
Sequencing and Analysis of Lumpy Skin Disease Virus Whole Genomes Reveals a New Viral Subgroup in West and Central Africa
 ,
,  , , , , , , , , , , and
, , , , , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Purification of LSDV DNA
2.3. Whole Genome Sequencing (WGS)
2.4. Genome Assembly and Mutation Detection
2.5. Genome Annotation, Diversity and Clustering
2.6. Phylogenetic, Temporal and Recombination Analysis
3. Results
3.1. Elevated SNP Diversity at the 5′ and 3′ Accessory Genome Regions
3.2. Model-Based Classification Finds Four Genetic Groups
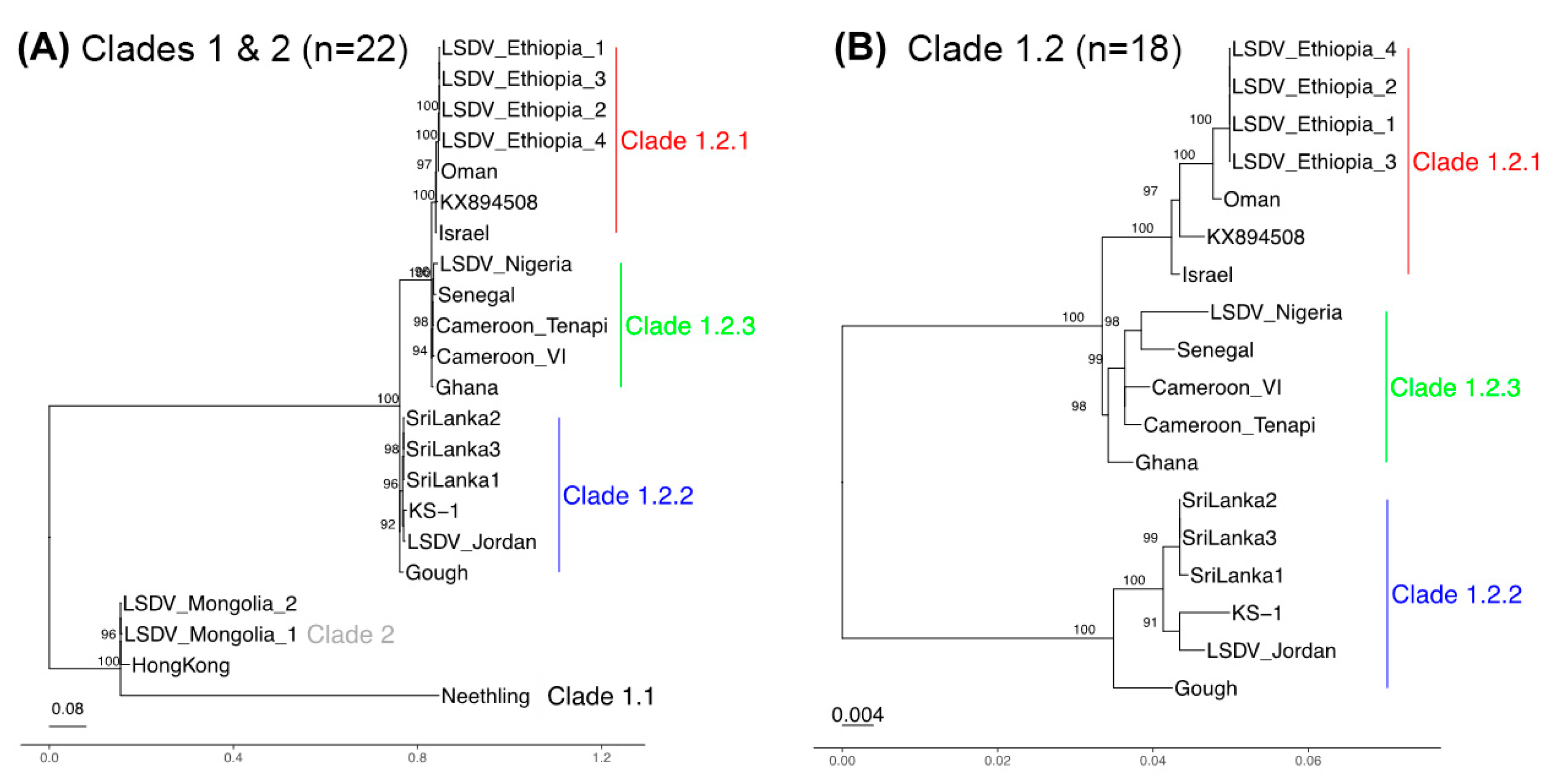
3.3. Phylogenetic Separation between and within Clades 1.1 and 1.2
3.4. A Genetically Distinct West and Central Africa Subgroup
3.5. Recombinant Origins of Hong Kong and the Mongolia Samples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Biswas, S.; Noyce, R.S.; Babiuk, L.A.; Lung, O.; Bulach, D.M.; Bowden, T.R.; Boyle, D.B.; Babiuk, S.; Evans, D.H. Extended sequencing of vaccine and wild-type capripoxvirus isolates provides insights into genes modulating virulence and host range. Transbound. Emerg. Dis. 2020, 67, 80–97. [Google Scholar] [CrossRef] [PubMed]
- Casal, J.; Allepuz, A.; Miteva, A.; Pite, L.; Tabakovsky, B.; Terzievski, D.; Alexandrov, T.; Beltran-Alcrudo, D. Economic cost of lumpy skin disease outbreaks in three Balkan countries: Albania, Bulgaria and the Former Yugoslav Republic of Macedonia (2016–2017). Transbound. Emerg. Dis. 2018, 65, 1680–1688. [Google Scholar] [CrossRef] [PubMed]
- Vinitchaikul, P.; Punyapornwithaya, V.; Seesupa, S.; Phuykhamsingha, S.; Arjkumpa, O.; Sansamur, C.; Jarassaeng, C. The first study on the impact of lumpy skin disease outbreaks on monthly milk production on dairy farms in Khon Kaen, Thailand. Vet. World 2023, 16, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Breman, F.C.; Haegeman, A.; Kresic, N.; Philips, W.; De Regge, N. Lumpy Skin Disease Virus Genome Sequence Analysis: Putative Spatio-Temporal Epidemiology, Single Gene versus Whole Genome Phylogeny and Genomic Evolution. Viruses 2023, 15, 1471. [Google Scholar] [CrossRef] [PubMed]
- Van Borm, S.; Dellicour, S.; Martin, D.P.; Lemey, P.; Agianniotaki, E.I.; Chondrokouki, E.D.; Vidanovic, D.; Vaskovic, N.; Petrovic, T.; Lazic, S.; et al. Complete genome reconstruction of the global and European regional dispersal history of the lumpy skin disease virus. J. Virol. 2023, 97, e01394-23. [Google Scholar] [CrossRef] [PubMed]
- Fay, P.C.; Cook, C.G.; Wijesiriwardana, N.; Tore, G.; Comtet, L.; Carpentier, A.; Shih, B.; Freimanis, G.; Haga, I.R.; Beard, P.M. Madin-Darby bovine kidney (MDBK) cells are a suitable cell line for the propagation and study of the bovine poxvirus lumpy skin disease virus. J. Virol. Methods 2020, 285, 113943. [Google Scholar] [CrossRef] [PubMed]
- Flannery, J.; Shih, B.; Haga, I.R.; Ashby, M.; Corla, A.; King, S.; Freimanis, G.; Polo, N.; Tse, A.C.; Brackman, C.J.; et al. A novel strain of lumpy skin disease virus causes clinical disease in cattle in Hong Kong. Transbound. Emerg. Dis. 2021, 69, e336–e343. [Google Scholar] [CrossRef] [PubMed]
- Hughes, L.; Wilkins, K.; Goldsmith, C.S.; Smith, S.; Hudson, P.; Patel, N.; Karem, K.; Damon, I.; Li, Y.; Olson, V.A.; et al. A rapid Orthopoxvirus purification protocol suitable for high-containment laboratories. J. Virol. Methods 2017, 243, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinformatics 2020, 70, e102. [Google Scholar] [CrossRef]
- Alonge, M.; Soyk, S.; Ramakrishnan, S.; Wang, X.; Goodwin, S.; Sedlazeck, F.J.; Lippman, Z.B.; Schatz, M.C. RaGOO: Fast and accurate reference-guided scaffolding of draft genomes. Genome Biol. 2019, 20, 224. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Pedersen, B.S.; Quinlan, A.R. Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics 2018, 34, 867–868. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. SNP-sites: Rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genom. 2016, 2, e000056. [Google Scholar] [CrossRef]
- Cook, D.E.; Andersen, E.C. VCF-kit: Assorted utilities for the variant call format. Bioinformatics 2017, 33, 1581–1582. [Google Scholar] [CrossRef]
- Tonkin-Hill, G.; Lees, J.A.; Bentley, S.D.; Frost, S.D.W.; Corander, J. Fast hierarchical Bayesian analysis of population structure. Nucleic Acids Res. 2019, 47, 5539–5549. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org (accessed on 21 December 2023).
- Team, R. RStudio: Integrated Development Environment for R. Available online: http://www.rstudio.com (accessed on 21 December 2023).
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H.; Hester, J.; Chang, W.; Bryan, J. devtools: Tools to Make Developing R Packages Easier. Available online: https://devtools.r-lib.org/ (accessed on 21 December 2023).
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Revell, L. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Rambaut, A. Figtree v1.4.4. Available online: https://github.com/rambaut/figtree/releases/tag/v1.4.4 (accessed on 21 December 2023).
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.G.; Lam, T.T.; Xu, S.; Dai, Z.; Zhou, L.; Feng, T.; Guo, P.; Dunn, C.W.; Jones, B.R.; Bradley, T.; et al. Treeio: An R Package for Phylogenetic Tree Input and Output with Richly Annotated and Associated Data. Mol. Biol. Evol. 2020, 37, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.M.; Ratmann, O.; Boni, M.F. Improved Algorithmic Complexity for the 3SEQ Recombination Detection Algorithm. Mol. Biol. Evol. 2018, 35, 247–251. [Google Scholar] [CrossRef]
- Watterson, G.A. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 1975, 7, 256–276. [Google Scholar] [CrossRef]
- Odonchimeg, M.; Erdenechimeg, D.; Tuvshinbayar, A.; Tsogtgerel, M.; Bazarragchaa, E.; Ulaankhuu, A.; Selenge, T.; Munkhgerel, D.; Munkhtsetseg, A.; Altanchimeg, A.; et al. Molecular identification and risk factor analysis of the first Lumpy skin disease outbreak in cattle in Mongolia. J. Vet. Med. Sci. 2022, 84, 1244–1252. [Google Scholar] [CrossRef]
- van Schalkwyk, A.; Kara, P.; Heath, L. Phylogenomic characterization of historic lumpy skin disease virus isolates from South Africa. Arch. Virol. 2022, 167, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Tuppurainen, E.S.; Pearson, C.R.; Bachanek-Bankowska, K.; Knowles, N.J.; Amareen, S.; Frost, L.; Henstock, M.R.; Lamien, C.E.; Diallo, A.; Mertens, P.P. Characterization of sheep pox virus vaccine for cattle against lumpy skin disease virus. Antivir. Res. 2014, 109, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yuan, Y.; Shao, J.; Sun, M.; He, W.; Chen, J.; Liu, Q. Genomic characterization of lumpy skin disease virus in southern China. Transbound. Emerg. Dis. 2021, 69, 2788–2799. [Google Scholar] [CrossRef]
- Mathijs, E.; Vandenbussche, F.; Nguyen, L.; Aerts, L.; Nguyen, T.; De Leeuw, I.; Quang, M.; Nguyen, H.D.; Philips, W.; Dam, T.V.; et al. Coding-Complete Sequences of Recombinant Lumpy Skin Disease Viruses Collected in 2020 from Four Outbreaks in Northern Vietnam. Microbiol. Resour. Announc. 2021, 10, e00897-21. [Google Scholar] [CrossRef] [PubMed]
- Sprygin, A.; Babin, Y.; Pestova, Y.; Kononova, S.; Wallace, D.B.; Van Schalkwyk, A.; Byadovskaya, O.; Diev, V.; Lozovoy, D.; Kononov, A. Analysis and insights into recombination signals in lumpy skin disease virus recovered in the field. PLoS ONE 2018, 13, e0207480. [Google Scholar] [CrossRef] [PubMed]
- Sprygin, A.; Van Schalkwyk, A.; Shumilova, I.; Nesterov, A.; Kononova, S.; Prutnikov, P.; Byadovskaya, O.; Kononov, A. Full-length genome characterization of a novel recombinant vaccine-like lumpy skin disease virus strain detected during the climatic winter in Russia, 2019. Arch. Virol. 2020, 165, 2675–2677. [Google Scholar] [CrossRef] [PubMed]
- Vandenbussche, F.; Mathijs, E.; Philips, W.; Saduakassova, M.; De Leeuw, I.; Sultanov, A.; Haegeman, A.; De Clercq, K. Recombinant LSDV Strains in Asia: Vaccine Spillover or Natural Emergence? Viruses 2022, 14, 1429. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.; Tuppurainen, E.; Adedeji, A.; Meseko, C.; Asala, O.; Adole, J.; Atai, R.; Dogonyaro, B.; Globig, A.; Hoffmann, D.; et al. Characterization of a Nigerian Lumpy Skin Disease Virus Isolate after Experimental Infection of Cattle. Pathogens 2021, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Bamouh, Z.; Hamdi, J.; Fellahi, S.; Khayi, S.; Jazouli, M.; Tadlaoui, K.O.; Fihri, O.F.; Tuppurainen, E.; Elharrak, M. Investigation of Post Vaccination Reactions of Two Live Attenuated Vaccines against Lumpy Skin Disease of Cattle. Vaccines 2021, 9, 621. [Google Scholar] [CrossRef]
- Gari, G.; Abie, G.; Gizaw, D.; Wubete, A.; Kidane, M.; Asgedom, H.; Bayissa, B.; Ayelet, G.; Oura, C.A.; Roger, F.; et al. Evaluation of the safety, immunogenicity and efficacy of three capripoxvirus vaccine strains against lumpy skin disease virus. Vaccine 2015, 33, 3256–3261. [Google Scholar] [CrossRef]
- Molla, W.; Frankena, K.; Gari, G.; de Jong, M.C.M. Field study on the use of vaccination to control the occurrence of lumpy skin disease in Ethiopian cattle. Prev. Vet. Med. 2017, 147, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Chander, Y.; Kumar, R.; Khandelwal, N.; Riyesh, T.; Chaudhary, K.; Shanmugasundaram, K.; Kumar, S.; Kumar, A.; Gupta, M.K.; et al. Isolation and characterization of lumpy skin disease virus from cattle in India. PLoS ONE 2021, 16, e0241022. [Google Scholar] [CrossRef] [PubMed]
- Sudhakar, S.B.; Mishra, N.; Kalaiyarasu, S.; Jhade, S.K.; Singh, V.P. Genetic and phylogenetic analysis of lumpy skin disease viruses (LSDV) isolated from the first and subsequent field outbreaks in India during 2019 reveals close proximity with unique signatures of historical Kenyan NI-2490/Kenya/KSGP-like field strains. Transbound. Emerg. Dis. 2021, 69, E451–E462. [Google Scholar] [CrossRef] [PubMed]
- Maw, M.T.; Khin, M.M.; Hadrill, D.; Meki, I.K.; Settypalli, T.B.K.; Kyin, M.M.; Myint, W.W.; Thein, W.Z.; Aye, O.; Palamara, E.; et al. First Report of Lumpy Skin Disease in Myanmar and Molecular Analysis of the Field Virus Isolates. Microorganisms 2022, 10, 897. [Google Scholar] [CrossRef]
- Abutarbush, S.M.; Hananeh, W.M.; Ramadan, W.; Al Sheyab, O.M.; Alnajjar, A.R.; Al Zoubi, I.G.; Knowles, N.J.; Bachanek-Bankowska, K.; Tuppurainen, E.S. Adverse Reactions to Field Vaccination against Lumpy Skin Disease in Jordan. Transbound. Emerg. Dis. 2016, 63, e213–e219. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Isolate Name | Abbreviation | Origin | Year 1 | Acc Number |
|---|---|---|---|---|
| LSDV/Gough/1959 | LSDV Gough | Unknown | 1959 | SRR27563725 |
| LSDV/Senegal/1997 | LSDV Senegal | Senegal | 1997 | SRR27563735 |
| LSDV/CameroonVI/2006 | LSDV Cameroon VI | Cameroon | 2006 | SRR27563734 |
| LSDV/Ghana/2006 | LSDV Ghana | Ghana | 2006 | SRR27563732 |
| LSDV/Israel/2007 | LSDV Israel | Israel | 2007 | SRR27563731 |
| LSDV/Ethiopia7_3/2019 | LSDV Ethiopia 4 | Ethiopia | 2019 | SRR28085235 |
| LSDV/Ethiopia3_2/2019 | LSDV Ethiopia 3 | Ethiopia | 2019 | SRR28085236 |
| LSDV/Ethiopia1_1/2019 | LSDV Ethiopia 1 | Ethiopia | 2019 | SRR28085238 |
| LSDV/Ethiopia21_4/2019 | LSDV Ethiopia 2 | Ethiopia | 2019 | SRR28085237 |
| LSDV/HongKong/2020 2 | LSDV Hong Kong | Hong Kong | 2020 | See ref [7] |
| LSDV/SriLanka1/2021 | LSDV Sri Lanka 1 | Sri Lanka | 2020 | SRR27563730 |
| LSDV/SriLanka2/2021 | LSDV Sri Lanka 2 | Sri Lanka | 2020 | SRR27563729 |
| LSDV/SriLanka3/2021 | LSDV Sri Lanka 3 | Sri Lanka | 2020 | SRR27563728 |
| LSDV/MongoliaM5_S6/2021 | LSDV Mongolia 1 | Mongolia | 2021 | SRR28085241 |
| LSDV/MongoliaM2_S5/2021 | LSDV Mongolia 2 | Mongolia | 2021 | SRR28085240 |
| LSDV/Nigeria_Bokkos_S10/2021 | LSDV Nigeria | Nigeria | 2021 3 | SRR28085239 |
| LSDV/KS_1/1995 | LSDV KS-1 | Kenya | 1995 | SRR27563727 |
| LSDV/Oman/2009 | LSDV Oman | Oman | 2009 | SRR27563726 |
| LSDV/Jordan/2013 | LSDV Jordan | Jordan | 2013 | SRR27563724 |
| LSDV/CameroonTenapi/UK | LSDV Cameroon Tenapi | Cameroon | Unknown | SRR27563723 |
| LSDV/Neethling/UK | LSDV Neethling | Unknown | Unknown | SRR27563733 |
| Region | Length | #SNPs | SNPs/Kb | Mean Number of Pairwise SNPs | Theta/Kb | Pi/Kb |
|---|---|---|---|---|---|---|
| Whole genome | 150,562 | 2023 | 13.5 | 452 | 4.5 | 3.0 |
| 5′ accessory region | 11,350 | 260 | 22.9 | 60 | 7.4 | 5.3 |
| Core genome | 93,060 | 939 | 10.1 | 213 | 3.3 | 2.3 |
| 3′ accessory region | 41,090 | 816 | 19.9 | 174 | 6.4 | 4.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haga, I.R.; Shih, B.B.; Tore, G.; Polo, N.; Ribeca, P.; Gombo-Ochir, D.; Shura, G.; Tserenchimed, T.; Enkhbold, B.; Purevtseren, D.; et al. Sequencing and Analysis of Lumpy Skin Disease Virus Whole Genomes Reveals a New Viral Subgroup in West and Central Africa. Viruses 2024, 16, 557. https://doi.org/10.3390/v16040557
Haga IR, Shih BB, Tore G, Polo N, Ribeca P, Gombo-Ochir D, Shura G, Tserenchimed T, Enkhbold B, Purevtseren D, et al. Sequencing and Analysis of Lumpy Skin Disease Virus Whole Genomes Reveals a New Viral Subgroup in West and Central Africa. Viruses. 2024; 16(4):557. https://doi.org/10.3390/v16040557
Chicago/Turabian StyleHaga, Ismar R., Barbara B. Shih, Gessica Tore, Noemi Polo, Paolo Ribeca, Delgerzul Gombo-Ochir, Gansukh Shura, Tsagaan Tserenchimed, Bazarragchaa Enkhbold, Dulam Purevtseren, and et al. 2024. "Sequencing and Analysis of Lumpy Skin Disease Virus Whole Genomes Reveals a New Viral Subgroup in West and Central Africa" Viruses 16, no. 4: 557. https://doi.org/10.3390/v16040557
APA StyleHaga, I. R., Shih, B. B., Tore, G., Polo, N., Ribeca, P., Gombo-Ochir, D., Shura, G., Tserenchimed, T., Enkhbold, B., Purevtseren, D., Ulziibat, G., Damdinjav, B., Yimer, L., Bari, F. D., Gizaw, D., Adedeji, A. J., Atai, R. B., Adole, J. A., Dogonyaro, B. B., ... Beard, P. M. (2024). Sequencing and Analysis of Lumpy Skin Disease Virus Whole Genomes Reveals a New Viral Subgroup in West and Central Africa. Viruses, 16(4), 557. https://doi.org/10.3390/v16040557