [PubMed]
- Trapani, J.A.; Dawson, M.; Apostolidis, V.A.; Browne, K.A. Genomic organization of IFI16, an interferon-inducible gene whose expression is associated with human myeloid cell differentiation: Correlation of predicted protein domains with exon organization. Immunogenetics 1994, 40, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Ansari, M.A.; Kumar, B.; Dutta, D.; Roy, A.; Chikoti, L.; Pisano, G.; Dutta, S.; Vahedi, S.; Veettil, M.V.; et al. Histone H2B-IFI16 Recognition of Nuclear Herpesviral Genome Induces Cytoplasmic Interferon-beta Responses. PLoS Pathog. 2016, 12, e1005967. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wei, F.; Zhang, Y.; Wang, T.; Gao, W.; Yu, S.; Sun, H.; Pu, J.; Sun, Y.; Wang, M.; et al. IFI16 directly senses viral RNA and enhances RIG-I transcription and activation to restrict influenza virus infection. Nat. Microbiol. 2021, 6, 932–945. [Google Scholar] [CrossRef]
- Johnson, K.E.; Bottero, V.; Flaherty, S.; Dutta, S.; Singh, V.V.; Chandran, B. IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog. 2014, 10, e1004503. [Google Scholar] [CrossRef]
- Cigno, I.L.; De Andrea, M.; Borgogna, C.; Albertini, S.; Landini, M.M.; Peretti, A.; Johnson, K.E.; Chandran, B.; Landolfo, S.; Gariglio, M. The Nuclear DNA Sensor IFI16 Acts as a Restriction Factor for Human Papillomavirus Replication through Epigenetic Modifications of the Viral Promoters. J. Virol. 2015, 89, 7506–7520. [Google Scholar] [CrossRef]
- Merkl, P.E.; Knipe, D.M. Role for a Filamentous Nuclear Assembly of IFI16, DNA, and Host Factors in Restriction of Herpesviral Infection. mBio 2019, 10, e02621-18. [Google Scholar] [CrossRef] [PubMed]
- Orzalli, M.H.; Conwell, S.E.; Berrios, C.; DeCaprio, J.A.; Knipe, D.M. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc. Natl. Acad. Sci. USA 2013, 110, E4492–E4501. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Dutta, D.; Iqbal, J.; Pisano, G.; Gjyshi, O.; Ansari, M.A.; Kumar, B.; Chandran, B. Nuclear Innate Immune DNA Sensor IFI16 Is Degraded during Lytic Reactivation of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV): Role of IFI16 in Maintenance of KSHV Latency. J. Virol. 2016, 90, 8822–8841. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Ghosh, A.; Kumar, B.; Chandran, B. IFI16, a nuclear innate immune DNA sensor, mediates epigenetic silencing of herpesvirus genomes by its association with H3K9 methyltransferases SUV39H1 and GLP. eLife 2019, 8, e49500. [Google Scholar] [CrossRef]
- Pisano, G.; Roy, A.; Ansari, M.A.; Kumar, B.; Chikoti, L.; Chandran, B. Interferon-gamma-inducible protein 16 (IFI16) is required for the maintenance of Epstein-Barr virus latency. Virol. J. 2017, 14, 221. [Google Scholar] [CrossRef]
- Gariano, G.R.; Dell’Oste, V.; Bronzini, M.; Gatti, D.; Luganini, A.; De Andrea, M.; Gribaudo, G.; Gariglio, M.; Landolfo, S. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog. 2012, 8, e1002498. [Google Scholar] [CrossRef]
- Monroe, K.M.; Yang, Z.; Johnson, J.R.; Geng, X.; Doitsh, G.; Krogan, N.J.; Greene, W.C. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science 2014, 343, 428–432. [Google Scholar] [CrossRef]
- Wichit, S.; Hamel, R.; Yainoy, S.; Gumpangseth, N.; Panich, S.; Phuadraksa, T.; Saetear, P.; Monteil, A.; Morales Vargas, R.; Missé, D. Interferon-inducible protein (IFI) 16 regulates Chikungunya and Zika virus infection in human skin fibroblasts. EXCLI J. 2019, 18, 467–476. [Google Scholar]
- Jakobsen, M.R.; Bak, R.O.; Andersen, A.; Berg, R.K.; Jensen, S.B.; Jin, T.; Laustsen, A.; Hansen, K.; Østergaard, L.; Fitzgerald, K.A.; et al. PNAS Plus: From the Cover: IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. USA 2013, 110, E4571–E4580. [Google Scholar] [CrossRef] [PubMed]
- Hotter, D.; Bosso, M.; Jønsson, K.L.; Krapp, C.; Stürzel, C.M.; Das, A.; Littwitz-Salomon, E.; Berkhout, B.; Russ, A.; Wittmann, S.; et al. IFI16 Targets the Transcription Factor Sp1 to Suppress HIV-1 Transcription and Latency Reactivation. Cell Host Microbe 2019, 25, 858–872.e13. [Google Scholar] [CrossRef] [PubMed]
- Sodroski, C.N.; Knipe, D.M. Nuclear interferon-stimulated gene product maintains heterochromatin on the herpes simplex viral genome to limit lytic infection. Proc. Natl. Acad. Sci. USA 2023, 120, e2310996120. [Google Scholar] [CrossRef]
- Howard, T.R.; Lum, K.K.; Kennedy, M.A.; Cristea, I.M. The Nuclear DNA Sensor IFI16 Indiscriminately Binds to and Diminishes Accessibility of the HSV-1 Genome to Suppress Infection. mSystems 2022, 7, e0019822. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, X.; Wang, Z.; Shu, W.; Li, L.; Li, Y.; Guo, Z.; Gao, B.; Xiong, S. Nuclear Sensor Interferon-Inducible Protein 16 Inhibits the Function of Hepatitis B Virus Covalently Closed Circular DNA by Integrating Innate Immune Activation and Epigenetic Suppression. Hepatology 2020, 71, 1154–1169. [Google Scholar] [CrossRef]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. USA 2012, 109, E3008–E3017. [Google Scholar] [CrossRef]
- Li, T.; Chen, J.; Cristea, I.M. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 2013, 14, 591–599. [Google Scholar] [CrossRef]
- Song, Y.; Wu, X.; Xu, Y.; Zhu, J.; Li, J.; Zou, Z.; Chen, L.; Zhang, B.; Hua, C.; Rui, H.; et al. HPV E7 inhibits cell pyroptosis by promoting TRIM21-mediated degradation and ubiquitination of the IFI16 inflammasome. Int. J. Biol. Sci. 2020, 16, 2924–2937. [Google Scholar] [CrossRef]
- Seczynska, M.; Lehner, P.J. The sound of silence: Mechanisms and implications of HUSH complex function. Trends Genet. 2023, 39, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Timms, R.T.; Tchasovnikarova, I.A.; Lehner, P.J. Position-effect variegation revisited: HUSHing up heterochromatin in human cells. BioEssays 2016, 38, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Timms, R.T.; Tchasovnikarova, I.A.; Antrobus, R.; Dougan, G.; Lehner, P.J. ATF7IP-Mediated Stabilization of the Histone Methyltransferase SETDB1 Is Essential for Heterochromatin Formation by the HUSH Complex. Cell Rep. 2016, 17, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Harten, S.K.; Bruxner, T.J.; Bharti, V.; Blewitt, M.; Nguyen, T.-M.-T.; Whitelaw, E.; Epp, T. The first mouse mutants of D14Abb1e (Fam208a) show that it is critical for early development. Mamm. Genome 2014, 25, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Soehn, A.S.; Pham, T.; Schaeferhoff, K.; Floss, T.; Weisenhorn, D.M.V.; Wurst, W.; Bonin, M.; Riess, O. Periphilin is strongly expressed in the murine nervous system and is indispensable for murine development. Genesis 2009, 47, 697–707. [Google Scholar] [CrossRef]
- Liu, S.; Brind’amour, J.; Karimi, M.M.; Shirane, K.; Bogutz, A.; Lefebvre, L.; Sasaki, H.; Shinkai, Y.; Lorincz, M.C. Setdb1 is required for germline development and silencing of H3K9me3-marked endogenous retroviruses in primordial germ cells. Genes Dev. 2014, 28, 2041–2055. [Google Scholar] [CrossRef]
- Elgin, S.C.; Reuter, G. Position-effect variegation, heterochromatin formation, and gene silencing in Drosophila. Cold Spring Harb. Perspect. Biol. 2013, 5, a017780. [Google Scholar] [CrossRef]
- Oh, J.-M.; Venters, C.C.; Di, C.; Pinto, A.M.; Wan, L.; Younis, I.; Cai, Z.; Arai, C.; So, B.R.; Duan, J.; et al. U1 snRNP regulates cancer cell migration and invasion in vitro. Nat. Commun. 2020, 11, 1. [Google Scholar] [CrossRef]
- Tunbak, H.; Enriquez-Gasca, R.; Tie, C.H.C.; Gould, P.A.; Mlcochova, P.; Gupta, R.K.; Fernandes, L.; Holt, J.; van der Veen, A.G.; Giampazolias, E.; et al. The HUSH complex is a gatekeeper of type I interferon through epigenetic regulation of LINE-1s. Nat. Commun. 2020, 11, 5387. [Google Scholar] [CrossRef]
- Hagelkruys, A.; Horrer, M.; Taubenschmid-Stowers, J.; Kavirayani, A.; Novatchkova, M.; Orthofer, M.; Pai, T.-P.; Cikes, D.; Zhuk, S.; Balmaña, M.; et al. The HUSH complex controls brain architecture and protocadherin fidelity. Sci. Adv. 2022, 8, eabo7247. [Google Scholar] [CrossRef] [PubMed]
- Tchasovnikarova, I.A.; Timms, R.T.; Matheson, N.J.; Wals, K.; Antrobus, R.; Göttgens, B.; Dougan, G.; Dawson, M.A.; Lehner, P.J. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science 2015, 348, 1481–1485. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Lee, C.H.; Swigut, T.; Grow, E.; Gu, B.; Bassik, M.C.; Wysocka, J. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature 2018, 553, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, G.Z.; Cingöz, O.; Goff, S.P. NP220 mediates silencing of unintegrated retroviral DNA. Nature 2018, 564, 278–282. [Google Scholar] [CrossRef]
- McGeary, M.K.; Damsky, W.; Daniels, A.; Song, E.; Micevic, G.; Calderwood, C.; Lou, H.J.; Paradkar, S.; Kaech, S.; Calderwood, D.A.; et al. Setdb1-loss induces type-I interferons and immune clearance of melanoma. bioRxiv 2023. [Google Scholar] [CrossRef]
- Robbez-Masson, L.; Tie, C.H.; Conde, L.; Tunbak, H.; Husovsky, C.; Tchasovnikarova, I.A.; Timms, R.T.; Herrero, J.; Lehner, P.J.; Rowe, H.M. The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes. Genome Res. 2018, 28, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Vijayan, M.; Walton, E.M.; Stafford, V.G.; Fiflis, D.N.; Asokan, A. Epigenetic Silencing of Recombinant Adeno-associated Virus Genomes by NP220 and the HUSH Complex. J. Virol. 2022, 96, e0203921. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, L.; Guney, M.H.; Kim, K.; Goh, S.L.; McCauley, S.; Dauphin, A.; Diehl, W.E.; Luban, J. Primate immunodeficiency virus proteins Vpx and Vpr counteract transcriptional repression of proviruses by the HUSH complex. Nat. Microbiol. 2018, 3, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Chougui, G.; Munir-Matloob, S.; Matkovic, R.; Martin, M.M.; Morel, M.; Lahouassa, H.; Leduc, M.; Ramirez, B.C.; Etienne, L.; Margottin-Goguet, F. HIV-2/SIV viral protein X counteracts HUSH repressor complex. Nat. Microbiol. 2018, 3, 891–897. [Google Scholar] [CrossRef]
- Greenwood, E.; Williamson, J.; Sienkiewicz, A.; Naamati, A.; Matheson, N.; Lehner, P. Promiscuous Targeting of Cellular Proteins by Vpr Drives Systems-Level Proteomic Remodeling in HIV-1 Infection. Cell Rep. 2019, 27, 1579–1596.e7. [Google Scholar] [CrossRef]
- Vauthier, V.; Lasserre, A.; Morel, M.; Versapuech, M.; Berlioz-Torrent, C.; Zamborlini, A.; Margottin-Goguet, F.; Matkovic, R. HUSH-mediated HIV silencing is independent of TASOR phosphorylation on threonine 819. Retrovirology 2022, 19, 23. [Google Scholar] [CrossRef]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piątkowski, P.; Baginski, B.; Wirecki, T.K.; De Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef]
- Li, S.; Mason, C.E. The pivotal regulatory landscape of RNA modifications. Annu. Rev. Genom. Hum. Genet. 2014, 15, 127–150. [Google Scholar] [CrossRef]
- Netzband, R.; Pager, C.T. Epitranscriptomic marks: Emerging modulators of RNA virus gene expression. Wiley Interdiscip. Rev. RNA 2020, 11, e1576. [Google Scholar] [CrossRef] [PubMed]
- George, C.X.; John, L.; Samuel, C.E. An RNA editor, adenosine deaminase acting on double-stranded RNA (ADAR1). J. Interf. Cytokine Res. 2014, 34, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.E. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 2011, 411, 180–193. [Google Scholar] [CrossRef]
- Lu, D.; Lu, J.; Liu, Q.; Zhang, Q. Emerging role of the RNA-editing enzyme ADAR1 in stem cell fate and function. Biomark. Res. 2023, 11, 61. [Google Scholar] [CrossRef]
- Pfaller, C.K.; George, C.X.; Samuel, C.E. Adenosine Deaminases Acting on RNA (ADARs) and Viral Infections. Annu. Rev. Virol. 2021, 8, 239–264. [Google Scholar] [CrossRef]
- Piontkivska, H.; Wales-McGrath, B.; Miyamoto, M.; Wayne, M.L. ADAR Editing in Viruses: An Evolutionary Force to Reckon with. Genome Biol. Evol. 2021, 13, evab240. [Google Scholar] [CrossRef]
- Ward, S.V.; George, C.X.; Welch, M.J.; Liou, L.-Y.; Hahm, B.; Lewicki, H.; De La Torre, J.C.; Samuel, C.E.; Oldstone, M.B. RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.B.; Cornu, T.I.; Redwine, J.; Dales, S.; Lewicki, H.; Holz, A.; Thomas, D.; Billeter, M.A.; Oldstone, M.B. Evidence that the hypermutated M protein of a subacute sclerosing panencephalitis measles virus actively contributes to the chronic progressive CNS disease. Virology 2001, 291, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Martínez, I.; Melero, J.A. A model for the generation of multiple A to G transitions in the human respiratory syncytial virus genome: Predicted RNA secondary structures as substrates for adenosine deaminases that act on RNA. J. Gen. Virol. 2002, 83, 1445–1455. [Google Scholar] [CrossRef]
- Rima, B.K.; Gatherer, D.; Young, D.F.; Norsted, H.; Randall, R.E.; Davison, A.J. Stability of the parainfluenza virus 5 genome revealed by deep sequencing of strains isolated from different hosts and following passage in cell culture. J. Virol. 2014, 88, 3826–3836. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.R.; Puig, M.; Darnell, M.E.R.; Mihalik, K.; Feinstone, S.M. New antiviral pathway that mediates hepatitis C virus replicon interferon sensitivity through ADAR1. J. Virol. 2005, 79, 6291–6298. [Google Scholar] [CrossRef] [PubMed]
- Garaigorta, U.; Chisari, F.V. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe 2009, 6, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, Z.J.; Prasad, A.N.; Ronk, A.J.; Kuzmin, I.V.; Ilinykh, P.A.; Andino, R.; Bukreyev, A. Species-Specific Evolution of Ebola Virus during Replication in Human and Bat Cells. Cell Rep. 2020, 32, 108028. [Google Scholar] [CrossRef]
- Vogel, O.A.; Han, J.; Liang, C.-Y.; Manicassamy, S.; Perez, J.T.; Manicassamy, B. The p150 Isoform of ADAR1 Blocks Sustained RLR signaling and Apoptosis during Influenza Virus Infection. PLoS Pathog. 2020, 16, e1008842. [Google Scholar] [CrossRef] [PubMed]
- Suspène, R.; Renard, M.; Henry, M.; Guétard, D.; Puyraimond-Zemmour, D.; Billecocq, A.; Bouloy, M.; Tangy, F.; Vartanian, J.-P.; Wain-Hobson, S. Inversing the natural hydrogen bonding rule to selectively amplify GC-rich ADAR-edited RNAs. Nucleic Acids Res. 2008, 36, e72. [Google Scholar] [CrossRef] [PubMed]
- Zahn, R.C.; Schelp, I.; Utermöhlen, O.; von Laer, D. A-to-G hypermutation in the genome of lymphocytic choriomeningitis virus. J. Virol. 2007, 81, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Memczak, S.; Wyler, E.; Torti, F.; Porath, H.T.; Orejuela, M.R.; Piechotta, M.; Levanon, E.Y.; Landthaler, M.; Dieterich, C.; et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015, 10, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Liu, Y.; Samuel, C.E. Adenovirus VAI RNA antagonizes the RNA-editing activity of the ADAR adenosine deaminase. Virology 1998, 245, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Yang, C.; Zhao, F.; Huang, Y.; Lin, Y.; Huang, C.; Ma, X.; Du, J.; Wang, Y.; Long, G.; et al. Double-stranded RNA deaminase ADAR1 promotes the Zika virus replication by inhibiting the activation of protein kinase PKR. J. Biol. Chem. 2019, 294, 18168–18180. [Google Scholar] [CrossRef]
- Nie, Y.; Hammond, G.L.; Yang, J.-H. Double-stranded RNA deaminase ADAR1 increases host susceptibility to virus infection. J. Virol. 2007, 81, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Clerzius, G.; Gélinas, J.-F.; Daher, A.; Bonnet, M.; Meurs, E.F.; Gatignol, A. ADAR1 interacts with PKR during human immunodeficiency virus infection of lymphocytes and contributes to viral replication. J. Virol. 2009, 83, 10119–10128. [Google Scholar] [CrossRef]
- Toth, A.M.; Li, Z.; Cattaneo, R.; Samuel, C.E. RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. J. Biol. Chem. 2009, 284, 29350–29356. [Google Scholar] [CrossRef]
- Doria, M.; Neri, F.; Gallo, A.; Farace, M.G.; Michienzi, A. Editing of HIV-1 RNA by the double-stranded RNA deaminase ADAR1 stimulates viral infection. Nucleic Acids Res. 2009, 37, 5848–5858. [Google Scholar] [CrossRef]
- Phuphuakrat, A.; Kraiwong, R.; Boonarkart, C.; Lauhakirti, D.; Lee, T.-H.; Auewarakul, P. Double-stranded RNA adenosine deaminases enhance expression of human immunodeficiency virus type 1 proteins. J. Virol. 2008, 82, 10864–10872. [Google Scholar] [CrossRef]
- Tang, Y.-D.; Na, L.; Fu, L.-H.; Yang, F.; Zhu, C.-H.; Tang, L.; Li, Q.; Wang, J.-Y.; Li, Z.; Wang, X.-F.; et al. Double-stranded RNA-specific adenosine deaminase 1 (ADAR1) promotes EIAV replication and infectivity. Virology 2015, 476, 364–371. [Google Scholar] [CrossRef]
- Eyler, D.E.; Franco, M.K.; Batool, Z.; Wu, M.Z.; Dubuke, M.L.; Dobosz-Bartoszek, M.; Jones, J.D.; Polikanov, Y.S.; Roy, B.; Koutmou, K.S. Pseudouridinylation of mRNA coding sequences alters translation. Proc. Natl. Acad. Sci. USA 2019, 116, 23068–23074. [Google Scholar] [CrossRef]
- McIntyre, W.; Netzband, R.; Bonenfant, G.; Biegel, J.M.; Miller, C.; Fuchs, G.; Henderson, E.; Arra, M.; Canki, M.; Fabris, D.; et al. Positive-sense RNA viruses reveal the complexity and dynamics of the cellular and viral epitranscriptomes during infection. Nucleic Acids Res. 2018, 46, 5776–5791. [Google Scholar] [CrossRef]
- Marceau, C.D.; Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Brewer, S.M.; Fuchs, G.; Swaminathan, K.; Mata, M.A.; Elias, J.E.; Sarnow, P.; et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 2016, 535, 159–163. [Google Scholar] [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Ping, X.-L.; Sun, B.-F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.-J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.-S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Zhen, D.; Wu, Y.; Zhang, Y.; Chen, K.; Song, B.; Xu, H.; Tang, Y.; Wei, Z.; Meng, J. m6A Reader: Epitranscriptome Target Prediction and Functional Characterization of N6-Methyladenosine (m6A) Readers. Front. Cell Dev. Biol. 2020, 8, 741. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, N.S.; McIntyre, A.B.R.; McFadden, M.J.; Roder, A.E.; Kennedy, E.M.; Gandara, J.A.; Hopcraft, S.E.; Quicke, K.M.; Vazquez, C.; Willer, J.; et al. N6 -Methyladenosine in Flaviviridae Viral RNA Genomes Regulates Infection. Cell Host Microbe 2016, 20, 654–665. [Google Scholar] [CrossRef]
- Lichinchi, G.; Zhao, B.S.; Wu, Y.; Lu, Z.; Qin, Y.; He, C.; Rana, T.M. Dynamics of Human and Viral RNA Methylation during Zika Virus Infection. Cell Host Microbe 2016, 20, 666–673. [Google Scholar] [CrossRef]
- Lichinchi, G.; Gao, S.; Saletore, Y.; Gonzalez, G.M.; Bansal, V.; Wang, Y.; Mason, C.E.; Rana, T.M. Dynamics of the human and viral m6A RNA methylomes during HIV-1 infection of T cells. Nat. Microbiol. 2016, 1, 16011. [Google Scholar] [CrossRef]
- Lu, W.; Tirumuru, N.; St. Gelais, C.; Koneru, P.C.; Liu, C.; Kvaratskhelia, M.; He, C.; Wu, L. N6-Methyladenosine–binding proteins suppress HIV-1 infectivity and viral production. J. Biol. Chem. 2018, 293, 12992–13005. [Google Scholar] [CrossRef]
- Jurczyszak, D.; Zhang, W.; Terry, S.N.; Kehrer, T.; González, M.C.B.; McGregor, E.; Mulder, L.C.F.; Eckwahl, M.J.; Pan, T.; Simon, V. HIV protease cleaves the antiviral m6A reader protein YTHDF3 in the viral particle. PLoS Pathog. 2020, 16, e1008305. [Google Scholar] [CrossRef]
- Kennedy, E.M.; Bogerd, H.P.; Kornepati, A.V.R.; Kang, D.; Ghoshal, D.; Marshall, J.B.; Poling, B.C.; Tsai, K.; Gokhale, N.S.; Horner, S.M.; et al. Posttranscriptional m 6 A Editing of HIV-1 mRNAs Enhances Viral Gene Expression. Cell Host Microbe 2016, 19, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Tirumuru, N.; Zhao, B.S.; Lu, W.; Lu, Z.; He, C.; Wu, L. N(6)-methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. eLife 2016, 5, e15528. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Bogerd, H.P.; Kennedy, E.M.; Emery, A.; Swanstrom, R.; Cullen, B.R. Epitranscriptomic addition of m6A regulates HIV-1 RNA stability and alternative splicing. Genes Dev. 2021, 35, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Chelmicki, T.; Roger, E.; Teissandier, A.; Dura, M.; Bonneville, L.; Rucli, S.; Dossin, F.; Fouassier, C.; Lameiras, S.; Bourc’his, D. m6A RNA methylation regulates the fate of endogenous retroviruses. Nature 2021, 591, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.; Farnham, P.J. KAP1 protein: An enigmatic master regulator of the genome. J. Biol. Chem. 2011, 286, 26267–26276. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, M.; Amato, R.; Sgura, A.; Antoccia, A.; Berardinelli, F. The Multiple Facets of ATRX Protein. Cancers 2021, 13, 2211. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.R.; Munkhjargal, A.; Kim, M.-J.; Park, S.Y.; Jung, E.; Ryu, J.-H.; Yang, Y.; Lim, J.-S.; Kim, Y. The functional roles of PML nuclear bodies in genome maintenance. Mutat. Res. Mol. Mech. Mutagen. 2018, 809, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.S.; Kao, H.Y. PML: Regulation and multifaceted function beyond tumor suppression. Cell Biosci. 2018, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Zhang, S.; Gao, X.; Gao, X.; Xu, X.; Lv, Y.; Zhang, Y.; Zhu, Z.; Zhang, C.; Li, Q.; et al. Roles of Kruppel-associated Box (KRAB)-associated Co-repressor KAP1 Ser-473 Phosphorylation in DNA Damage Response. J. Biol. Chem. 2012, 287, 18937–18952. [Google Scholar] [CrossRef]
- Jang, S.M.; Kauzlaric, A.; Quivy, J.-P.; Pontis, J.; Rauwel, B.; Coluccio, A.; Offner, S.; Duc, J.; Turelli, P.; Almouzni, G.; et al. KAP1 facilitates reinstatement of heterochromatin after DNA replication. Nucleic Acids Res. 2018, 46, 8788–8802. [Google Scholar] [CrossRef] [PubMed]
- Cuchet-Lourenço, D.; Anderson, G.; Sloan, E.; Orr, A.; Everett, R.D. The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J. Virol. 2013, 87, 13422–13432. [Google Scholar] [CrossRef] [PubMed]
- Diner, B.A.; Lum, K.K.; Javitt, A.; Cristea, I.M. Interactions of the Antiviral Factor Interferon Gamma-Inducible Protein 16 (IFI16) Mediate Immune Signaling and Herpes Simplex Virus-1 Immunosuppression. Mol. Cell. Proteom. 2015, 14, 2341–2356. [Google Scholar] [CrossRef]
- Morrone, S.R.; Wang, T.; Constantoulakis, L.M.; Hooy, R.M.; Delannoy, M.J.; Sohn, J. Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc. Natl. Acad. Sci. USA 2013, 111, E62–E71. [Google Scholar] [CrossRef]
- Diner, B.A.; Lum, K.K.; Toettcher, J.E.; Cristea, I.M. Viral DNA Sensors IFI16 and Cyclic GMP-AMP Synthase Possess Distinct Functions in Regulating Viral Gene Expression, Immune Defenses, and Apoptotic Responses during Herpesvirus Infection. mBio 2016, 7, e01553-16. [Google Scholar] [CrossRef]
- Merkl, P.E.; Orzalli, M.H.; Knipe, D.M. Mechanisms of Host IFI16, PML, and Daxx Protein Restriction of Herpes Simplex Virus 1 Replication. J. Virol. 2018, 92, e00057-18. [Google Scholar] [CrossRef]
- Taura, M.; Song, E.; Ho, Y.-C.; Iwasaki, A. Apobec3A maintains HIV-1 latency through recruitment of epigenetic silencing machinery to the long terminal repeat. Proc. Natl. Acad. Sci. USA 2019, 116, 2282–2289. [Google Scholar] [CrossRef]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef]
- Nakaya, Y.; Stavrou, S.; Blouch, K.; Tattersall, P.; Ross, S.R. In Vivo Examination of Mouse APOBEC3- and Human APOBEC3A- and APOBEC3G-Mediated Restriction of Parvovirus and Herpesvirus Infection in Mouse Models. J. Virol. 2016, 90, 8005–8012. [Google Scholar] [CrossRef]
- Suspène, R.; Aynaud, M.-M.; Koch, S.; Pasdeloup, D.; Labetoulle, M.; Gaertner, B.; Vartanian, J.-P.; Meyerhans, A.; Wain-Hobson, S. Genetic editing of herpes simplex virus 1 and Epstein-Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J. Virol. 2011, 85, 7594–7602. [Google Scholar] [CrossRef]
- Warren, C.J.; Xu, T.; Guo, K.; Griffin, L.M.; Westrich, J.A.; Lee, D.; Lambert, P.F.; Santiago, M.L.; Pyeon, D. APOBEC3A functions as a restriction factor of human papillomavirus. J. Virol. 2015, 89, 688–702. [Google Scholar] [CrossRef]
- Carpenter, M.A.; Li, M.; Rathore, A.; Lackey, L.; Law, E.K.; Land, A.M.; Leonard, B.; Shandilya, S.M.D.; Bohn, M.-F.; Schiffer, C.A.; et al. Methylcytosine and normal cytosine deamination by the foreign DNA restriction enzyme APOBEC3A. J. Biol. Chem. 2012, 287, 34801–34808. [Google Scholar] [CrossRef]
- Chahwan, R.; Wontakal, S.N.; Roa, S. Crosstalk between genetic and epigenetic information through cytosine deamination. Trends Genet. 2010, 26, 443–448. [Google Scholar] [CrossRef]
- Wijesinghe, P.; Bhagwat, A.S. Efficient deamination of 5-methylcytosines in DNA by human APOBEC3A, but not by AID or APOBEC3G. Nucleic Acids Res. 2012, 40, 9206–9217. [Google Scholar] [CrossRef] [PubMed]
- Kan, R.L.; Chen, J.; Sallam, T. Crosstalk between epitranscriptomic and epigenetic mechanisms in gene regulation. Trends Genet. 2022, 38, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Yue, M.; Wang, J.; Kumar, S.; Wechsler-Reya, R.J.; Zhang, Z.; Ogawa, Y.; Kellis, M.; Duester, G.; et al. N6-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat. Neurosci. 2018, 21, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xia, L.; Tan, K.; Ye, X.; Zuo, Z.; Li, M.; Xiao, R.; Wang, Z.; Liu, X.; Deng, M.; et al. N6-Methyladenosine co-transcriptionally directs the demethylation of histone H3K9me2. Nat. Genet. 2020, 52, 870–877. [Google Scholar] [CrossRef]
- Wu, C.; Chen, W.; He, J.; Jin, S.; Liu, Y.; Yi, Y.; Gao, Z.; Yang, J.; Yang, J.; Cui, J.; et al. Interplay of m 6 A and H3K27 trimethylation restrains inflammation during bacterial infection. Sci. Adv. 2020, 6, eaba0647. [Google Scholar] [CrossRef]
- Liu, J.; Gao, M.; He, J.; Wu, K.; Lin, S.; Jin, L.; Chen, Y.; Liu, H.; Shi, J.; Wang, X.; et al. The RNA m6A reader YTHDC1 silences retrotransposons and guards ES cell identity. Nature 2021, 591, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Duda, K.J.; Ching, R.W.; Jerabek, L.; Shukeir, N.; Erikson, G.; Engist, B.; Onishi-Seebacher, M.; Perrera, V.; Richter, F.; Mittler, G.; et al. m6A RNA methylation of major satellite repeat transcripts facilitates chromatin association and RNA:DNA hybrid formation in mouse heterochromatin. Nucleic Acids Res. 2021, 49, 5568–5587. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Li, J.; He, C.; Wen, J.; Ma, H.; Rong, B.; Diao, J.; Wang, L.; Wang, J.; Wu, F.; et al. METTL3 regulates heterochromatin in mouse embryonic stem cells. Nature 2021, 591, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Zhou, K.; Wu, T.; Zhao, B.S.; Sun, M.; Chen, Z.; Deng, X.; Xiao, G.; Auer, F.; et al. Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature 2019, 567, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Sahafnejad, Z.; Ramazi, S.; Allahverdi, A. An Update of Epigenetic Drugs for the Treatment of Cancers and Brain Diseases: A Comprehensive Review. Genes 2023, 14, 873. [Google Scholar] [CrossRef] [PubMed]
- Berdasco, M.; Esteller, M. Towards a druggable epitranscriptome: Compounds that target RNA modifications in cancer. Br. J. Pharmacol. 2022, 179, 2868–2889. [Google Scholar] [CrossRef]
- Feehley, T.; O’donnell, C.W.; Mendlein, J.; Karande, M.; McCauley, T. Drugging the epigenome in the age of precision medicine. Clin. Epigenet. 2023, 15, 6. [Google Scholar] [CrossRef]
- Nehme, Z.; Pasquereau, S.; Herbein, G. Control of viral infections by epigenetic-targeted therapy. Clin. Epigenet. 2019, 11, 55. [Google Scholar] [CrossRef]
- Arbuckle, J.H.; Gardina, P.J.; Gordon, D.N.; Hickman, H.D.; Yewdell, J.W.; Pierson, T.C.; Myers, T.G.; Kristie, T.M. Inhibitors of the Histone Methyltransferases EZH2/1 Induce a Potent Antiviral State and Suppress Infection by Diverse Viral Pathogens. mBio 2017, 8, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; He, M.; Zhou, F.; Ye, F.; Gao, S.J. Activation of Kaposi’s sarcoma-associated herpesvirus (KSHV) by inhibitors of class III histone deacetylases: Identification of sirtuin 1 as a regulator of the KSHV life cycle. J. Virol. 2014, 88, 6355–6367. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Z.; Phuc, T.; Xu, Z.; Yang, D.; Chen, Z.; Lin, Z.; Kendrick, S.; Dai, L.; Li, H.-Y.; et al. Oncolytic strategy using new bifunctional HDACs/BRD4 inhibitors against virus-associated lymphomas. PLoS Pathog. 2023, 19, e1011089. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, A.; Ghosh, A. Epigenetic Restriction Factors (eRFs) in Virus Infection. Viruses 2024, 16, 183. https://doi.org/10.3390/v16020183
Roy A, Ghosh A. Epigenetic Restriction Factors (eRFs) in Virus Infection. Viruses. 2024; 16(2):183. https://doi.org/10.3390/v16020183
Chicago/Turabian StyleRoy, Arunava, and Anandita Ghosh. 2024. "Epigenetic Restriction Factors (eRFs) in Virus Infection" Viruses 16, no. 2: 183. https://doi.org/10.3390/v16020183
APA StyleRoy, A., & Ghosh, A. (2024). Epigenetic Restriction Factors (eRFs) in Virus Infection. Viruses, 16(2), 183. https://doi.org/10.3390/v16020183