

STINGing Defenses: Unmasking the Mechanisms of DNA Oncovirus-Mediated Immune Escape




Abstract
:1. Introduction
1.1. Oncogenic DNA Viruses
1.1.1. Herpesviruses
1.1.2. Papillomaviruses
1.1.3. Hepatitis B Virus
1.2. Cancer Immunosurveillance
1.3. STING Pathway
1.3.1. cGAS
1.3.2. cGAMP
1.3.3. STING
1.3.4. TBK1/IRF3/NF-κB
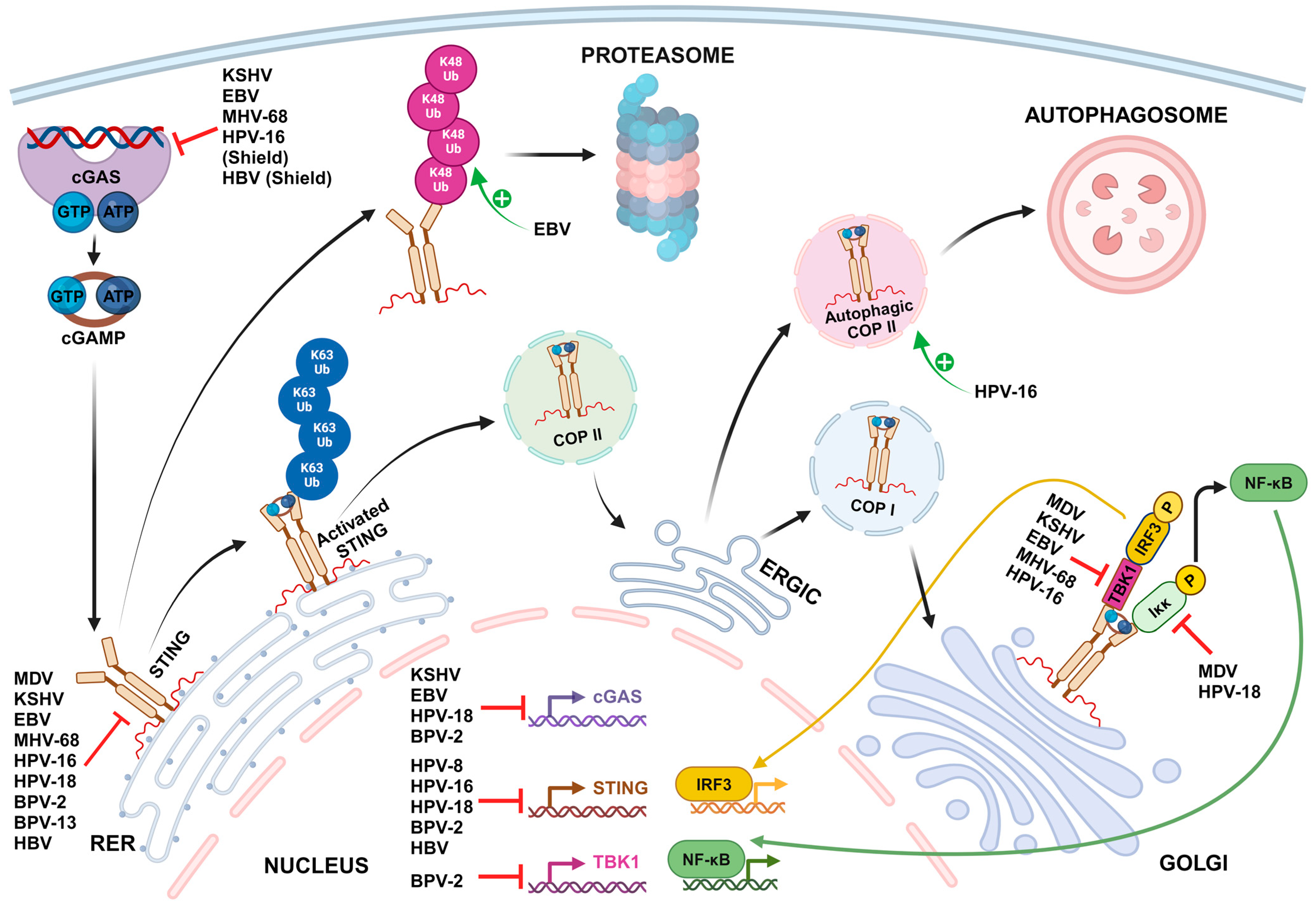
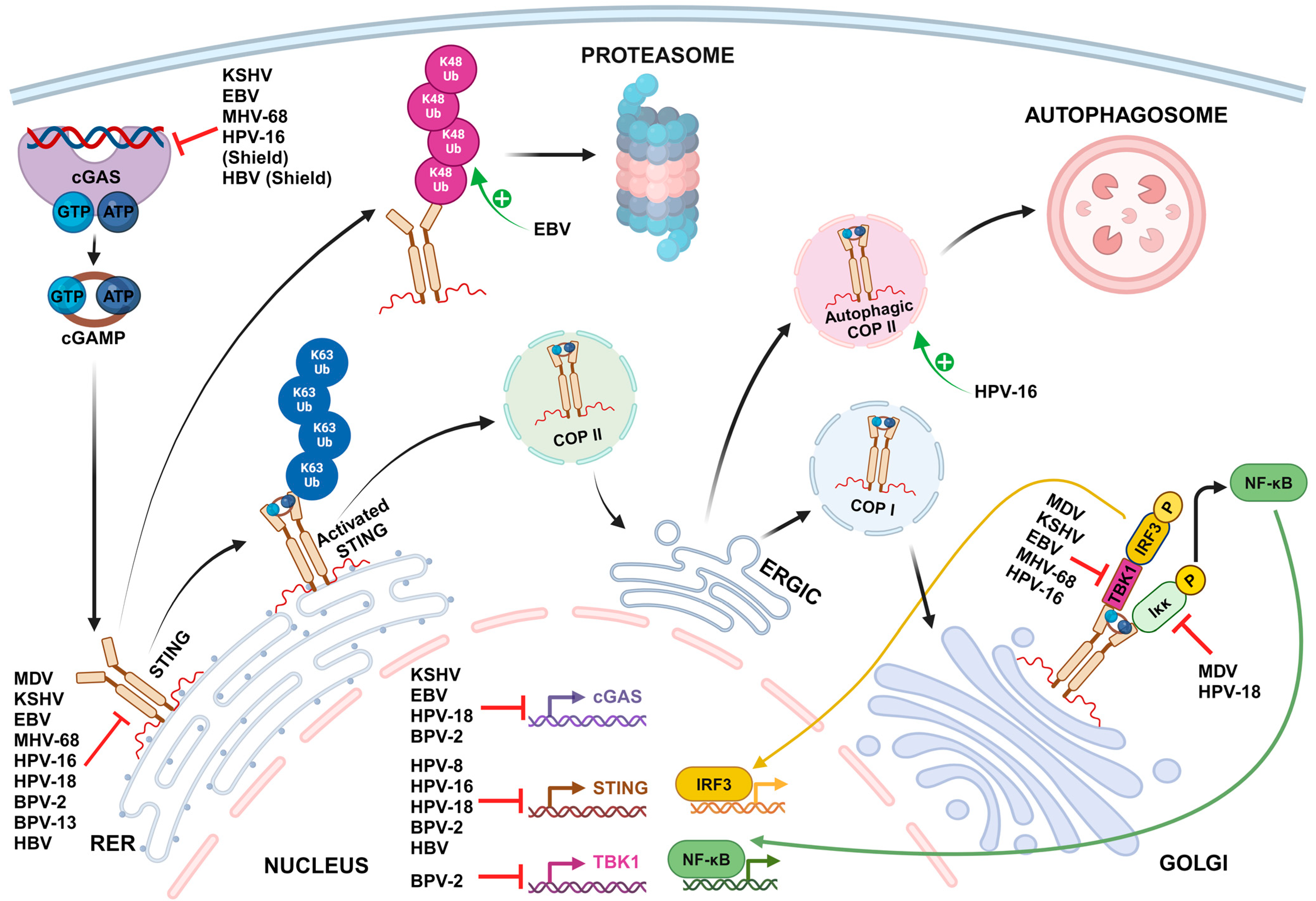
2. Mechanisms of STING-Mediated Immune Evasion by Oncogenic DNA Viruses
2.1. Shielding the Viral Genome from cGAS Sensing
2.2. Transcriptional and Post-Transcriptional Inhibition of cGAS-STING Pathway Gene Expression
2.3. Inhibition of cGAS DNA Binding and Activation
2.4. STING Inhibition through Direct Binding
2.5. Inhibition of TBK1 and cGAS-STING-Mediated Activation of Transcription Factors
3. Targeting the cGAS/STING Associated Immune Evasion Mechanisms as a Therapeutical Approach
Author Contributions
Funding
Conflicts of Interest
References
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global Burden of Cancers Attributable to Infections in 2012: A Synthetic Analysis. Lancet Glob. Health 2016, 4, e609–e616. [Google Scholar] [CrossRef]
- Chang, Y.; Moore, P.S.; Weiss, R.A. Human Oncogenic Viruses: Nature and Discovery. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160264. [Google Scholar] [CrossRef]
- Mattisson, K. Über Die Sogenannte Kapillaranalyse Vom Magensaft Nach Dr. I. Holmgren. Arch. Für Verdauungskrankheiten 2009, 19, 226–231. [Google Scholar] [CrossRef]
- zur Hausen, H. The Search for Infectious Causes of Human Cancers: Where and Why. Virology 2009, 392, 1–10. [Google Scholar] [CrossRef]
- Moore, P.S.; Chang, Y. Why Do Viruses Cause Cancer? Highlights of the First Century of Human Tumour Virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef]
- Mui, U.N.; Haley, C.T.; Tyring, S.K. Viral Oncology: Molecular Biology and Pathogenesis. J. Clin. Med. 2017, 6, 111. [Google Scholar] [CrossRef]
- Tang, K.-W.; Larsson, E. Tumour Virology in the Era of High-Throughput Genomics. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160265. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Jhunjhunwala, S.; Liu, J.; Haverty, P.M.; Kennemer, M.I.; Guan, Y.; Lee, W.; Carnevali, P.; Stinson, J.; Johnson, S.; et al. The Effects of Hepatitis B Virus Integration into the Genomes of Hepatocellular Carcinoma Patients. Genome Res. 2012, 22, 593–601. [Google Scholar] [CrossRef]
- Hu, Z.; Zhu, D.; Wang, W.; Li, W.; Jia, W.; Zeng, X.; Ding, W.; Yu, L.; Wang, X.; Wang, L.; et al. Genome-Wide Profiling of HPV Integration in Cervical Cancer Identifies Clustered Genomic Hot Spots and a Potential Microhomology-Mediated Integration Mechanism. Nat. Genet. 2015, 47, 158–163. [Google Scholar] [CrossRef]
- Krump, N.A.; You, J. Molecular Mechanisms of Viral Oncogenesis in Humans. Nat. Rev. Microbiol. 2018, 16, 684–698. [Google Scholar] [CrossRef]
- Gatherer, D.; Depledge, D.P.; Hartley, C.A.; Szpara, M.L.; Vaz, P.K.; Benkő, M.; Brandt, C.R.; Bryant, N.A.; Dastjerdi, A.; Doszpoly, A.; et al. ICTV Virus Taxonomy Profile: Herpesviridae 2021. J. Gen. Virol. 2021, 102, 001673. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.J. Epstein–Barr Virus and Cancer. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 29–53. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. Herpesvirus Latency. J. Clin. Investig. 2020, 130, 3361–3369. [Google Scholar] [CrossRef]
- Grinde, B. Herpesviruses: Latency and Reactivation—Viral Strategies and Host Response. J. Oral Microbiol. 2013, 5, 22766. [Google Scholar] [CrossRef]
- Biggs, P.M.; Nair, V. The Long View: 40 Years of Marek’s Disease Research and Avian Pathology. Avian Pathol. 2012, 41, 3–9. [Google Scholar] [CrossRef]
- Bertzbach, L.D.; Conradie, A.M.; You, Y.; Kaufer, B.B. Latest Insights into Marek’s Disease Virus Pathogenesis and Tumorigenesis. Cancers 2020, 12, 647. [Google Scholar] [CrossRef]
- McPherson, M.C.; Delany, M.E. Virus and Host Genomic, Molecular, and Cellular Interactions during Marek’s Disease Pathogenesis and Oncogenesis. Poult. Sci. 2016, 95, 412–429. [Google Scholar] [CrossRef]
- Khan, G.; Fitzmaurice, C.; Naghavi, M.; Ahmed, L.A. Global and Regional Incidence, Mortality and Disability-Adjusted Life-Years for Epstein-Barr Virus-Attributable Malignancies, 1990–2017. BMJ Open 2020, 10, e037505. [Google Scholar] [CrossRef]
- Münz, C. Latency and Lytic Replication in Epstein–Barr Virus-Associated Oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef]
- Abbott, R.J.; Pachnio, A.; Pedroza-Pacheco, I.; Leese, A.M.; Begum, J.; Long, H.M.; Croom-Carter, D.; Stacey, A.; Moss, P.A.H.; Hislop, A.D.; et al. Asymptomatic Primary Infection with Epstein-Barr Virus: Observations on Young Adult Cases. J. Virol. 2017, 91, e00382-17. [Google Scholar] [CrossRef]
- Cohen, J.I.; Fauci, A.S.; Varmus, H.; Nabel, G.J. Epstein-Barr Virus: An Important Vaccine Target for Cancer Prevention. Sci. Transl. Med. 2011, 3, 107fs7. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s Sarcoma–Associated Herpesvirus-Like DNA Sequences in AIDS-Related Body-Cavity–Based Lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi Sarcoma. Nat. Rev. Dis. Primers 2019, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Chinula, L.; Moses, A.; Gopal, S. HIV-Associated Malignancies in Sub-Saharan Africa: Progress, Challenges, and Opportunities. Curr. Opin. HIV AIDS 2017, 12, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of Herpesvirus-Like DNA Sequences in AIDS-Sssociated Kaposi’s Sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.-F.; Clauvel, J.-P.; Raphael, M.; Degos, L.; et al. Kaposi’s Sarcoma-Associated Herpesvirus-Like DNA Sequences in Multicentric Castleman’s Disease. Blood 1995, 86, 1276–1280. [Google Scholar] [CrossRef] [PubMed]
- Mariggiò, G.; Koch, S.; Schulz, T.F. Kaposi Sarcoma Herpesvirus Pathogenesis. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160275. [Google Scholar] [CrossRef] [PubMed]
- Dow, D.E.; Cunningham, C.K.; Buchanan, A.M. A Review of Human Herpesvirus 8, the Kaposi’s Sarcoma-Associated Herpesvirus, in the Pediatric Population. J. Pediatr. Infect. Dis. Soc. 2014, 3, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Giffin, L.; Damania, B. Chapter Two—KSHV: Pathways to Tumorigenesis and Persistent Infection. In Advances in Virus Research; Maramorosch, K., Murphy, F.A., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 88, pp. 111–159. [Google Scholar] [CrossRef]
- De Paoli, P.; Carbone, A. Kaposi’s Sarcoma Herpesvirus: Twenty years after its discovery. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 1288–1294. Available online: https://www.europeanreview.org/article/10575 (accessed on 15 January 2024).
- Douglas, J.L.; Gustin, J.K.; Moses, A.V.; Dezube, B.J.; Pantanowitz, L. Kaposi Sarcoma Pathogenesis: A Triad of Viral Infection, Oncogenesis and Chronic Inflammation. Transl. Biomed. 2010, 1, 172. [Google Scholar]
- Egawa, N.; Egawa, K.; Griffin, H.; Doorbar, J. Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia. Viruses 2015, 7, 3863–3890. [Google Scholar] [CrossRef] [PubMed]
- de Villiers, E.-M.; Fauquet, C.; Broker, T.R.; Bernard, H.-U.; zur Hausen, H. Classification of Papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Araldi, R.P.; Assaf, S.M.R.; de Carvalho, R.F.; de Carvalho, M.A.C.R.; de Souza, J.M.; Magnelli, R.F.; Módolo, D.G.; Roperto, F.P.; de Cassia Stocco, R.; Beçak, W. Papillomaviruses: A Systematic Review. Genet. Mol. Biol. 2017, 40, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Frias-De-Diego, A.; Jara, M.; Escobar, L.E. Papillomavirus in Wildlife. Front. Ecol. Evol. 2019, 7, 406. [Google Scholar] [CrossRef]
- de Sanjosé, S.; Diaz, M.; Castellsagué, X.; Clifford, G.; Bruni, L.; Muñoz, N.; Bosch, F.X. Worldwide Prevalence and Genotype Distribution of Cervical Human Papillomavirus DNA in Women with Normal Cytology: A Meta-Analysis. Lancet Infect. Dis. 2007, 7, 453–459. [Google Scholar] [CrossRef]
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; et al. Global Burden of Human Papillomavirus and Related Diseases. Vaccine 2012, 30, F12–F23. [Google Scholar] [CrossRef]
- Bruni, L.; Albero, G.; Rowley, J.; Alemany, L.; Arbyn, M.; Giuliano, A.R.; Markowitz, L.E.; Broutet, N.; Taylor, M. Global and Regional Estimates of Genital Human Papillomavirus Prevalence among Men: A Systematic Review and Meta-Analysis. Lancet Glob. Health 2023, 11, e1345–e1362. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.C.; Schiffman, M.; Herrero, R.; Wacholder, S.; Hildesheim, A.; Castle, P.E.; Solomon, D.; Burk, R.; On behalf of the Proyecto Epidemiológico Guanacaste Group. Rapid Clearance of Human Papillomavirus and Implications for Clinical Focus on Persistent Infections. JNCI J. Natl. Cancer Inst. 2008, 100, 513–517. [Google Scholar] [CrossRef]
- de Sanjosé, S.; Serrano, B.; Tous, S.; Alejo, M.; Lloveras, B.; Quirós, B.; Clavero, O.; Vidal, A.; Ferrándiz-Pulido, C.; Pavón, M.Á.; et al. Burden of Human Papillomavirus (HPV)-Related Cancers Attributable to HPVs 6/11/16/18/31/33/45/52 and 58. JNCI Cancer Spectr. 2018, 2, pky045. [Google Scholar] [CrossRef]
- Milano, G.; Guarducci, G.; Nante, N.; Montomoli, E.; Manini, I. Human Papillomavirus Epidemiology and Prevention: Is There Still a Gender Gap? Vaccines 2023, 11, 1060. [Google Scholar] [CrossRef]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The Biology and Life-Cycle of Human Papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.K.; Aimagambetova, G.; Ukybassova, T.; Kongrtay, K.; Azizan, A. Human Papillomavirus Infection and Cervical Cancer: Epidemiology, Screening, and Vaccination—Review of Current Perspectives. J. Oncol. 2019, 2019, e3257939. [Google Scholar] [CrossRef]
- Monsonego, J.; Cox, J.T.; Behrens, C.; Sandri, M.; Franco, E.L.; Yap, P.-S.; Huh, W. Prevalence of High-Risk Human Papilloma Virus Genotypes and Associated Risk of Cervical Precancerous Lesions in a Large U.S. Screening Population: Data from the ATHENA Trial. Gynecol. Oncol. 2015, 137, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; Ghissassi, F.E.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A Review of Human Carcinogens—Part B: Biological Agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. Keratinocyte Differentiation-Dependent Human Papillomavirus Gene Regulation. Viruses 2017, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, N.; Satsuka, A.; Kawate, A.; Sakai, H. Productive Lifecycle of Human Papillomaviruses That Depends upon Squamous Epithelial Differentiation. Front. Microbiol. 2012, 3, 152. [Google Scholar] [CrossRef] [PubMed]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of Human Papillomavirus Infection Requires Cell Cycle Progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef]
- Campos, S.K. Subcellular Trafficking of the Papillomavirus Genome during Initial Infection: The Remarkable Abilities of Minor Capsid Protein L2. Viruses 2017, 9, 370. [Google Scholar] [CrossRef]
- Ferreira, A.R.; Ramalho, A.C.; Marques, M.; Ribeiro, D. The Interplay between Antiviral Signalling and Carcinogenesis in Human Papillomavirus Infections. Cancers 2020, 12, 646. [Google Scholar] [CrossRef]
- Hoppe-Seyler, K.; Bossler, F.; Braun, J.A.; Herrmann, A.L.; Hoppe-Seyler, F. The HPV E6/E7 Oncogenes: Key Factors for Viral Carcinogenesis and Therapeutic Targets. Trends Microbiol. 2018, 26, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Scheurer, M.E.; Tortolero-Luna, G.; Adler-Storthz, K. Human Papillomavirus Infection: Biology, Epidemiology, and Prevention. Int. J. Gynecol. Cancer 2005, 15, 727–746. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human Papillomavirus Oncoproteins: Pathways to Transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Bocaneti, F.; Altamura, G.; Corteggio, A.; Velescu, E.; Roperto, F.; Borzacchiello, G. Bovine Papillomavirus: New Insights into an Old Disease. Transbound. Emerg. Dis. 2016, 63, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Daudt, C.; Da Silva, F.R.C.; Lunardi, M.; Alves, C.B.D.T.; Weber, M.N.; Cibulski, S.P.; Alfieri, A.F.; Alfieri, A.A.; Canal, C.W. Papillomaviruses in Ruminants: An Update. Transbound. Emerg. Dis. 2018, 65, 1381–1395. [Google Scholar] [CrossRef] [PubMed]
- Medeiros-Fonseca, B.; Abreu-Silva, A.L.; Medeiros, R.; Oliveira, P.A.; Gil da Costa, R.M. Pteridium spp. and Bovine Papillomavirus: Partners in Cancer. Front. Vet. Sci. 2021, 8, 758720. [Google Scholar] [CrossRef] [PubMed]
- Roperto, S.; Russo, V.; Ozkul, A.; Sepici-Dincel, A.; Maiolino, P.; Borzacchiello, G.; Marcus, I.; Esposito, I.; Riccardi, M.G.; Roperto, F. Bovine Papillomavirus Type 2 Infects the Urinary Bladder of Water Buffalo (Bubalus bubalis) and Plays a Crucial Role in Bubaline Urothelial Carcinogenesis. J. Gen. Virol. 2013, 94, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Resendes, A.R.; Roperto, S.; Trapani, F.; Urraro, C.; Rodrigues, A.; Roperto, F.; Borzacchiello, G. Association of Bovine Papillomavirus Type 2 (BPV-2) and Urinary Bladder Tumours in Cattle from the Azores Archipelago. Res. Vet. Sci. 2011, 90, 526–529. [Google Scholar] [CrossRef]
- Roperto, S.; Borzacchiello, G.; Brun, R.; Leonardi, L.; Maiolino, P.; Martano, M.; Paciello, O.; Papparella, S.; Restucci, B.; Russo, V.; et al. A Review of Bovine Urothelial Tumours and Tumour-Like Lesions of the Urinary Bladder. J. Comp. Pathol. 2010, 142, 95–108. [Google Scholar] [CrossRef]
- Venuti, A.; Paolini, F.; Nasir, L.; Corteggio, A.; Roperto, S.; Campo, M.S.; Borzacchiello, G. Papillomavirus E5: The Smallest Oncoprotein with Many Functions. Mol. Cancer 2011, 10, 140. [Google Scholar] [CrossRef]
- Global Progress Report on HIV, Viral Hepatitis and Sexually Transmitted Infections. 2021. Available online: https://www.who.int/publications-detail-redirect/9789240027077 (accessed on 17 January 2024).
- Seeger, C.; Mason, W.S. Molecular Biology of Hepatitis B Virus Infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef]
- Tuttleman, J.S.; Pourcel, C.; Summers, J. Formation of the Pool of Covalently Closed Circular Viral DNA in Hepadnavirus-Infected Cells. Cell 1986, 47, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M. HBV cccDNA: Viral Persistence Reservoir and Key Obstacle for a Cure of Chronic Hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef]
- Seto, W.-K.; Lo, Y.-R.; Pawlotsky, J.-M.; Yuen, M.-F. Chronic Hepatitis B Virus Infection. Lancet 2018, 392, 2313–2324. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.-N.; Kuo, C.-F.; Ou, J.-H.J. Mechanisms of Hepatitis B Virus Persistence. Trends Microbiol. 2018, 26, 33–42. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Isogawa, M.; Chisari, F.V. Host–Virus Interactions in Hepatitis B Virus Infection. Curr. Opin. Immunol. 2015, 36, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Ferrari, C. Adaptive Immunity in HBV Infection. J. Hepatol. 2016, 64, S71–S83. [Google Scholar] [CrossRef]
- Shi, Y.; Zheng, M. Hepatitis B Virus Persistence and Reactivation. BMJ 2020, 370, m2200. [Google Scholar] [CrossRef]
- Iannacone, M.; Guidotti, L.G. Immunobiology and Pathogenesis of Hepatitis B Virus Infection. Nat. Rev. Immunol. 2022, 22, 19–32. [Google Scholar] [CrossRef]
- Tang, L.S.Y.; Covert, E.; Wilson, E.; Kottilil, S. Chronic Hepatitis B Infection: A Review. JAMA 2018, 319, 1802–1813. [Google Scholar] [CrossRef]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-Induced Hepatocellular Carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef] [PubMed]
- Prüll, C.-R. Arthur M Silverstein, Paul Ehrlich’s Receptor Immunology: The Magnificent Obsession, San Diego and London, Academic Press, 2002, Pp. Xix, 202, Illus., US$75.00 (Hardback 0-12-643765-3). Med. Hist. 2003, 47, 266–267. [Google Scholar] [CrossRef]
- Thomas, L. Cellular and Humoral Aspects of the Hypersensitive States: A Symposium at the New York Academy of Medicine. J. Am. Med. Assoc. 1959, 170, 883. [Google Scholar] [CrossRef]
- Burnet, M. Cancer; a Biological Approach. I. The Processes of Control. Br. Med. J. 1957, 1, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Burnet, F.M. The Concept of Immunological Surveillance. Prog. Exp. Tumor Res. 1970, 13, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. The Concept of Immune Surveillance against Tumors: The First Theories. Oncotarget 2016, 8, 7175–7180. [Google Scholar] [CrossRef] [PubMed]
- Old, L.J.; Boyse, E.A. Immunology of Experimental Tumors. Annu. Rev. Med. 1964, 15, 167–186. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef] [PubMed]
- Bredholt, G.; Mannelqvist, M.; Stefansson, I.M.; Birkeland, E.; Bø, T.H.; Øyan, A.M.; Trovik, J.; Kalland, K.-H.; Jonassen, I.; Salvesen, H.B.; et al. Tumor Necrosis Is an Important Hallmark of Aggressive Endometrial Cancer and Associates with Hypoxia, Angiogenesis and Inflammation Responses. Oncotarget 2015, 6, 39676–39691. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Savitsky, D.; Tamura, T.; Taniguchi, T. Regulation of the Cytosolic DNA-Sensing System in Innate Immunity: A Current View. Curr. Opin. Immunol. 2009, 21, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Astudillo-de la Vega, H.; Ruiz-Garcia, E.; Lopez-Camarillo, C.; de la Garza-Salazar, J.G.; Meneses-Garcia, A.; Benitez-Bribiesca, L. Malignant Transforming Mechanisms of Human Papillomavirus. In Cervical Cancer; de la Garza-Salazar, J.G., Morales-Vásquez, F., Meneses-Garcia, A., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 35–56. [Google Scholar] [CrossRef]
- Vossen, M.T.; Westerhout, E.M.; Söderberg-Nauclér, C.; Wiertz, E.J. Viral Immune Evasion: A Masterpiece of Evolution. Immunogenetics 2002, 54, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Schreiber, R.D. Cancer Immunoediting: Antigens, Mechanisms, and Implications to Cancer Immunotherapy. Ann. N. Y. Acad. Sci. 2013, 1284, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Barbalat, R.; Ewald, S.E.; Mouchess, M.L.; Barton, G.M. Nucleic Acid Recognition by the Innate Immune System. Annu. Rev. Immunol. 2011, 29, 185–214. [Google Scholar] [CrossRef]
- Ma, Z.; Damania, B. The cGAS-STING Defense Pathway and Its Counteraction by Viruses. Cell Host Microbe 2016, 19, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. cGAS Produces a 2′-5′-Linked Cyclic Dinucleotide Second Messenger That Activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef]
- Sun, H.; Huang, Y.; Mei, S.; Xu, F.; Liu, X.; Zhao, F.; Yin, L.; Zhang, D.; Wei, L.; Wu, C.; et al. A Nuclear Export Signal Is Required for cGAS to Sense Cytosolic DNA. Cell Rep. 2021, 34, 108586. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Chen, Z.J. DNA-Induced Liquid Phase Condensation of cGAS Activates Innate Immune Signaling. Science 2018, 361, 704–709. [Google Scholar] [CrossRef]
- Zhao, M.; Xia, T.; Xing, J.-Q.; Yin, L.-H.; Li, X.-W.; Pan, J.; Liu, J.-Y.; Sun, L.-M.; Wang, M.; Li, T.; et al. The Stress Granule Protein G3BP1 Promotes Pre-Condensation of cGAS to Allow Rapid Responses to DNA. EMBO Rep. 2022, 23, e53166. [Google Scholar] [CrossRef]
- Casella, G.; Rasouli, J.; Mason, K.; Boehm, A.; Kumar, G.; Hwang, D.; Thome, R.; Ishikawa, L.; Zhang, G.-X.; Ciric, B.; et al. A Serine Protease Inhibitor Suppresses Autoimmune Neuroinflammation by Activating the STING/IFN-β Axis in Macrophages. Cell. Mol. Immunol. 2020, 17, 1278–1280. [Google Scholar] [CrossRef]
- Barnett, K.C.; Coronas-Serna, J.M.; Zhou, W.; Ernandes, M.J.; Cao, A.; Kranzusch, P.J.; Kagan, J.C. Phosphoinositide Interactions Position cGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell 2019, 176, 1432–1446.e11. [Google Scholar] [CrossRef] [PubMed]
- Guey, B.; Wischnewski, M.; Decout, A.; Makasheva, K.; Kaynak, M.; Sakar, M.S.; Fierz, B.; Ablasser, A. BAF Restricts cGAS on Nuclear DNA to Prevent Innate Immune Activation. Science 2020, 369, 823–828. [Google Scholar] [CrossRef]
- Pathare, G.R.; Decout, A.; Glück, S.; Cavadini, S.; Makasheva, K.; Hovius, R.; Kempf, G.; Weiss, J.; Kozicka, Z.; Guey, B.; et al. Structural Mechanism of cGAS Inhibition by the Nucleosome. Nature 2020, 587, 668–672. [Google Scholar] [CrossRef]
- Hu, M.-M.; Yang, Q.; Xie, X.-Q.; Liao, C.-Y.; Lin, H.; Liu, T.-T.; Yin, L.; Shu, H.-B. Sumoylation Promotes the Stability of the DNA Sensor cGAS and the Adaptor STING to Regulate the Kinetics of Response to DNA Virus. Immunity 2016, 45, 555–569. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Yu, H.; Zheng, X.; Peng, R.; Wang, Q.; Zhou, Y.; Wang, R.; Wang, J.; Qu, B.; Shen, N.; et al. SENP7 Potentiates cGAS Activation by Relieving SUMO-Mediated Inhibition of Cytosolic DNA Sensing. PLoS Pathog. 2017, 13, e1006156. [Google Scholar] [CrossRef]
- Chen, M.; Meng, Q.; Qin, Y.; Liang, P.; Tan, P.; He, L.; Zhou, Y.; Chen, Y.; Huang, J.; Wang, R.-F.; et al. TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor P62 to Promote Innate Immune Responses. Mol. Cell 2016, 64, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Jiang, F.; Kong, L.; Li, B.; Yang, Y.; Zhang, L.; Liu, B.; Zheng, Y.; Gao, C. Cutting Edge: USP27X Deubiquitinates and Stabilizes the DNA Sensor cGAS to Regulate Cytosolic DNA–Mediated Signaling. J. Immunol. 2019, 203, 2049–2054. [Google Scholar] [CrossRef]
- Shi, C.; Yang, X.; Liu, Y.; Li, H.; Chu, H.; Li, G.; Yin, H. ZDHHC18 Negatively Regulates cGAS-Mediated Innate Immunity through Palmitoylation. EMBO J. 2022, 41, e109272. [Google Scholar] [CrossRef]
- Xia, P.; Ye, B.; Wang, S.; Zhu, X.; Du, Y.; Xiong, Z.; Tian, Y.; Fan, Z. Glutamylation of the DNA Sensor cGAS Regulates Its Binding and Synthase Activity in Antiviral Immunity. Nat. Immunol. 2016, 17, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, I.; Behl, B.; Mendonca, M.; Shrivastava, G.; Russo, A.J.; Menoret, A.; Ghosh, A.; Vella, A.T.; Vanaja, S.K.; Sarkar, S.N.; et al. Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity 2018, 49, 413–426.e5. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Tan, H.-Y.; Feng, Y.-G.; Zhang, C.; Chen, F.; Feng, Y. microRNA-23a in Human Cancer: Its Roles, Mechanisms and Therapeutic Relevance. Cancers 2019, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Chu, L.; Li, Y.; Wang, Q.; Zhu, J.; Wang, C.; Cui, S. miR-23a/b Suppress cGAS-Mediated Innate and Autoimmunity. Cell. Mol. Immunol. 2021, 18, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Diner, E.J.; Burdette, D.L.; Wilson, S.C.; Monroe, K.M.; Kellenberger, C.A.; Hyodo, M.; Hayakawa, Y.; Hammond, M.C.; Vance, R.E. The Innate Immune DNA Sensor cGAS Produces a Noncanonical Cyclic Dinucleotide That Activates Human STING. Cell Rep. 2013, 3, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Cordova, A.F.; Ritchie, C.; Böhnert, V.; Li, L. Human SLC46A2 Is the Dominant cGAMP Importer in Extracellular cGAMP-Sensing Macrophages and Monocytes. ACS Cent. Sci. 2021, 7, 1073–1088. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, Y.; Zhang, X.; An, X.; Weng, M.; Shi, J.; Wang, S.; Liu, C.; Luo, S.; Zheng, T. An Alternatively Spliced STING Isoform Localizes in the Cytoplasmic Membrane and Directly Senses Extracellular cGAMP. J. Clin. Investig. 2022, 132, e144339. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Duran, M.A.; Dhanota, N.; Chatila, W.K.; Bettigole, S.E.; Kwon, J.; Sriram, R.K.; Humphries, M.P.; Salto-Tellez, M.; James, J.A.; et al. Metastasis and Immune Evasion from Extracellular cGAMP Hydrolysis. Cancer Discov. 2021, 11, 1212–1227. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, H.; Wu, J.; Zhang, X.; Sun, L.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Containing Mixed Phosphodiester Linkages Is An Endogenous High-Affinity Ligand for STING. Mol. Cell 2013, 51, 226–235. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Hayashi, T.; Takahara, K.; Satoh, T.; Lee, H.; Matsunaga, K.; Kageyama, S.; Omori, H.; Noda, T.; et al. Atg9a Controls dsDNA-Driven Dynamic Translocation of STING and the Innate Immune Response. Proc. Natl. Acad. Sci. USA 2009, 106, 20842–20846. [Google Scholar] [CrossRef]
- Jia, M.; Qin, D.; Zhao, C.; Chai, L.; Yu, Z.; Wang, W.; Tong, L.; Lv, L.; Wang, Y.; Rehwinkel, J.; et al. Redox Homeostasis Maintained by GPX4 Facilitates STING Activation. Nat. Immunol. 2020, 21, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-D.; Xiong, T.-C.; Yao, S.-Q.; Wei, M.-C.; Chen, M.; Lin, D.; Zhong, B. RNF115 Plays Dual Roles in Innate Antiviral Responses by Catalyzing Distinct Ubiquitination of MAVS and MITA. Nat. Commun. 2020, 11, 5536. [Google Scholar] [CrossRef] [PubMed]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS–STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, X.; Veleeparambil, M.; Kessler, P.M.; Willard, B.; Chattopadhyay, S.; Sen, G.C. EGFR-Mediated Tyrosine Phosphorylation of STING Determines Its Trafficking Route and Cellular Innate Immunity Functions. EMBO J. 2020, 39, e104106. [Google Scholar] [CrossRef]
- Ni, G.; Ma, Z.; Wong, J.P.; Zhang, Z.; Cousins, E.; Major, M.B.; Damania, B. PPP6C Negatively Regulates STING-Dependent Innate Immune Responses. mBio 2020, 11, e01728-20. [Google Scholar] [CrossRef] [PubMed]
- Ran, Y.; Xiong, M.; Xu, Z.; Luo, W.; Wang, S.; Wang, Y.-Y. YIPF5 Is Essential for Innate Immunity to DNA Virus and Facilitates COPII-Dependent STING Trafficking. J. Immunol. 2019, 203, 1560–1570. [Google Scholar] [CrossRef] [PubMed]
- Putri, D.D.P.; Kawasaki, T.; Murase, M.; Sueyoshi, T.; Deguchi, T.; Ori, D.; Suetsugu, S.; Kawai, T. PtdIns3P Phosphatases MTMR3 and MTMR4 Negatively Regulate Innate Immune Responses to DNA through Modulating STING Trafficking. J. Biol. Chem. 2019, 294, 8412–8423. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Jiang, Q.; Guan, Y.; Gao, P.; Zhang, R.; Zhao, Z.; Jiang, Z. Golgi Apparatus-Synthesized Sulfated Glycosaminoglycans Mediate Polymerization and Activation of the cGAMP Sensor STING. Immunity 2021, 54, 962–975.e8. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Konno, H.; Akiba, T.; Uemura, T.; Waguri, S.; Kobayashi, T.; Barber, G.N.; Arai, H.; Taguchi, T. Activation of STING Requires Palmitoylation at the Golgi. Nat. Commun. 2016, 7, 11932. [Google Scholar] [CrossRef]
- Guo, H.; König, R.; Deng, M.; Riess, M.; Mo, J.; Zhang, L.; Petrucelli, A.; Yoh, S.M.; Barefoot, B.; Samo, M.; et al. NLRX1 Sequesters STING to Negatively Regulate the Interferon Response, Thereby Facilitating the Replication of HIV-1 and DNA Viruses. Cell Host Microbe 2016, 19, 515–528. [Google Scholar] [CrossRef]
- Konno, H.; Konno, K.; Barber, G.N. Cyclic Dinucleotides Trigger ULK1 (ATG1) Phosphorylation of STING to Prevent Sustained Innate Immune Signaling. Cell 2013, 155, 688–698. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Abe, T.; Barber, G.N. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF-κB Activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef]
- Zhong, B.; Zhang, L.; Lei, C.; Li, Y.; Mao, A.-P.; Yang, Y.; Wang, Y.-Y.; Zhang, X.-L.; Shu, H.-B. The Ubiquitin Ligase RNF5 Regulates Antiviral Responses by Mediating Degradation of the Adaptor Protein MITA. Immunity 2009, 30, 397–407. [Google Scholar] [CrossRef]
- Guo, Y.; Jiang, F.; Kong, L.; Wu, H.; Zhang, H.; Chen, X.; Zhao, J.; Cai, B.; Li, Y.; Ma, C.; et al. OTUD5 Promotes Innate Antiviral and Antitumor Immunity through Deubiquitinating and Stabilizing STING. Cell. Mol. Immunol. 2021, 18, 1945–1955. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, L.; Shen, J.; Zhai, Y.; Jiang, Q.; Yi, M.; Deng, X.; Ruan, Z.; Fang, R.; Chen, Z.; et al. The STING Phase-Separator Suppresses Innate Immune Signalling. Nat. Cell Biol. 2021, 23, 330–340. [Google Scholar] [CrossRef]
- Tsuchida, T.; Zou, J.; Saitoh, T.; Kumar, H.; Abe, T.; Matsuura, Y.; Kawai, T.; Akira, S. The Ubiquitin Ligase TRIM56 Regulates Innate Immune Responses to Intracellular Double-Stranded DNA. Immunity 2010, 33, 765–776. [Google Scholar] [CrossRef]
- Zhao, B.; Du, F.; Xu, P.; Shu, C.; Sankaran, B.; Bell, S.L.; Liu, M.; Lei, Y.; Gao, X.; Fu, X.; et al. A Conserved PLPLRT/SD Motif of STING Mediates the Recruitment and Activation of TBK1. Nature 2019, 569, 718–722. [Google Scholar] [CrossRef]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.; Chen, Z.J. Structural Basis of STING Binding with and Phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wei, X.; Wang, X.; Liu, T.; Zhao, Y.; Chen, L.; Luo, Y.; Du, H.; Li, Y.; Liu, T.; et al. TBK1-METTL3 Axis Facilitates Antiviral Immunity. Cell Rep. 2022, 38, 110373. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Sun, H.; You, F.; Sun, W.; Zhou, X.; Chen, L.; Yang, J.; Wang, Y.; Tang, H.; Guan, Y.; et al. Activation of STAT6 by STING Is Critical for Antiviral Innate Immunity. Cell 2011, 147, 436–446. [Google Scholar] [CrossRef]
- Ning, X.; Wang, Y.; Jing, M.; Sha, M.; Lv, M.; Gao, P.; Zhang, R.; Huang, X.; Feng, J.-M.; Jiang, Z. Apoptotic Caspases Suppress Type I Interferon Production via the Cleavage of cGAS, MAVS, and IRF3. Mol. Cell 2019, 74, 19–31.e7. [Google Scholar] [CrossRef]
- Tian, M.; Liu, W.; Zhang, Q.; Huang, Y.; Li, W.; Wang, W.; Zhao, P.; Huang, S.; Song, Y.; Shereen, M.A.; et al. MYSM1 Represses Innate Immunity and Autoimmunity through Suppressing the cGAS-STING Pathway. Cell Rep. 2020, 33, 108297. [Google Scholar] [CrossRef]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic Analysis of the Host Response to Hepatitis B Virus Infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6669–6674. [Google Scholar] [CrossRef]
- Wieland, S.F.; Chisari, F.V. Stealth and Cunning: Hepatitis B and Hepatitis C Viruses. J. Virol. 2005, 79, 9369–9380. [Google Scholar] [CrossRef]
- Fletcher, S.P.; Chin, D.J.; Ji, Y.; Iniguez, A.L.; Taillon, B.; Swinney, D.C.; Ravindran, P.; Cheng, D.T.; Bitter, H.; Lopatin, U.; et al. Transcriptomic Analysis of the Woodchuck Model of Chronic Hepatitis B. Hepatology 2012, 56, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Luangsay, S.; Gruffaz, M.; Isorce, N.; Testoni, B.; Michelet, M.; Faure-Dupuy, S.; Maadadi, S.; Ait-Goughoulte, M.; Parent, R.; Rivoire, M.; et al. Early Inhibition of Hepatocyte Innate Responses by Hepatitis B Virus. J. Hepatol. 2015, 63, 1314–1322. [Google Scholar] [CrossRef]
- Suslov, A.; Boldanova, T.; Wang, X.; Wieland, S.; Heim, M.H. Hepatitis B Virus Does Not Interfere With Innate Immune Responses in the Human Liver. Gastroenterology 2018, 154, 1778–1790. [Google Scholar] [CrossRef]
- Dunn, C.; Peppa, D.; Khanna, P.; Nebbia, G.; Jones, M.; Brendish, N.; Lascar, R.M.; Brown, D.; Gilson, R.J.; Tedder, R.J.; et al. Temporal Analysis of Early Immune Responses in Patients with Acute Hepatitis B Virus Infection. Gastroenterology 2009, 137, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Stacey, A.R.; Norris, P.J.; Qin, L.; Haygreen, E.A.; Taylor, E.; Heitman, J.; Lebedeva, M.; DeCamp, A.; Li, D.; Grove, D.; et al. Induction of a Striking Systemic Cytokine Cascade Prior to Peak Viremia in Acute Human Immunodeficiency Virus Type 1 Infection, in Contrast to More Modest and Delayed Responses in Acute Hepatitis B and C Virus Infections. J. Virol. 2009, 83, 3719–3733. [Google Scholar] [CrossRef] [PubMed]
- Mutz, P.; Metz, P.; Lempp, F.A.; Bender, S.; Qu, B.; Schöneweis, K.; Seitz, S.; Tu, T.; Restuccia, A.; Frankish, J.; et al. HBV Bypasses the Innate Immune Response and Does Not Protect HCV from Antiviral Activity of Interferon. Gastroenterology 2018, 154, 1791–1804.e22. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Xia, Y.; Serti, E.; Block, P.D.; Chung, M.; Chayama, K.; Rehermann, B.; Liang, T.J. Hepatitis B Virus Evades Innate Immunity of Hepatocytes but Activates Cytokine Production by Macrophages. Hepatology 2017, 66, 1779–1793. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.K.; Nandakumar, R.; Stadler, D.; Malo, A.; Valls, R.M.; Wang, F.; Reinert, L.S.; Dagnæs-Hansen, F.; Hollensen, A.K.; Mikkelsen, J.G.; et al. Lack of Immunological DNA Sensing in Hepatocytes Facilitates Hepatitis B Virus Infection. Hepatology 2016, 64, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Dansako, H.; Ueda, Y.; Okumura, N.; Satoh, S.; Sugiyama, M.; Mizokami, M.; Ikeda, M.; Kato, N. The Cyclic GMP-AMP Synthetase–STING Signaling Pathway Is Required for Both the Innate Immune Response against HBV and the Suppression of HBV Assembly. FEBS J. 2016, 283, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhao, K.; Su, X.; Lu, L.; Zhao, H.; Zhang, X.; Wang, Y.; Wu, C.; Chen, J.; Zhou, Y.; et al. MITA/STING and Its Alternative Splicing Isoform MRP Restrict Hepatitis B Virus Replication. PLoS ONE 2017, 12, e0169701. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Tang, L.; Shu, S.; Sehgal, M.; Sheraz, M.; Liu, B.; Zhao, Q.; Cheng, J.; Zhao, X.; Zhou, T.; et al. Activation of Stimulator of Interferon Genes in Hepatocytes Suppresses the Replication of Hepatitis B Virus. Antimicrob. Agents Chemother. 2017, 61, e00771-17. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Hao, R.; Liu, D.; Liu, X.; Wu, S.; Guo, S.; Wang, Y.; Tien, P.; Guo, D. Inhibition of Hepatitis B Virus Replication by Activation of the cGAS-STING Pathway. J. Gen. Virol. 2016, 97, 3368–3378. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach-Rivière, L.; Bergez, M.; Mönch, S.; Qu, B.; Riess, M.; Vondran, F.W.R.; Liese, J.; Hornung, V.; Urban, S.; König, R. Hepatitis B Virus DNA Is a Substrate for the cGAS/STING Pathway but Is Not Sensed in Infected Hepatocytes. Viruses 2020, 12, 592. [Google Scholar] [CrossRef] [PubMed]
- Verrier, E.R.; Yim, S.-A.; Heydmann, L.; El Saghire, H.; Bach, C.; Turon-Lagot, V.; Mailly, L.; Durand, S.C.; Lucifora, J.; Durantel, D.; et al. Hepatitis B Virus Evasion From Cyclic Guanosine Monophosphate–Adenosine Monophosphate Synthase Sensing in Human Hepatocytes. Hepatology 2018, 68, 1695–1709. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-C.; Kao, J.-H. Persistence of Hepatitis B Virus Covalently Closed Circular DNA in Hepatocytes: Molecular Mechanisms and Clinical Significance. Emerg. Microbes Infect. 2014, 3, 1–7. [Google Scholar] [CrossRef]
- Cui, X.; Clark, D.N.; Liu, K.; Xu, X.-D.; Guo, J.-T.; Hu, J. Viral DNA-Dependent Induction of Innate Immune Response to Hepatitis B Virus in Immortalized Mouse Hepatocytes. J. Virol. 2015, 90, 486–496. [Google Scholar] [CrossRef]
- Uhlorn, B.L.; Jackson, R.; Li, S.; Bratton, S.M.; Doorslaer, K.V.; Campos, S.K. Vesicular Trafficking Permits Evasion of cGAS/STING Surveillance during Initial Human Papillomavirus Infection. PLoS Pathog. 2020, 16, e1009028. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Roden, R.B.S. L2, the Minor Capsid Protein of Papillomavirus. Virology 2013, 445, 175–186. [Google Scholar] [CrossRef] [PubMed]
- DiGiuseppe, S.; Bienkowska-Haba, M.; Sapp, M. Human Papillomavirus Entry: Hiding in a Bubble. J. Virol. 2016, 90, 8032–8035. [Google Scholar] [CrossRef]
- DiGiuseppe, S.; Bienkowska-Haba, M.; Guion, L.G.; Sapp, M. Cruising the Cellular Highways: How Human Papillomavirus Travels from the Surface to the Nucleus. Virus Res. 2017, 231, 1–9. [Google Scholar] [CrossRef] [PubMed]
- De Falco, F.; Cutarelli, A.; Catoi, A.F.; Uberti, B.D.; Cuccaro, B.; Roperto, S. Bovine Delta Papillomavirus E5 Oncoprotein Negatively Regulates the cGAS-STING Signaling Pathway in Cattle in a Spontaneous Model of Viral Disease. Front. Immunol. 2022, 13, 937736. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Donnelly, C.R.; Gong, W.; Heath, B.R.; Hao, Y.; Donnelly, L.A.; Moghbeli, T.; Tan, Y.S.; Lin, X.; Bellile, E.; et al. HPV16 Drives Cancer Immune Escape via NLRX1-Mediated Degradation of STING. J. Clin. Investig. 2020, 130, 1635–1652. [Google Scholar] [CrossRef]
- Albertini, S.; Lo Cigno, I.; Calati, F.; De Andrea, M.; Borgogna, C.; Dell’Oste, V.; Landolfo, S.; Gariglio, M. HPV18 Persistence Impairs Basal and DNA Ligand–Mediated IFN-β and IFN-Λ1 Production through Transcriptional Repression of Multiple Downstream Effectors of Pattern Recognition Receptor Signaling. J. Immunol. 2018, 200, 2076–2089. [Google Scholar] [CrossRef]
- Lo Cigno, I.; Calati, F.; Borgogna, C.; Zevini, A.; Albertini, S.; Martuscelli, L.; De Andrea, M.; Hiscott, J.; Landolfo, S.; Gariglio, M. Human Papillomavirus E7 Oncoprotein Subverts Host Innate Immunity via SUV39H1-Mediated Epigenetic Silencing of Immune Sensor Genes. J. Virol. 2020, 94, e01812-19. [Google Scholar] [CrossRef]
- Sunthamala, N.; Thierry, F.; Teissier, S.; Pientong, C.; Kongyingyoes, B.; Tangsiriwatthana, T.; Sangkomkamhang, U.; Ekalaksananan, T. E2 Proteins of High Risk Human Papillomaviruses Down-Modulate STING and IFN-κ Transcription in Keratinocytes. PLoS ONE 2014, 9, e91473. [Google Scholar] [CrossRef]
- Rattay, S.; Hufbauer, M.; Hagen, C.; Putschli, B.; Coch, C.; Akgül, B.; Hartmann, G. Human Beta Papillomavirus Type 8 E1 and E2 Proteins Suppress the Activation of the RIG-I-Like Receptor MDA5. Viruses 2022, 14, 1361. [Google Scholar] [CrossRef]
- Li, Y.; He, M.; Wang, Z.; Duan, Z.; Guo, Z.; Wang, Z.; Gong, R.; Chu, T.; Cai, J.; Gao, B. STING Signaling Activation Inhibits HBV Replication and Attenuates the Severity of Liver Injury and HBV-Induced Fibrosis. Cell. Mol. Immunol. 2022, 19, 92–107. [Google Scholar] [CrossRef]
- Zhao, L.; Yuan, H.; Wang, Y.; Geng, Y.; Yun, H.; Zheng, W.; Yuan, Y.; Lv, P.; Hou, C.; Zhang, H.; et al. HBV Confers Innate Immune Evasion through Triggering HAT1/Acetylation of H4K5/H4K12/miR-181a-5p or KPNA2/cGAS-STING/IFN-I Signaling. J. Med. Virol. 2023, 95, e28966. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Jiang, L.; Chen, S.; Hu, Q.; Huang, Y.; Wu, Y.; Chen, W. HBx Inhibits DNA Sensing Signaling Pathway via Ubiquitination and Autophagy of cGAS. Virol. J. 2022, 19, 55. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Feng, J.; Liu, Y.; Zhao, M.; Yuan, Y.; Yuan, H.; Yun, H.; Sun, M.; Bu, Y.; Liu, L.; et al. HAT1 Signaling Confers to Assembly and Epigenetic Regulation of HBV cccDNA Minichromosome. Theranostics 2019, 9, 7345–7358. [Google Scholar] [CrossRef]
- Parthun, M.R. Histone Acetyltransferase 1: More than Just an Enzyme? Biochim. Biophys. Acta BBA—Gene Regul. Mech. 2012, 1819, 256–263. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear cGAS Suppresses DNA Repair and Promotes Tumorigenesis. Nature 2018, 563, 131–136. [Google Scholar] [CrossRef]
- Zheng, B.; Yu, Y.; Pan, Z.; Feng, Y.; Zhao, H.; Han, Q.; Zhang, J. HBsAg Dampened STING Associated Activation of NK Cells in HBeAg-Negative CHB Patients. Int. J. Mol. Sci. 2021, 22, 7643. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-S.; Zhao, Q.; Zhang, J.; Wang, J.-W.; Qian, Y.; Fan, Y.-C.; Wang, K. Methylation Status of the Stimulator of Interferon Genes Promoter in Patients with Chronic Hepatitis B. Medicine 2018, 97, e13904. [Google Scholar] [CrossRef]
- Xing, J.; Zhang, A.; Zhang, H.; Wang, J.; Li, X.C.; Zeng, M.-S.; Zhang, Z. TRIM29 Promotes DNA Virus Infections by Inhibiting Innate Immune Response. Nat. Commun. 2017, 8, 945. [Google Scholar] [CrossRef]
- Wu, J.; Li, W.; Shao, Y.; Avey, D.; Fu, B.; Gillen, J.; Hand, T.; Ma, S.; Liu, X.; Miley, W.; et al. Inhibition of cGAS DNA Sensing by a Herpesvirus Virion Protein. Cell Host Microbe 2015, 18, 333–344. [Google Scholar] [CrossRef]
- Zhang, G.; Chan, B.; Samarina, N.; Abere, B.; Weidner-Glunde, M.; Buch, A.; Pich, A.; Brinkmann, M.M.; Schulz, T.F. Cytoplasmic Isoforms of Kaposi Sarcoma Herpesvirus LANA Recruit and Antagonize the Innate Immune DNA Sensor cGAS. Proc. Natl. Acad. Sci. USA 2016, 113, E1034–E1043. [Google Scholar] [CrossRef]
- Bhowmik, D.; Du, M.; Tian, Y.; Ma, S.; Wu, J.; Chen, Z.; Yin, Q.; Zhu, F. Cooperative DNA Binding Mediated by KicGAS/ORF52 Oligomerization Allows Inhibition of DNA-Induced Phase Separation and Activation of cGAS. Nucleic Acids Res. 2021, 49, 9389–9403. [Google Scholar] [CrossRef]
- Broussard, G.; Ni, G.; Zhang, Z.; Li, Q.; Cano, P.; Dittmer, D.P.; Damania, B. Barrier-to-Autointegration Factor 1 Promotes Gammaherpesvirus Reactivation from Latency. Nat. Commun. 2023, 14, 434. [Google Scholar] [CrossRef]
- Tabtieng, T.; Lent, R.C.; Kaku, M.; Monago Sanchez, A.; Gaglia, M.M. Caspase-Mediated Regulation and Cellular Heterogeneity of the cGAS/STING Pathway in Kaposi’s Sarcoma-Associated Herpesvirus Infection. mBio 2022, 13, e02446-22. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Jacobs, S.R.; West, J.A.; Stopford, C.; Zhang, Z.; Davis, Z.; Barber, G.N.; Glaunsinger, B.A.; Dittmer, D.P.; Damania, B. Modulation of the cGAS-STING DNA Sensing Pathway by Gammaherpesviruses. Proc. Natl. Acad. Sci. USA 2015, 112, E4306–E4315. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Jia, X.; He, Y.; Ma, X.; Qi, X.; Li, W.; Gao, S.-J.; Yan, Q.; Lu, C. Immune Evasion Strategy Involving Propionylation by the KSHV Interferon Regulatory Factor 1 (vIRF1). PLoS Pathog. 2023, 19, e1011324. [Google Scholar] [CrossRef]
- Lou, M.; Huang, D.; Zhou, Z.; Shi, X.; Wu, M.; Rui, Y.; Su, J.; Zheng, W.; Yu, X.-F. DNA Virus Oncoprotein HPV18 E7 Selectively Antagonizes cGAS-STING-Triggered Innate Immune Activation. J. Med. Virol. 2023, 95, e28310. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Tian, H.; Deng, H. PPM1G Restricts Innate Immune Signaling Mediated by STING and MAVS and Is Hijacked by KSHV for Immune Evasion. Sci. Adv. 2020, 6, eabd0276. [Google Scholar] [CrossRef]
- Li, K.; Liu, Y.; Xu, Z.; Zhang, Y.; Luo, D.; Gao, Y.; Qian, Y.; Bao, C.; Liu, C.; Zhang, Y.; et al. Avian Oncogenic Herpesvirus Antagonizes the cGAS-STING DNA-Sensing Pathway to Mediate Immune Evasion. PLoS Pathog. 2019, 15, e1007999. [Google Scholar] [CrossRef]
- Santhakumar, D.; Rubbenstroth, D.; Martinez-Sobrido, L.; Munir, M. Avian Interferons and Their Antiviral Effectors. Front. Immunol. 2017, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Cheng, A.; Wang, M. Innate Sensing of Viruses by Pattern Recognition Receptors in Birds. Vet. Res. 2013, 44, 82. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Murata, T.; Kanda, T.; Isomura, H.; Narita, Y.; Sugimoto, A.; Kawashima, D.; Tsurumi, T. Epstein-Barr Virus Deubiquitinase Downregulates TRAF6-Mediated NF-κB Signaling during Productive Replication. J. Virol. 2013, 87, 4060–4070. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Whitehurst, C.B.; Pagano, J.S. The Rad6/18 Ubiquitin Complex Interacts with the Epstein-Barr Virus Deubiquitinating Enzyme, BPLF1, and Contributes to Virus Infectivity. J. Virol. 2014, 88, 6411–6422. [Google Scholar] [CrossRef] [PubMed]
- Lui, W.-Y.; Bharti, A.; Wong, N.-H.M.; Jangra, S.; Botelho, M.G.; Yuen, K.-S.; Jin, D.-Y. Suppression of cGAS- and RIG-I-Mediated Innate Immune Signaling by Epstein-Barr Virus Deubiquitinase BPLF1. PLoS Pathog. 2023, 19, e1011186. [Google Scholar] [CrossRef] [PubMed]
- Speck, S.H.; Virgin, H.W. Host and Viral Genetics of Chronic Infection: A Mouse Model of Gamma-Herpesvirus Pathogenesis. Curr. Opin. Microbiol. 1999, 2, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Schattgen, S.A.; Pisitkun, P.; Jorgensen, J.P.; Hilterbrand, A.T.; Wang, L.J.; West, J.A.; Hansen, K.; Horan, K.A.; Jakobsen, M.R.; et al. Evasion of Innate Cytosolic DNA Sensing by a Gammaherpesvirus Facilitates Establishment of Latent Infection. J. Immunol. 2015, 194, 1819–1831. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.; Chen, J.; Li, Y.; Wang, W.; Du, X.; Song, W.; Zhang, W.; Lin, L.; Yuan, Z. Hepatitis B Virus Polymerase Disrupts K63-Linked Ubiquitination of STING to Block Innate Cytosolic DNA-Sensing Pathways. J. Virol. 2015, 89, 2287–2300. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA Tumor Virus Oncogenes Antagonize the cGAS-STING DNA-Sensing Pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef]
- Miyauchi, S.; Kim, S.S.; Jones, R.N.; Zhang, L.; Guram, K.; Sharma, S.; Schoenberger, S.P.; Cohen, E.E.W.; Califano, J.A.; Sharabi, A.B. Human Papillomavirus E5 Suppresses Immunity via Inhibition of the Immunoproteasome and STING Pathway. Cell Rep. 2023, 42, 112508. [Google Scholar] [CrossRef]
- Kang, H.-R.; Cheong, W.-C.; Park, J.-E.; Ryu, S.; Cho, H.-J.; Youn, H.; Ahn, J.-H.; Song, M.J. Murine Gammaherpesvirus 68 Encoding Open Reading Frame 11 Targets TANK Binding Kinase 1 To Negatively Regulate the Host Type I Interferon Response. J. Virol. 2014, 88, 6832–6846. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Li, K.; Zhang, Y.; Liu, Y.; Liu, C.; Zhang, Y.; Gao, Y.; Qi, X.; Cui, H.; Wang, Y.; et al. Inhibition of DNA-Sensing Pathway by Marek’s Disease Virus VP23 Protein through Suppression of Interferon Regulatory Factor 7 Activation. J. Virol. 2019, 93, e01934-18. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gao, L.; Xu, Z.; Luo, D.; Zhang, Y.; Gao, Y.; Liu, C.; Zhang, Y.; Qi, X.; Cui, H.; et al. Marek’s Disease Virus RLORF4 Inhibits Type I Interferon Production by Antagonizing NF-κB Activation. J. Virol. 2019, 93, e01037-19. [Google Scholar] [CrossRef]
- Zheng, B.; Yang, Y.; Han, Q.; Yin, C.; Pan, Z.; Zhang, J. STAT3 Directly Regulates NKp46 Transcription in NK Cells of HBeAg-Negative CHB Patients. J. Leukoc. Biol. 2019, 106, 987–996. [Google Scholar] [CrossRef]
- Karimi-Googheri, M.; Daneshvar, H.; Khaleghinia, M.; Bidaki, R.; Kazemi Arababadi, M. Decreased Expressions of STING but Not IRF3 Molecules in Chronic HBV Infected Patients. Arch. Iran. Med. 2015, 18, 351–354. [Google Scholar]
- Memorial Sloan Kettering Cancer Center. A Phase I Trial of CRD3874-SI, a STING Agonist, in Patients with Advanced/Metastatic Sarcoma and Merkel Cell Carcinoma; Clinical trial registration NCT06021626; Clinicaltrials.gov: Bethesda, MD, USA, 2023. Available online: https://clinicaltrials.gov/study/NCT06021626 (accessed on 31 December 2023).
- Ramos, J.C. A Pilot Safety Trial of STING-Dependent Activators (STAVs) and Stimulated Dendritic Cells for Aggressive Relapsed/Refractory Leukemias; Clinical trial registration NCT05321940; Clinicaltrials.gov: Bethesda, MD, USA, 2023. Available online: https://clinicaltrials.gov/study/NCT05321940 (accessed on 31 December 2023).
- Eisai Inc. An Open-Label, Multicenter Phase 1/1b Study of Intratumorally Administered STING Agonist E7766 in Subjects with Advanced Solid Tumors or Lymphomas—INSTAL-101; Clinical trial registration NCT04144140; Clinicaltrials.gov: Bethesda, MD, USA, 2023. Available online: https://clinicaltrials.gov/study/NCT04144140 (accessed on 31 December 2023).
- Eisai Inc. INtravesical Phase 1/1b Study of STING Agonist E7766 in NMIBC Including Subjects Unresponsive to BCG Therapy, INPUT-102; Clinical trial registration NCT04109092; Clinicaltrials.gov: Bethesda, MD, USA, 2020. Available online: https://clinicaltrials.gov/study/NCT04109092 (accessed on 31 December 2023).
- Lu, S.; Concha-Benavente, F.; Shayan, G.; Srivastava, R.M.; Gibson, S.P.; Wang, L.; Gooding, W.E.; Ferris, R.L. STING Activation Enhances Cetuximab-Mediated NK Cell Activation and DC Maturation and Correlates with HPV+ Status in Head and Neck Cancer. Oral Oncol. 2018, 78, 186–193. [Google Scholar] [CrossRef]
- Srivastava, R.M.; Lee, S.C.; Andrade Filho, P.A.; Lord, C.A.; Jie, H.-B.; Davidson, H.C.; López-Albaitero, A.; Gibson, S.P.; Gooding, W.E.; Ferrone, S.; et al. Cetuximab-Activated Natural Killer and Dendritic Cells Collaborate to Trigger Tumor Antigen–Specific T-Cell Immunity in Head and Neck Cancer Patients. Clin. Cancer Res. 2013, 19, 1858–1872. [Google Scholar] [CrossRef]
- Baird, J.R.; Feng, Z.; Xiao, H.D.; Friedman, D.; Cottam, B.; Fox, B.A.; Kramer, G.; Leidner, R.S.; Bell, R.B.; Young, K.H.; et al. STING Expression and Response to Treatment with STING Ligands in Premalignant and Malignant Disease. PLoS ONE 2017, 12, e0187532. [Google Scholar] [CrossRef]
- Dorta-Estremera, S.; Hegde, V.L.; Slay, R.B.; Sun, R.; Yanamandra, A.V.; Nicholas, C.; Nookala, S.; Sierra, G.; Curran, M.A.; Sastry, K.J. Targeting Interferon Signaling and CTLA-4 Enhance the Therapeutic Efficacy of Anti-PD-1 Immunotherapy in Preclinical Model of HPV+ Oral Cancer. J. Immunother. Cancer 2019, 7, 252. [Google Scholar] [CrossRef]
- Guo, F.; Han, Y.; Zhao, X.; Wang, J.; Liu, F.; Xu, C.; Wei, L.; Jiang, J.-D.; Block, T.M.; Guo, J.-T.; et al. STING Agonists Induce an Innate Antiviral Immune Response against Hepatitis B Virus. Antimicrob. Agents Chemother. 2015, 59, 1273–1281. [Google Scholar] [CrossRef]
- Pimkova Polidarova, M.; Vanekova, L.; Brehova, P.; Dejmek, M.; Vavrina, Z.; Birkus, G.; Brazdova, A. Synthetic Stimulator of Interferon Genes (STING) Agonists Induce a Cytokine-Mediated Anti-Hepatitis B Virus Response in Nonparenchymal Liver Cells. ACS Infect. Dis. 2023, 9, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Dansako, H.; Ueda, Y.; Satoh, S.; Kato, N. Daunorubicin, a Topoisomerase II Poison, Suppresses Viral Production of Hepatitis B Virus by Inducing cGAS-Dependent Innate Immune Response. Biochem. Biophys. Res. Commun. 2018, 504, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, R.W. Novel Compounds as STING Modulators for Treating Hepatitis B Virus Infections. ACS Med. Chem. Lett. 2020, 11, 2372–2373. [Google Scholar] [CrossRef] [PubMed]
- Skouboe, M.K.; Knudsen, A.; Reinert, L.S.; Boularan, C.; Lioux, T.; Perouzel, E.; Thomsen, M.K.; Paludan, S.R. STING Agonists Enable Antiviral Cross-Talk between Human Cells and Confer Protection against Genital Herpes in Mice. PLoS Pathog. 2018, 14, e1006976. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Family | Virus | Host | Associated Cancers | Target Cells |
|---|---|---|---|---|
| Herpesviridae | Marek’s disease virus | Chicken | T cell lymphoma | T lymphocytes |
| Kaposi sarcoma-associated virus | Human | Kaposi sarcoma Primary effusion lymphoma Multicentric Castleman’s disease | B lymphocytes, Endothelial cells | |
| Epstein–Barr virus | Human | Burkitt’s lymphoma Hodgkin’s lymphoma Other lymphomas Nasopharyngeal carcinoma Gastric carcinoma | B lymphocytes Epithelial cells | |
| Papillomaviridae | Human papillomavirus types 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59 | Human | Cervical cancer Oropharyngeal carcinoma Other anogenital cancers | Skin and mucosa epithelial cells |
| Bovine papillomavirus types 1, 2, 4, 13 | Bovine | Urinary bladder cancer Oral squamous cell carcinoma | Skin and mucosa epithelial cells | |
| Hepadnaviridae | Hepatitis B virus | Human | Hepatocellular carcinoma | Hepatocytes |
| Polyomaviridae | Merkel cell polyomavirus * | Human | Merkel cell carcinoma | Dermal fibroblasts |
| Family | Virus | Viral Protein | Evasion Mechanism | Experimental System | References |
|---|---|---|---|---|---|
| Shielding the viral genome from cGAS sensing | |||||
| Papillomaviridae | Human papillomavirus type 16 | Minor capsid protein L2 | L2-mediated trafficking of viral DNA into vesicular membranes for delivery to the host cell nucleus | Immortalized human keratinocyte cell line HaCaT and Primary human foreskin keratinocytes | [155] |
| Hepadnaviridae | Hepatitis B virus | Nucleocapsid | Hiding viral DNA and replication intermediates inside the nucleocapsid | Immortalized mouse hepatocyte cell line AML12HBV10 | [154] |
| Immortalized human hepatocyte cell line HepG2-hNTCP and Primary human hepatocytes (PHHs) | [151,152] | ||||
| Transcriptional and post-transcriptional inhibition of cGAS-STING pathway gene expression | |||||
| Herpesviridae | Epstein–Barr virus | - | Upregulation of host E3 ubiquitin ligase TRIM29, which then interacts with the c-di-GMP-binding domain of STING and induces its ubiquitination at Lys370 site by K48-mediated linkage for protein degradation | Human healthy airway epithelial cell line BEAS-2B, EBV-negative nasopharyngeal epithelial cell line NP69 and Human nasopharyngeal carcinoma cells CNE1 | [173] |
| Papillomaviridae | Human papillomavirus types 16 and 18 | Early protein E2 | Significant reduction of STING mRNAs levels. The transactivation amino-terminal domain of E2 is involved in the suppressive effect. | Human primary keratinocytes transduced by HPV E2 | [163] |
| Human papillomavirus type 18 | Oncoprotein E7 | Transcriptional activation of the host chromatin repressor SUV39H1, which then promotes epigenetic silencing of cGAS and STING genes | Immortalized human keratinocyte cell line NIKS stably harboring a high viral load of HPV18 episomal genomes (NIKSmcHPV18 cells) and Immortalized cervical carcinoma-derived cell line harboring integrated HPV18 DNA (HeLa cells) | [161,162] | |
| Human papillomavirus type 16 | Oncoprotein E7 | Interaction with NLRX1, a host protein complex scaffold recruiting autophagy-promoting molecules, to accelerate STING turnover through an autophagy-dependent mechanism | HPV16-positive head and neck squamous cell carcinoma (HNSCC) cell lines (93VU147T, UMSCC47 and SCC90 cells) and HPV-negative HNSCC cell line FaDu HPV16 E6/E7-expressing HNSCC mouse model, MOC2-E6/E7 | [160] | |
| Bovine papillomavirus types 2 and 13 | - | Significant reduction of cGAS, STING and TBK1 mRNAs in BPV-infected cells | BPV-infected bladder mucosa samples from cows with bladder neoplasms | [159] | |
| Hepadnaviridae | Hepatitis B virus | HBx | Decrease of cGAS protein levels by direct binding to cGAS and promoting cGAS autophagy and K48-linked ubiquitination | Human hepatocellular carcinoma cell lines (HEK293T, SMMC-7721 and LO2 cells) transfected with an HBx plasmid | [167] |
| Sp1 upregulates the expression HAT1. HAT1 increases miRNA levels of miR-181a-5p by modulating acetylation in the miR-181a-5p promoter. MiR-181a-5p in turn binds to the cGAS mRNA 3′UTR, decreasing cGAS mRNA and protein levels. | Immortalized human hepatocyte cell lines (HepG2-hNTCP, Huh7 and HepG2 cells), PHHs and Human liver chimeric mice | [166,168] | |||
| Sp1 upregulates the expression HAT1. HAT1 increases miRNA levels of miR-181a-5p by modulating acetylation in the miR-181a-5p promoter. MiR-181a-5p in turn binds to the cGAS mRNA 3′UTR, decreasing cGAS mRNA and protein levels. | HepG2-hNTCP, Huh7 and HepG2 cells, PHHs and Human liver chimeric mice | [166,170] | |||
| HBsAg | Inhibition of STAT3 and subsequent downregulation of STING expression in NK cells of patients with chronic hepatitis B (CHB) | NK Cells from HBeAg-Negative CHB Patients and Human NK cell line (NK-92 cells) | [171,197] | ||
| - | Hypermethylation of the STING gene promoter inducing significantly lower levels of STING mRNA in peripheral blood mononuclear cells (PBMCs) of CHB patients | Isolated PBMCs of CHB patients | [172,198] | ||
| Inhibition of cGAS DNA binding and activation | |||||
| Herpesviridae | Kaposi sarcoma-associated herpesvirus | Tegument protein ORF52/KicGAS | KicGAS self-oligomerizes and forms liquid droplets upon binding to DNA, thus inhibiting the DNA-induced phase separation and activation of cGAS | HEK293T cells stably expressing STING, THP1 Lucia™ ISG cells (InvivoGen), which express luciferase from a gene under the control of an IRF3-inducible promoter and Human primary lymphatic endothelial cells | [174,176] |
| LANA | Direct binding of cytoplasmic isoforms of LANA to cGAS antagonizes cGAS function | Primary effusion lymphoma-derived B-cell line BCBL-1, HEK 293T and HeLa cells, HuAR2T.rKSHV.219, a conditionally immortalized endothelial cell line persistently infected with recombinant virus rKSHV.219 | [175] | ||
| - | Upregulation of barrier-to-autointegration factor 1 (BAF) expression, a host protein that induces the degradation of cGAS through the proteasomal pathway | Kaposi’s sarcoma-derived cell line SLK, iSLK.219 cell line, which is latently infected with recombinant virus rKSHV.219, TREx-BCBL1-RTA cell line, a KSHV-infected BCBL-1 cell line | [177] | ||
| - | Activation of caspase-8 during lytic reactivation, which indirectly inhibits cGAS enzymatic activity | iSLK.219 cell line, BC3 cells, a KSHV-infected B cell line derived from a primary effusion lymphoma patient | [178] | ||
| Epstein–Barr virus | Tegument protein BLRF2 (KSHV ORF52 homolog) | Binding to both DNA and cGAS and inhibition of cGAS enzymatic activity | HEK293T cells stably expressing STING | [174,176] | |
| - | Upregulation of BAF expression, a host protein that induces the degradation of cGAS through the proteasomal pathway | Human gastric adenocarcinoma-derived cell line AGS and AGS-EBV cell line latently infected with GFP-expressing recombinant EBV | [177] | ||
| Murine gammaherpesvirus 68 | Tegument protein ORF52 | Binding to both DNA and cGAS and inhibition of cGAS enzymatic activity | HEK293T cells stably expressing STING | [174,176] | |
| STING inhibition through direct binding | |||||
| Herpesviridae | Marek’s disease virus | Oncoprotein Meq | Binding to both STING and IRF7, impeding the assembly of the STING-TBK1-IRF7 complex and the subsequent activation of TBK1 and IRF7, but not that of NF-κB | Immortalized chicken fibroblast cell line DF-1, chicken embryo fibroblasts (CEFs) and chickens | [183] |
| Kaposi sarcoma-associated herpesvirus | vIRF1 | Direct binding to STING through multiple domains and subsequent disruption of the TBK1-STING interaction, preventing STING phosphorylation and activation | Human umbilical vein endothelial cells (HUVECs), Immortalized endothelial cells EA.hy926 and rKSHV.219 iSLK cell line | [179] | |
| vIRF1 promotes its own propionylation, which is required for effective binding to STING | HEK293T and EA.hy926 cell lines, KSHV infected cell line iSLK-RGB | [180] | |||
| vIRF1 binding to STING inhibits STING-triggered IRF3 activation but not NF-κB activation | HEK293T cells | [181] | |||
| Tegument protein ORF33 | Binding to the CBD domain of STING and recruitment of the host protein phosphatase PPM1G to dephosphorylate p-STING and subsequently impair the recruitment of downstream IRF3 | rKSHV.219 iSLK, THP-1 and HEK293 cell lines | [182] | ||
| Epstein–Barr virus | Tegument protein BPLF1 | Removing of all types of ubiquitin moieties on STING, therefore suppressing STING activation and recruitment of TBK1 | HEK293 cells overexpressing BPLF1, cGAS and STING, HEK293-M81 cells constitutively carrying EBV M81 strain | [188] | |
| Murine gammaherpesvirus 68 | Tegument protein ORF64 (EBV BPLF1 homolog) | Antagonism of the STING pathway through a mechanism dependent on the deubiquitinase activity of ORF64, a homolog of EBV BPLF1 | Murine dendritic cells, Wild-type and STINGgt/gt mice | [190] | |
| Papillomaviridae | Human papillomavirus type 18 | Oncoprotein E7 | Binding to STING inhibits NF-κB activation and p65 nuclear accumulation, but not IRF3 activation | HeLa cells overexpressing STING, Primary mouse embryonic fibroblasts transduced with retroviral expression vectors containing HPV18 E7, HEK293T cells | [181,192] |
| Human papillomavirus type 16 | E5 | Direct binding to STING and subsequent inhibition of downstream signaling | Human HNSCC cell line CAL-27 | [193] | |
| Bovine papillomavirus types 2 and 13 | Oncoprotein E5 | Formation of a ternary complex composed of E5/STING/IFI16, which blocks interaction of IFI16 with STING | BPV-infected bladder mucosa samples from cows with bladder neoplasms | [159] | |
| Hepadnaviridae | Hepatitis B virus | Polymerase Pol | Direct binding to STING and subsequent disruption of its K63-linked ubiquitination | Huh7, HEK 293 and HepG2 derivative HepaAD38 cells overexpressing STING, PH5CH8 cells, differentiated proliferative human hepatoma-derived cells HepaRG and PHHs | [191] |
| Inhibition of TBK1 and cGAS-STING-mediated activation of transcription factors | |||||
| Herpesviridae | Marek’s disease virus | Capsid protein VP23 | Interaction with IRF7 and disruption of its binding to TBK1, leading to the inhibition of IRF7 phosphorylation and nuclear translocation | CEFs, chicken macrophage HD11 cells, DF-1 cells | [195] |
| RLORF4 | Binding to the Rel homology domains of the NF-κB subunits p65 and p50, interrupting their translocation to the nuclei and thereby inhibiting IFN beta production. | DF-1 cells, HEK293T cells, CEFs, chickens | [196] | ||
| Epstein–Barr virus | Tegument protein BPLF1 | Removing of all types of ubiquitin moieties on TBK1, leading to inactivation of the kinase inhibition of TBK1-induced IRF3 dimerization | HEK293 cells overexpressing BPLF1, cGAS, STING, TBK1 and IRF3 | [188] | |
| Murine gammaherpesvirus 68 | Tegument protein ORF11 | Direct binding to TBK1, reducing the interaction between TBK1 and IRF3 and subsequently inhibiting IRF3 activation | HEK293T cells, Murine embryonic fibroblasts and Raw264.7 macrophage cells | [194] | |
| Papillomaviridae | Human papillomavirus type 16 | Oncoprotein E7 | Decrease in phosphorylation of TBK1 with increased levels of HPV16 E7 | 93VU147T, UMSCC47, and FaDu cells overexpressing HPV16 E7 | [160] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-López, M.F.; Muslin, C.; Kyriakidis, N.C. STINGing Defenses: Unmasking the Mechanisms of DNA Oncovirus-Mediated Immune Escape. Viruses 2024, 16, 574. https://doi.org/10.3390/v16040574
Martínez-López MF, Muslin C, Kyriakidis NC. STINGing Defenses: Unmasking the Mechanisms of DNA Oncovirus-Mediated Immune Escape. Viruses. 2024; 16(4):574. https://doi.org/10.3390/v16040574
Chicago/Turabian StyleMartínez-López, Mayra F, Claire Muslin, and Nikolaos C. Kyriakidis. 2024. "STINGing Defenses: Unmasking the Mechanisms of DNA Oncovirus-Mediated Immune Escape" Viruses 16, no. 4: 574. https://doi.org/10.3390/v16040574
APA StyleMartínez-López, M. F., Muslin, C., & Kyriakidis, N. C. (2024). STINGing Defenses: Unmasking the Mechanisms of DNA Oncovirus-Mediated Immune Escape. Viruses, 16(4), 574. https://doi.org/10.3390/v16040574