
Human Oncogenic Viruses: Characteristics and Prevention Strategies—Lessons Learned from Human Papillomaviruses


Abstract
:1. Introduction
2. Infectious Agents and Cancer
3. Oncogenic Viruses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Family | Virus | Year of Discovery | Genome kb | Tropism | Associated Neoplasia | Cases n (%) |
|---|---|---|---|---|---|---|
| Papillomaviridae | HPV16, 18, 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59 | 1983 [21] | dsDNA 8.0 kb | Keratinocytes | Cervical, anal, vulva, vagina, penis and head and neck cancers | 730,000 1 (31.1%) |
| Hepadnaviridae | HBV | 1965 [22] | Partial dsDNA 3.3 kb | Hepatocytes | Hepatocellular carcinoma | 380,000 1 (16.4%) |
| Flaviviridae | HCV | 1989 [23] | Positive-sense RNA strand 9.6 kb | Hepatocytes | Hepatocellular carcinoma and other non-Hodgkin lymphomas | 170,000 1 (7.4%) |
| Herpesviridae | EBV (HHV-4) | 1964 [24] | dsDNA 172 kb | Epithelial cells and B cells | Hodgkin lymphoma, Burkitt lymphoma and nasopharyngeal carcinoma | 156,600 2 (6.8%) |
| Herpesviridae | KSHV (HHV-8) | 1994 [25] | dsDNA 140 kb | Epithelial cells and B cells | Kaposi sarcoma | 42,000 2 (1.8%) |
| Retroviridae | HTLV-1 | 1980 [26] | Positive-sense RNA strand 9.0 kb | T and B cells | Adult T-cell leukaemia and lymphoma | 3600 2 (0.16%) |
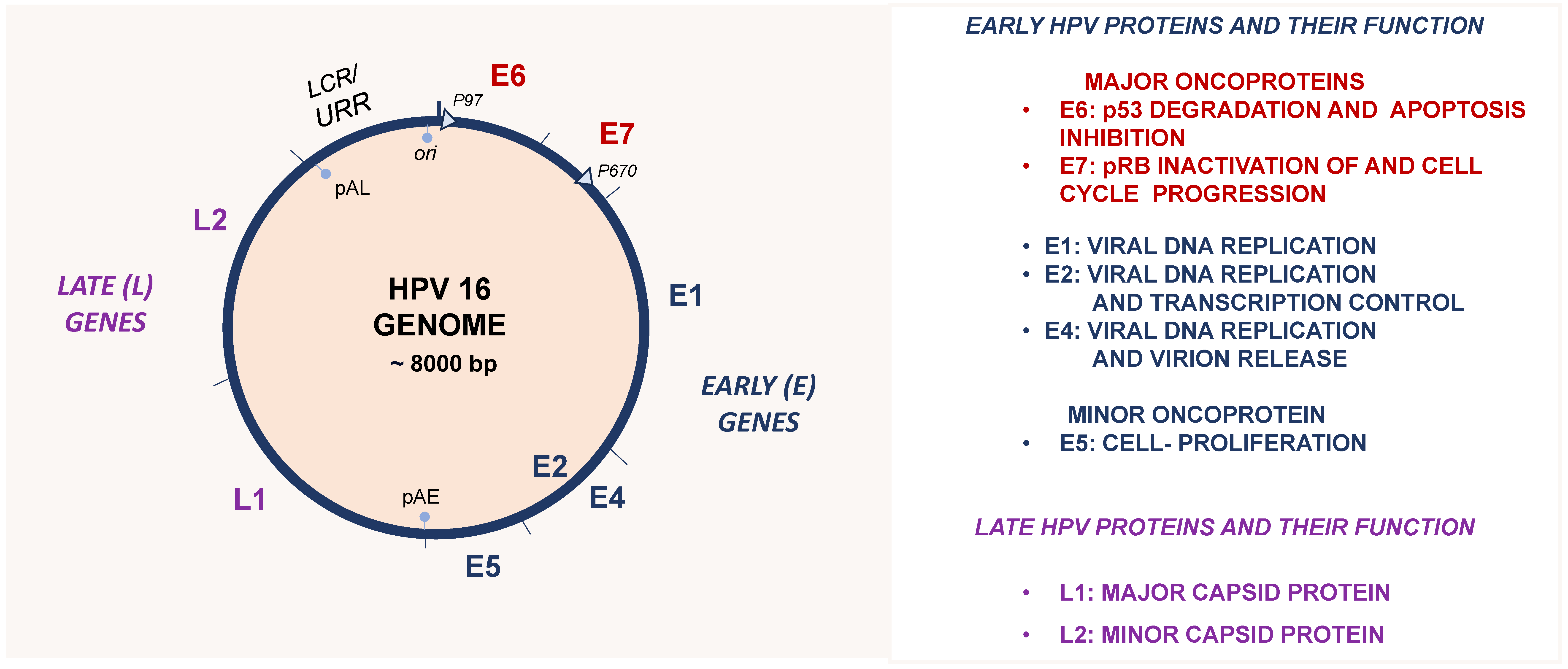
4. Human Papillomaviruses
5. Epidemiology and Natural History of HPV Infection
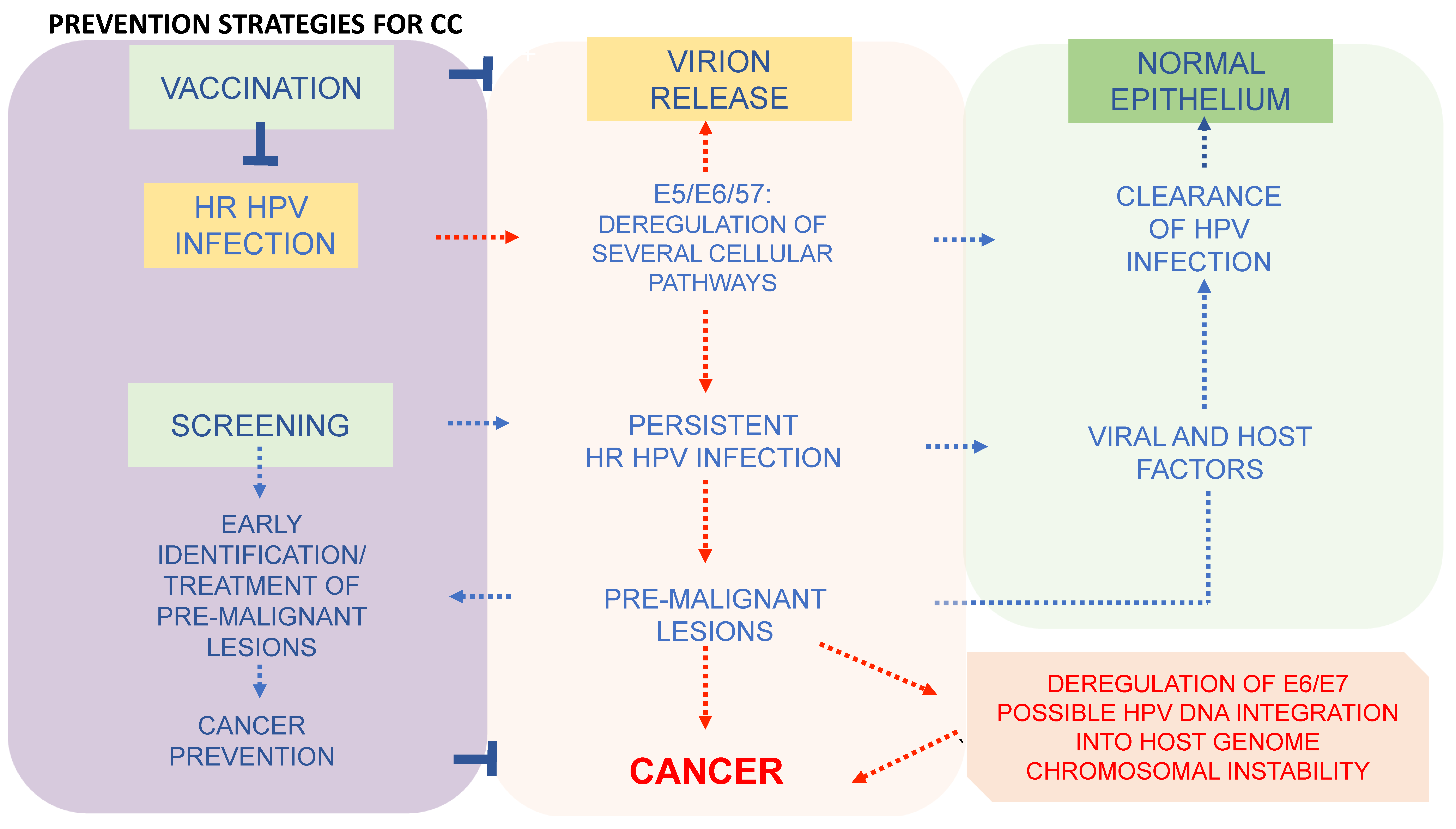
6. Prevention Strategies
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- de Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Vignat, J.; Lorenzoni, V.; Eslahi, M.; Ginsburg, O.; Lauby-Secretan, B.; Arbyn, M.; Basu, P.; Bray, F.; Vaccarella, S. Global estimates of incidence and mortality of cervical cancer in 2020: A baseline analysis of the WHO Global Cervical Cancer Elimination Initiative. Lancet Glob. Health 2023, 11, e197–e206. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group. Biological agents. IARC Monogr. Eval. Carcinog Risks Hum. 2012, 100 Pt B, 1–441. [Google Scholar]
- zur Hausen, H. The search for infectious causes of human cancers: Where and why (Nobel lecture). Angew. Chem. Int. Ed. Engl. 2009, 48, 5798–5808. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Stelzle, D.; Tanaka, L.F.; Lee, K.K.; Ibrahim Khalil, A.; Baussano, I.; Shah, A.S.V.; McAllister, D.A.; Gottlieb, S.L.; Klug, S.J.; Winkler, A.S.; et al. Estimates of the global burden of cervical cancer associated with HIV. Lancet Glob. Health 2021, 9, e161–e169. [Google Scholar] [CrossRef] [PubMed]
- Grulich, A.E.; van Leeuwen, M.T.; Falster, M.O.; Vajdic, C.M. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: A meta-analysis. Lancet 2007, 370, 59–67. [Google Scholar] [CrossRef]
- de Martel, C.; Shiels, M.S.; Franceschi, S.; Simard, E.P.; Vignat, J.; Hall, H.I.; Engels, E.A.; Plummer, M. Cancers attributable to infections among adults with HIV in the United States. Aids 2015, 29, 2173–2181. [Google Scholar] [CrossRef]
- Deshmukh, A.A.; Damgacioglu, H.; Georges, D.; Sonawane, K.; Ferlay, J.; Bray, F.; Clifford, G.M. Global burden of HPV-attributable squamous cell carcinoma of the anus in 2020, according to sex and HIV status: A worldwide analysis. Int. J. Cancer 2023, 152, 417–428. [Google Scholar] [CrossRef]
- Khalil, A.I.; Franceschi, S.; de Martel, C.; Bray, F.; Clifford, G.M. Burden of Kaposi sarcoma according to HIV status: A systematic review and global analysis. Int. J. Cancer 2022, 150, 1948–1957. [Google Scholar] [CrossRef]
- WHO. Global Health Sector Strategy on Viral Hepatitis 2016–2021; Global Hepatitis Programme Department of HIV/AIDS; WHO: Geneva, Switzerland, 2016; p. 53. [Google Scholar]
- Falcaro, M.; Castañon, A.; Ndlela, B.; Checchi, M.; Soldan, K.; Lopez-Bernal, J.; Elliss-Brookes, L.; Sasieni, P. The effects of the national HPV vaccination programme in England, UK, on cervical cancer and grade 3 cervical intraepithelial neoplasia incidence: A register-based observational study. Lancet 2021, 398, 2084–2092. [Google Scholar] [CrossRef]
- Drolet, M.; Bénard, É.; Pérez, N.; Brisson, M.; HPV Vaccination Impact Study Group. Population-level impact and herd effects following the introduction of human papillomavirus vaccination programmes: Updated systematic review and meta-analysis. Lancet 2019, 394, 497–509. [Google Scholar] [CrossRef]
- Cui, F.; Blach, S.; Manzengo Mingiedi, C.; Gonzalez, M.A.; Sabry Alaama, A.; Mozalevskis, A.; Séguy, N.; Rewari, B.B.; Chan, P.L.; Le, L.V.; et al. Global reporting of progress towards elimination of hepatitis B and hepatitis C. Lancet Gastroenterol. Hepatol. 2023, 8, 332–342. [Google Scholar] [CrossRef]
- Butel, J.S. Viral carcinogenesis: Revelation of molecular mechanisms and etiology of human disease. Carcinogenesis 2000, 21, 405–426. [Google Scholar] [CrossRef]
- Wallace, N.A.; Galloway, D.A. Chapter 8—Viral Oncogenesis: Infections that Can Lead to Cancer. In Viral Pathogenesis, 3rd ed.; Katze, M.G., Korth, M.J., Law, G.L., Nathanson, N., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 95–105. [Google Scholar] [CrossRef]
- Vogt, P.K. Retroviral oncogenes: A historical primer. Nat. Rev. Cancer 2012, 12, 639–648. [Google Scholar] [CrossRef]
- Kristiansen, E.; Jenkins, A.; Holm, R. Coexistence of episomal and integrated HPV16 DNA in squamous cell carcinoma of the cervix. J. Clin. Pathol. 1994, 47, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Dürst, M.; Gissmann, L.; Ikenberg, H.; zur Hausen, H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc. Natl. Acad. Sci. USA 1983, 80, 3812–3815. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, B.S.; Alter, H.J.; Visnich, S. A “NEW” ANTIGEN IN LEUKEMIA SERA. JAMA 1965, 191, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [CrossRef]
- Epstein, M.A.; Henle, G.; Achong, B.G.; Barr, Y.M. Morphological and biological studies on A virus in cultured lymphoblasts from burkitt’s lymphoma. J. Exp. Med. 1965, 121, 761–770. [Google Scholar] [CrossRef]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef]
- Poiesz, B.J.; Ruscetti, F.W.; Gazdar, A.F.; Bunn, P.A.; Minna, J.D.; Gallo, R.C. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. USA 1980, 77, 7415–7419. [Google Scholar] [CrossRef]
- Prado, J.C.M.; Monezi, T.A.; Amorim, A.T.; Lino, V.; Paladino, A.; Boccardo, E. Human polyomaviruses and cancer: An overview. Clinics 2018, 73 (Suppl. S1), e558s. [Google Scholar] [CrossRef]
- Moens, U.; Prezioso, C.; Pietropaolo, V. Functional Domains of the Early Proteins and Experimental and Epidemiological Studies Suggest a Role for the Novel Human Polyomaviruses in Cancer. Front. Microbiol. 2022, 13, 834368. [Google Scholar] [CrossRef]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbé, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17077. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A. Molecular Pathogenesis of Merkel Cell Carcinoma. Annu. Rev. Pathol. 2021, 16, 69–91. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Malaria and some polyomaviruses (SV40, BK, JC, and Markel cell viruses). IARC Monogr. Eval. Carcinog Risks Hum. 2014, 104, 9–350. [Google Scholar]
- Donà, M.G.; Gheit, T.; Chiantore, M.V.; Vescio, M.F.; Luzi, F.; Rollo, F.; Accardi, L.; Cota, C.; Galati, L.; Romeo, G.; et al. Prevalence of 13 polyomaviruses in actinic keratosis and matched healthy skin samples of immunocompetent individuals. Infect. Agents Cancer 2022, 17, 59. [Google Scholar] [CrossRef]
- Bouvard, V.; Baan, R.A.; Grosse, Y.; Lauby-Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Straif, K. Carcinogenicity of malaria and of some polyomaviruses. Lancet Oncol. 2012, 13, 339–340. [Google Scholar] [CrossRef]
- Lambert, P. Oncogenic Viruses. In Encyclopedia of Microbiology, 3rd ed.; Schaechter, M., Ed.; Academic Press: Oxford, UK, 2009; pp. 421–429. [Google Scholar] [CrossRef]
- Grulich, A.E.; Vajdic, C.M. The epidemiology of cancers in human immunodeficiency virus infection and after organ transplantation. Semin. Oncol. 2015, 42, 247–257. [Google Scholar] [CrossRef]
- Chang, Y.; Moore, P.S.; Weiss, R.A. Human oncogenic viruses: Nature and discovery. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Gaglia, M.M.; Munger, K. More than just oncogenes: Mechanisms of tumorigenesis by human viruses. Curr. Opin. Virol. 2018, 32, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Zella, D.; Gallo, R.C. Viruses and Bacteria Associated with Cancer: An Overview. Viruses 2021, 13, 1039. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Estêvão, D.; Costa, N.R.; Gil da Costa, R.M.; Medeiros, R. Hallmarks of HPV carcinogenesis: The role of E6, E7 and E5 oncoproteins in cellular malignancy. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 153–162. [Google Scholar] [CrossRef]
- Viarisio, D.; Gissmann, L.; Tommasino, M. Human papillomaviruses and carcinogenesis: Well-established and novel models. Curr. Opin. Virol. 2017, 26, 56–62. [Google Scholar] [CrossRef]
- Veillette, A.; Pérez-Quintero, L.A.; Latour, S. X-linked lymphoproliferative syndromes and related autosomal recessive disorders. Curr. Opin. Allergy Clin. Immunol. 2013, 13, 614–622. [Google Scholar] [CrossRef]
- Accardi, R.; Gruffat, H.; Sirand, C.; Fusil, F.; Gheit, T.; Hernandez-Vargas, H.; Le Calvez-Kelm, F.; Traverse-Glehen, A.; Cosset, F.L.; Manet, E.; et al. The mycotoxin aflatoxin B1 stimulates Epstein-Barr virus-induced B-cell transformation in in vitro and in vivo experimental models. Carcinogenesis 2015, 36, 1440–1451. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Primers 2019, 5, 9. [Google Scholar] [CrossRef]
- Vitiello, G.A.F.; Ferreira, W.A.S.; Cordeiro de Lima, V.C.; Medina, T.D.S. Antiviral Responses in Cancer: Boosting Antitumor Immunity Through Activation of Interferon Pathway in the Tumor Microenvironment. Front. Immunol. 2021, 12, 782852. [Google Scholar] [CrossRef] [PubMed]
- Leo, P.J.; Madeleine, M.M.; Wang, S.; Schwartz, S.M.; Newell, F.; Pettersson-Kymmer, U.; Hemminki, K.; Hallmans, G.; Tiews, S.; Steinberg, W.; et al. Defining the genetic susceptibility to cervical neoplasia—A genome-wide association study. PLoS Genet. 2017, 13, e1006866. [Google Scholar] [CrossRef]
- Bowden, S.J.; Bodinier, B.; Kalliala, I.; Zuber, V.; Vuckovic, D.; Doulgeraki, T.; Whitaker, M.D.; Wielscher, M.; Cartwright, R.; Tsilidis, K.K.; et al. Genetic variation in cervical preinvasive and invasive disease: A genome-wide association study. Lancet Oncol. 2021, 22, 548–557. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Gheit, T. Mucosal and Cutaneous Human Papillomavirus Infections and Cancer Biology. Front. Oncol. 2019, 9, 355. [Google Scholar] [CrossRef]
- Tommasino, M. The human papillomavirus family and its role in carcinogenesis. Semin. Cancer Biol. 2014, 26, 13–21. [Google Scholar] [CrossRef]
- de Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef]
- Chen, Z.; Schiffman, M.; Herrero, R.; Desalle, R.; Burk, R.D. Human papillomavirus (HPV) types 101 and 103 isolated from cervicovaginal cells lack an E6 open reading frame (ORF) and are related to gamma-papillomaviruses. Virology 2007, 360, 447–453. [Google Scholar] [CrossRef]
- Nobre, R.J.; Herráez-Hernández, E.; Fei, J.W.; Langbein, L.; Kaden, S.; Grone, H.J.; de Villiers, E.M. E7 oncoprotein of novel human papillomavirus type 108 lacking the E6 gene induces dysplasia in organotypic keratinocyte cultures. J. Virol. 2009, 83, 2907–2916. [Google Scholar] [CrossRef]
- Ghittoni, R.; Accardi, R.; Hasan, U.; Gheit, T.; Sylla, B.; Tommasino, M. The biological properties of E6 and E7 oncoproteins from human papillomaviruses. Virus Genes. 2010, 40, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, R.; Das, S.S.; Biswal, S.S.; Nath, A.; Das, D.; Basu, A.; Malik, S.; Kumar, L.; Kar, S.; Singh, S.K.; et al. Mechanistic role of HPV-associated early proteins in cervical cancer: Molecular pathways and targeted therapeutic strategies. Crit. Rev. Oncol. Hematol. 2022, 174, 103675. [Google Scholar] [CrossRef]
- Wetherill, L.F.; Holmes, K.K.; Verow, M.; Müller, M.; Howell, G.; Harris, M.; Fishwick, C.; Stonehouse, N.; Foster, R.; Blair, G.E.; et al. High-Risk Human Papillomavirus E5 Oncoprotein Displays Channel-Forming Activity Sensitive to Small-Molecule Inhibitors. J. Virol. 2012, 86, 5341–5351. [Google Scholar] [CrossRef]
- Venuti, A.; Paolini, F.; Nasir, L.; Corteggio, A.; Roperto, S.; Campo, M.S.; Borzacchiello, G. Papillomavirus E5: The smallest oncoprotein with many functions. Mol. Cancer 2011, 10, 140. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhao, L.; Yang, R.; Xu, T. Advances in molecular mechanism of HPV16 E5 oncoprotein carcinogenesis. Arch. Biochem. Biophys. 2023, 745, 109716. [Google Scholar] [CrossRef]
- Lambert, P.F. Transgenic Mouse Models of Tumor Virus Action. Annu. Rev. Virol. 2016, 3, 473–489. [Google Scholar] [CrossRef] [PubMed]
- Cerasuolo, A.; Annunziata, C.; Tortora, M.; Starita, N.; Stellato, G.; Greggi, S.; Maglione, M.G.; Ionna, F.; Losito, S.; Botti, G.; et al. Comparative analysis of HPV16 gene expression profiles in cervical and in oropharyngeal squamous cell carcinoma. Oncotarget 2017, 8, 34070–34081. [Google Scholar] [CrossRef]
- Lorenzon, L.; Mazzetta, F.; Venuti, A.; Frega, A.; Torrisi, M.R.; French, D. In vivo HPV 16 E5 mRNA: Expression pattern in patients with squamous intra-epithelial lesions of the cervix. J. Clin. Virol. 2011, 52, 79–83. [Google Scholar] [CrossRef]
- Martinez-Zapien, D.; Ruiz, F.X.; Poirson, J.; Mitschler, A.; Ramirez, J.; Forster, A.; Cousido-Siah, A.; Masson, M.; Vande Pol, S.; Podjarny, A.; et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 2016, 529, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Banks, L. Molecular mechanisms underlying human papillomavirus E6 and E7 oncoprotein-induced cell transformation. Mutat. Res. Rev. Mutat. Res. 2017, 772, 23–35. [Google Scholar] [CrossRef]
- Duensing, S.; Münger, K. Mechanisms of genomic instability in human cancer: Insights from studies with human papillomavirus oncoproteins. Int. J. Cancer 2004, 109, 157–162. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.; Levine, A. p53 Research: The past thirty years and the next thirty years. Cold Spring Harb. Perspect. Biol. 2010, 2, a000893. [Google Scholar] [CrossRef] [PubMed]
- Zilfou, J.T.; Lowe, S.W. Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Biol. 2009, 1, a001883. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.C.; Moody, C.A. Modulation of the DNA damage response during the life cycle of human papillomaviruses. Virus Res. 2017, 231, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Travé, G.; Zanier, K. HPV-mediated inactivation of tumor suppressor p53. Cell Cycle 2016, 15, 2231–2232. [Google Scholar] [CrossRef]
- Klingelhutz, A.J.; Foster, S.A.; McDougall, J.K. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature 1996, 380, 79–82. [Google Scholar] [CrossRef]
- Stöppler, H.; Hartmann, D.P.; Sherman, L.; Schlegel, R. The human papillomavirus type 16 E6 and E7 oncoproteins dissociate cellular telomerase activity from the maintenance of telomere length. J. Biol. Chem. 1997, 272, 13332–13337. [Google Scholar] [CrossRef]
- Stewart, S.A.; Weinberg, R.A. Telomerase and human tumorigenesis. Semin. Cancer Biol. 2000, 10, 399–406. [Google Scholar] [CrossRef]
- Veldman, T.; Liu, X.; Yuan, H.; Schlegel, R. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc. Natl. Acad. Sci. USA 2003, 100, 8211–8216. [Google Scholar] [CrossRef] [PubMed]
- Katzenellenbogen, R.A. Activation of telomerase by HPVs. Virus Res. 2017, 231, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Gewin, L.; Myers, H.; Kiyono, T.; Galloway, D.A. Identification of a novel telomerase repressor that interacts with the human papillomavirus type-16 E6/E6-AP complex. Genes. Dev. 2004, 18, 2269–2282. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; Burk, R.D. Association between hTERT activation by HPV E6 proteins and oncogenic risk. Virology 2012, 433, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Ganti, K.; Broniarczyk, J.; Manoubi, W.; Massimi, P.; Mittal, S.; Pim, D.; Szalmas, A.; Thatte, J.; Thomas, M.; Tomaić, V.; et al. The Human Papillomavirus E6 PDZ Binding Motif: From Life Cycle to Malignancy. Viruses 2015, 7, 3530–3551. [Google Scholar] [CrossRef] [PubMed]
- Accardi, R.; Rubino, R.; Scalise, M.; Gheit, T.; Shahzad, N.; Thomas, M.; Banks, L.; Indiveri, C.; Sylla, B.S.; Cardone, R.A.; et al. E6 and E7 from human papillomavirus type 16 cooperate to target the PDZ protein Na/H exchange regulatory factor 1. J. Virol. 2011, 85, 8208–8216. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Banks, L. The biology of papillomavirus PDZ associations: What do they offer papillomaviruses? Curr. Opin. Virol. 2021, 51, 119–126. [Google Scholar] [CrossRef]
- Banks, L.; Pim, D.; Thomas, M. Human tumour viruses and the deregulation of cell polarity in cancer. Nat. Rev. Cancer 2012, 12, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S.; Lee, L.Y.; Duensing, A.; Basile, J.; Piboonniyom, S.; Gonzalez, S.; Crum, C.P.; Munger, K. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc. Natl. Acad. Sci. USA 2000, 97, 10002–10007. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S.; Münger, K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002, 62, 7075–7082. [Google Scholar] [PubMed]
- Egawa, N. Papillomaviruses and cancer: Commonalities and differences in HPV carcinogenesis at different sites of the body. Int. J. Clin. Oncol. 2023, 28, 956–964. [Google Scholar] [CrossRef]
- Kombe Kombe, A.J.; Li, B.; Zahid, A.; Mengist, H.M.; Bounda, G.A.; Zhou, Y.; Jin, T. Epidemiology and Burden of Human Papillomavirus and Related Diseases, Molecular Pathogenesis, and Vaccine Evaluation. Front. Public. Health 2020, 8, 552028. [Google Scholar] [CrossRef] [PubMed]
- Basukala, O.; Banks, L. The Not-So-Good, the Bad and the Ugly: HPV E5, E6 and E7 Oncoproteins in the Orchestration of Carcinogenesis. Viruses 2021, 13, 1892. [Google Scholar] [CrossRef]
- Della Fera, A.N.; Warburton, A.; Coursey, T.L.; Khurana, S.; McBride, A.A. Persistent Human Papillomavirus Infection. Viruses 2021, 13, 321. [Google Scholar] [CrossRef]
- Egawa, N.; Egawa, K.; Griffin, H.; Doorbar, J. Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia. Viruses 2015, 7, 3863–3890. [Google Scholar] [CrossRef]
- Silva, L.L.D.; Teles, A.M.; Santos, J.M.O.; Souza de Andrade, M.; Medeiros, R.; Faustino-Rocha, A.I.; Oliveira, P.A.; Dos Santos, A.P.A.; Ferreira Lopes, F.; Braz, G.; et al. Malignancy Associated with Low-Risk HPV6 and HPV11: A Systematic Review and Implications for Cancer Prevention. Cancers 2023, 15, 4068. [Google Scholar] [CrossRef]
- Alemany, L.; Saunier, M.; Alvarado-Cabrero, I.; Quirós, B.; Salmeron, J.; Shin, H.R.; Pirog, E.C.; Guimerà, N.; Hernandez-Suarez, G.; Felix, A.; et al. Human papillomavirus DNA prevalence and type distribution in anal carcinomas worldwide. Int. J. Cancer 2015, 136, 98–107. [Google Scholar] [CrossRef]
- Alemany, L.; Cubilla, A.; Halec, G.; Kasamatsu, E.; Quirós, B.; Masferrer, E.; Tous, S.; Lloveras, B.; Hernández-Suarez, G.; Lonsdale, R.; et al. Role of Human Papillomavirus in Penile Carcinomas Worldwide. Eur. Urol. 2016, 69, 953–961. [Google Scholar] [CrossRef]
- Kang, Y.J.; Smith, M.; Canfell, K. Anal cancer in high-income countries: Increasing burden of disease. PLoS ONE 2018, 13, e0205105. [Google Scholar] [CrossRef] [PubMed]
- Islami, F.; Ferlay, J.; Lortet-Tieulent, J.; Bray, F.; Jemal, A. International trends in anal cancer incidence rates. Int. J. Epidemiol. 2017, 46, 924–938. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of Human Papillomavirus-Positive Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. 2015, 33, 3235–3242. [Google Scholar] [CrossRef]
- Agrawal, Y.; Koch, W.M.; Xiao, W.; Westra, W.H.; Trivett, A.L.; Symer, D.E.; Gillison, M.L. Oral human papillomavirus infection before and after treatment for human papillomavirus 16-positive and human papillomavirus 16-negative head and neck squamous cell carcinoma. Clin. Cancer Res. 2008, 14, 7143–7150. [Google Scholar] [CrossRef]
- Galati, L.; Chiocca, S.; Duca, D.; Tagliabue, M.; Simoens, C.; Gheit, T.; Arbyn, M.; Tommasino, M. HPV and head and neck cancers: Towards early diagnosis and prevention. Tumour Virus Res. 2022, 14, 200245. [Google Scholar] [CrossRef]
- Christiansen, I.K.; Sandve, G.K.; Schmitz, M.; Dürst, M.; Hovig, E. Transcriptionally active regions are the preferred targets for chromosomal HPV integration in cervical carcinogenesis. PLoS ONE 2015, 10, e0119566. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; de Sanjosé, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2016, 2, 16086. [Google Scholar] [CrossRef]
- Östör, A.G. Natural History of Cervical Intraepithelial Neoplasia: A Critical Review. Int. J. Gynecol. Pathol. 1993, 12, 186–192. [Google Scholar] [CrossRef]
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef]
- Gao, G.; Johnson, S.H.; Vasmatzis, G.; Pauley, C.E.; Tombers, N.M.; Kasperbauer, J.L.; Smith, D.I. Common fragile sites (CFS) and extremely large CFS genes are targets for human papillomavirus integrations and chromosome rearrangements in oropharyngeal squamous cell carcinoma. Genes. Chromosomes Cancer 2017, 56, 59–74. [Google Scholar] [CrossRef]
- Warburton, A.; Markowitz, T.E.; Katz, J.P.; Pipas, J.M.; McBride, A.A. Recurrent integration of human papillomavirus genomes at transcriptional regulatory hubs. NPJ Genom. Med. 2021, 6, 101. [Google Scholar] [CrossRef]
- Morgan, I.M.; DiNardo, L.J.; Windle, B. Integration of Human Papillomavirus Genomes in Head and Neck Cancer: Is It Time to Consider a Paradigm Shift? Viruses 2017, 9, 208. [Google Scholar] [CrossRef]
- Da Silva, M.L.R.; De Albuquerque, B.H.D.R.; Allyrio, T.A.D.M.F.; De Almeida, V.D.; Cobucci, R.N.D.O.; Bezerra, F.L.; Andrade, V.S.; Lanza, D.C.F.; De Azevedo, J.C.V.; De Araújo, J.M.G.; et al. The role of HPV-induced epigenetic changes in cervical carcinogenesis (Review). Biomed. Rep. 2021, 15, 60. [Google Scholar] [CrossRef]
- Stanley, M. Prophylactic HPV vaccines: Prospects for eliminating ano-genital cancer. Br. J. Cancer 2007, 96, 1320–1323. [Google Scholar] [CrossRef]
- Muñoz, N.; Kjaer, S.K.; Sigurdsson, K.; Iversen, O.E.; Hernandez-Avila, M.; Wheeler, C.M.; Perez, G.; Brown, D.R.; Koutsky, L.A.; Tay, E.H.; et al. Impact of human papillomavirus (HPV)-6/11/16/18 vaccine on all HPV-associated genital diseases in young women. J. Natl. Cancer Inst. 2010, 102, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Lehtinen, M.; Paavonen, J.; Wheeler, C.M.; Jaisamrarn, U.; Garland, S.M.; Castellsagué, X.; Skinner, S.R.; Apter, D.; Naud, P.; Salmerón, J.; et al. Overall efficacy of HPV-16/18 AS04-adjuvanted vaccine against grade 3 or greater cervical intraepithelial neoplasia: 4-year end-of-study analysis of the randomised, double-blind PATRICIA trial. Lancet Oncol. 2012, 13, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Dillner, J.; Kjaer, S.K.; Wheeler, C.M.; Sigurdsson, K.; Iversen, O.E.; Hernandez-Avila, M.; Perez, G.; Brown, D.R.; Koutsky, L.A.; Tay, E.H.; et al. Four year efficacy of prophylactic human papillomavirus quadrivalent vaccine against low grade cervical, vulvar, and vaginal intraepithelial neoplasia and anogenital warts: Randomised controlled trial. BMJ 2010, 341, c3493. [Google Scholar] [CrossRef] [PubMed]
- Patel, C.; Brotherton, J.M.; Pillsbury, A.; Jayasinghe, S.; Donovan, B.; Macartney, K.; Marshall, H. The impact of 10 years of human papillomavirus (HPV) vaccination in Australia: What additional disease burden will a nonavalent vaccine prevent? Euro Surveill. 2018, 23. [Google Scholar] [CrossRef] [PubMed]
- Palefsky, J.M.; Giuliano, A.R.; Goldstone, S.; Moreira, E.D., Jr.; Aranda, C.; Jessen, H.; Hillman, R.; Ferris, D.; Coutlee, F.; Stoler, M.H.; et al. HPV vaccine against anal HPV infection and anal intraepithelial neoplasia. N. Engl. J. Med. 2011, 365, 1576–1585. [Google Scholar] [CrossRef]
- Goldstone, S.E.; Giuliano, A.R.; Palefsky, J.M.; Lazcano-Ponce, E.; Penny, M.E.; Cabello, R.E.; Moreira, E.D., Jr.; Baraldi, E.; Jessen, H.; Ferenczy, A.; et al. Efficacy, immunogenicity, and safety of a quadrivalent HPV vaccine in men: Results of an open-label, long-term extension of a randomised, placebo-controlled, phase 3 trial. Lancet Infect. Dis. 2022, 22, 413–425. [Google Scholar] [CrossRef]
- Mühr, L.S.A.; Dillner, J. Biosimilar second-generation human papillomavirus vaccines. Lancet Infect. Dis. 2023, 23, 1215–1216. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.-C.; Zhong, G.-H.; Huang, W.-J.; Chu, K.; Zhang, L.; Bi, Z.-F.; Zhu, K.-X.; Chen, Q.; Zheng, T.-Q.; Zhang, M.-L.; et al. Head-to-head immunogenicity comparison of an Escherichia coli-produced 9-valent human papillomavirus vaccine and Gardasil 9 in women aged 18–26 years in China: A randomised blinded clinical trial. Lancet Infect. Dis. 2023, 23, 1313–1322. [Google Scholar] [CrossRef]
- Sharma, H.; Parekh, S.; Pujari, P.; Shewale, S.; Desai, S.; Bhatla, N.; Joshi, S.; Pimple, S.; Kawade, A.; Balasubramani, L.; et al. Immunogenicity and safety of a new quadrivalent HPV vaccine in girls and boys aged 9–14 years versus an established quadrivalent HPV vaccine in women aged 15–26 years in India: A randomised, active-controlled, multicentre, phase 2/3 trial. Lancet Oncol. 2023, 24, 1321–1333. [Google Scholar] [CrossRef]
- Bouvard, V.; Wentzensen, N.; Mackie, A.; Berkhof, J.; Brotherton, J.; Giorgi-Rossi, P.; Kupets, R.; Smith, R.; Arrossi, S.; Bendahhou, K.; et al. The IARC Perspective on Cervical Cancer Screening. N. Engl. J. Med. 2021, 385, 1908–1918. [Google Scholar] [CrossRef]
- Poljak, M. Towards cervical cancer eradication: Joint force of HPV vaccination and HPV-based cervical cancer screening. Clin. Microbiol. Infect. 2015, 21, 806–807. [Google Scholar] [CrossRef] [PubMed]
- von Karsa, L.; Arbyn, M.; De Vuyst, H.; Dillner, J.; Dillner, L.; Franceschi, S.; Patnick, J.; Ronco, G.; Segnan, N.; Suonio, E.; et al. European guidelines for quality assurance in cervical cancer screening. Summary of the supplements on HPV screening and vaccination. Papillomavirus Res. 2015, 1, 22–31. [Google Scholar] [CrossRef]
- Wright, T.C., Jr.; Stoler, M.H.; Behrens, C.M.; Apple, R.; Derion, T.; Wright, T.L. The ATHENA human papillomavirus study: Design, methods, and baseline results. Am. J. Obstet. Gynecol. 2012, 206, 46.e1–46.e11. [Google Scholar] [CrossRef]
- Ronco, G.; Dillner, J.; Elfström, K.M.; Tunesi, S.; Snijders, P.J.; Arbyn, M.; Kitchener, H.; Segnan, N.; Gilham, C.; Giorgi-Rossi, P.; et al. Efficacy of HPV-based screening for prevention of invasive cervical cancer: Follow-up of four European randomised controlled trials. Lancet 2014, 383, 524–532. [Google Scholar] [CrossRef]
- Cuzick, J.; Bergeron, C.; Doeberitz, M.v.K.; Gravitt, P.; Jeronimo, J.; Lorincz, A.T.; Meijer, C.J.; Sankaranarayanan, R.; Snijders, P.J.; Szarewski, A. New technologies and procedures for cervical cancer screening. Vaccine 2012, 30 (Suppl. S5), F107–F116. [Google Scholar] [CrossRef]
- Maver, P.J.; Poljak, M. Primary HPV-based cervical cancer screening in Europe: Implementation status, challenges, and future plans. Clin. Microbiol. Infect. 2020, 26, 579–583. [Google Scholar] [CrossRef]
- Arbyn, M.; Ronco, G.; Anttila, A.; Meijer, C.J.; Poljak, M.; Ogilvie, G.; Koliopoulos, G.; Naucler, P.; Sankaranarayanan, R.; Peto, J. Evidence regarding human papillomavirus testing in secondary prevention of cervical cancer. Vaccine 2012, 30 (Suppl. S5), F88–F99. [Google Scholar] [CrossRef]
- WHO Team. Global Strategy to Accelerate the Elimination of Cervical Cancer as a Public Health Problem; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Apaydin, K.Z.; Nguyen, A.; Borba, C.P.C.; Shtasel, D.L.; Ulery, S.; Mayer, K.H.; Keuroghlian, A.S. Factors associated with anal cancer screening follow-up by high-resolution anoscopy. Sex. Transm. Infect. 2019, 95, 83–86. [Google Scholar] [CrossRef]
- Darragh, T.M.; Winkler, B. Anal cancer and cervical cancer screening: Key differences. Cancer Cytopathol. 2011, 119, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Vyas, N.S.; Campbell, C.M.P.; Mathew, R.; Abrahamsen, M.; Van der Kooi, K.; Jukic, D.M.; Stoler, M.H.; Villa, L.L.; da Silva, R.C.; Lazcano-Ponce, E.; et al. Role of histological findings and pathologic diagnosis for detection of human papillomavirus infection in men. J. Med. Virol. 2015, 87, 1777–1787. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Lewis, J.S., Jr.; Chen, Z. Current status of clinical testing for human papillomavirus in oropharyngeal squamous cell carcinoma. J. Pathol. Clin. Res. 2018, 4, 213–226. [Google Scholar] [CrossRef] [PubMed]
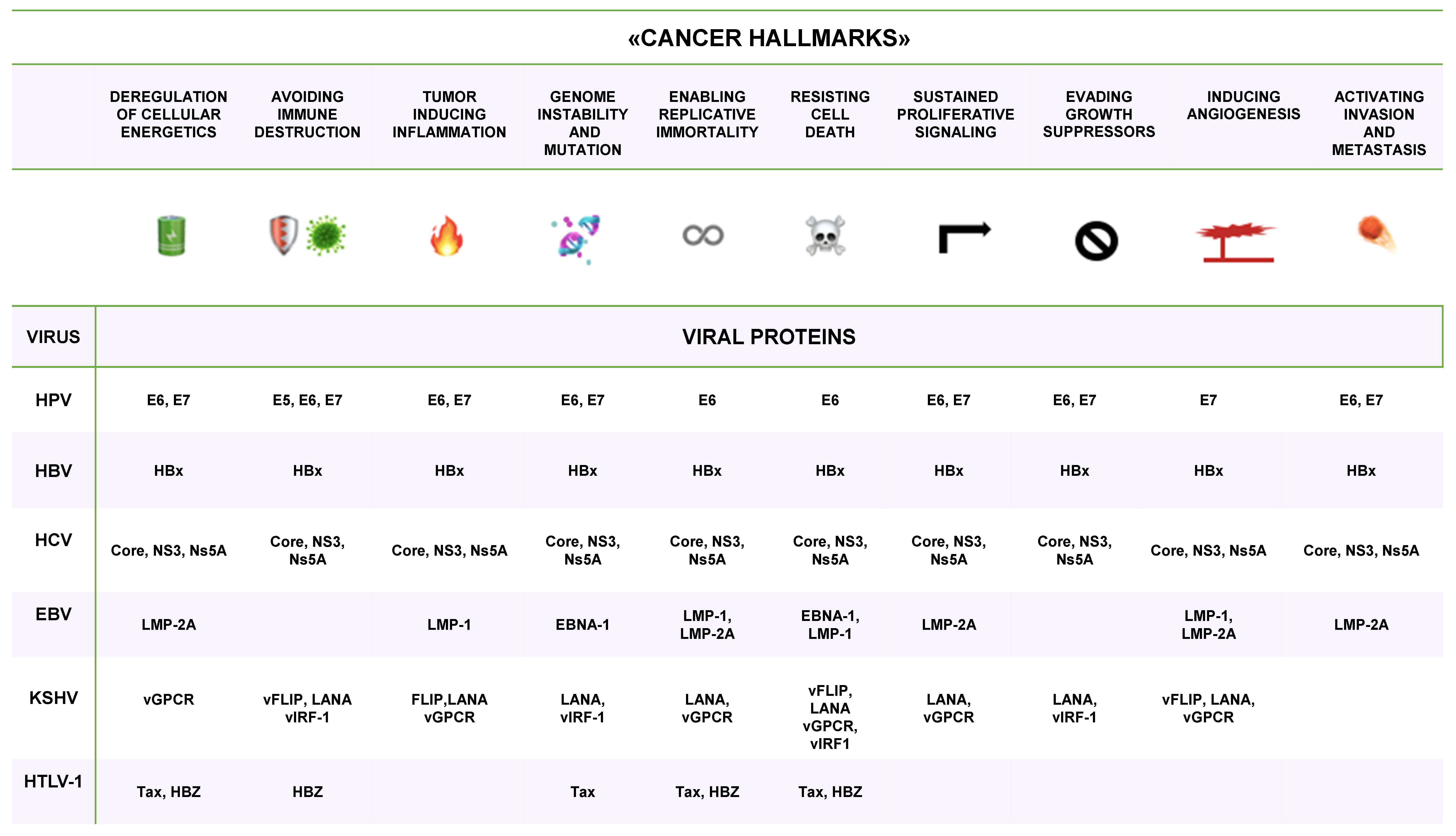


| VIRUS | ONCOPROTEIN | HOST TARGET |
|---|---|---|
| HPV | E6 | p53, mTOR, hTERT |
| E7 | pRB | |
| E5 | EGFR, ET1, COX-2 | |
| HBV | HBx | p53, pRB, Wnt, Src, DNMTs, Ras, PI3K, JNK, NF-κB, ERK, TGFβ, HDACs |
| HCV | Core, NS3, Ns5A | p53, PARP, hTERT, TGFβ, HDACs |
| EBV (HHV-4) | EBNA-1 | |
| LMP-1 | NF-κB | |
| LMP-2 | PI3K, AKT, mTOR, ERK | |
| KSHV (HHV-8) | Vflip | NF-κB, CREB, PI3K, DDR |
| LANA | p53, pRB, HIF, Notch, Wnt | |
| Vgpcr | PI3K, AKT, mTOR, ERK, p38, JNK, NF-κB | |
| Virf-1 | αIFN, p53, ATM, Bim | |
| HTLV-1 | Tax | NF-κB, CREB, PI3K, DDR |
| HBZ | c-jun, E2F |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galati, L.; Chiantore, M.V.; Marinaro, M.; Di Bonito, P. Human Oncogenic Viruses: Characteristics and Prevention Strategies—Lessons Learned from Human Papillomaviruses. Viruses 2024, 16, 416. https://doi.org/10.3390/v16030416
Galati L, Chiantore MV, Marinaro M, Di Bonito P. Human Oncogenic Viruses: Characteristics and Prevention Strategies—Lessons Learned from Human Papillomaviruses. Viruses. 2024; 16(3):416. https://doi.org/10.3390/v16030416
Chicago/Turabian StyleGalati, Luisa, Maria Vincenza Chiantore, Mariarosaria Marinaro, and Paola Di Bonito. 2024. "Human Oncogenic Viruses: Characteristics and Prevention Strategies—Lessons Learned from Human Papillomaviruses" Viruses 16, no. 3: 416. https://doi.org/10.3390/v16030416
APA StyleGalati, L., Chiantore, M. V., Marinaro, M., & Di Bonito, P. (2024). Human Oncogenic Viruses: Characteristics and Prevention Strategies—Lessons Learned from Human Papillomaviruses. Viruses, 16(3), 416. https://doi.org/10.3390/v16030416