
Community Structure, Drivers, and Potential Functions of Different Lifestyle Viruses in Chaohu Lake

Abstract
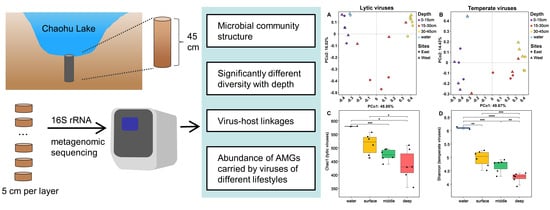
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
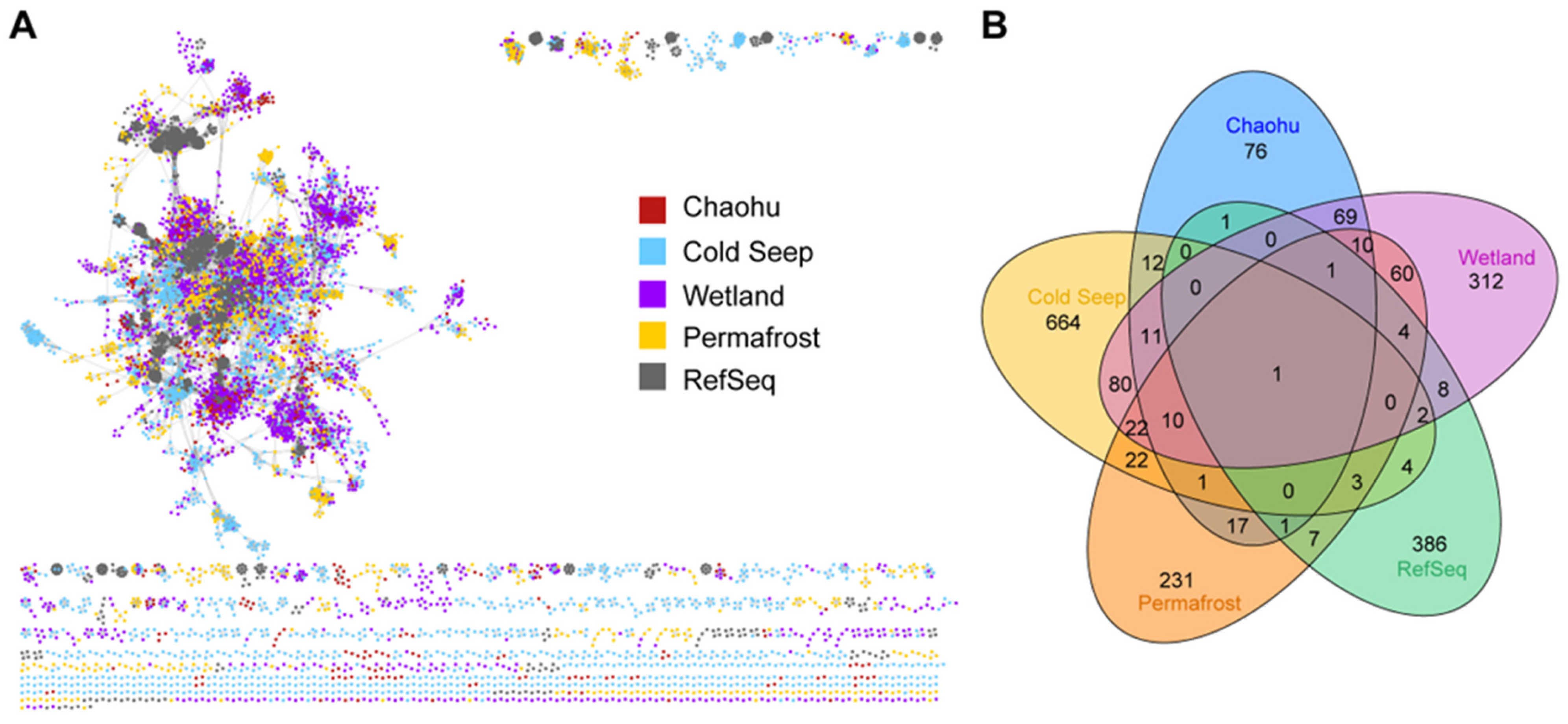{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. Sample Physicochemical Measurement
2.3. DNA Extraction, Sequencing, and Metagenome Assembly
2.4. Identification of Viral Contigs
2.5. Taxonomy Assignment
2.6. Abundance of Viruses
2.7. Virus–Host Prediction
2.8. Identification of Auxiliary Metabolic Genes
2.9. Statistical Analyses
3. Results
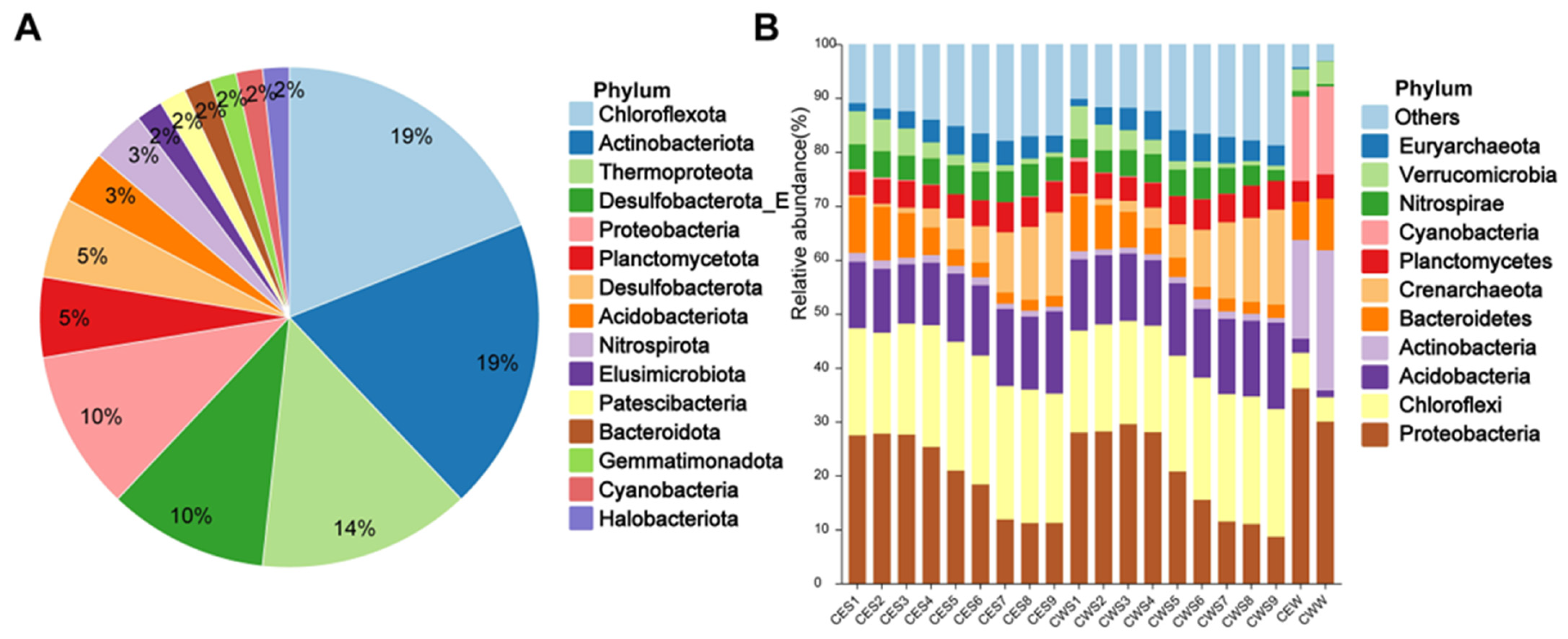
3.1. Community Structure of Prokaryotic Microorganisms in Chaohu Lake
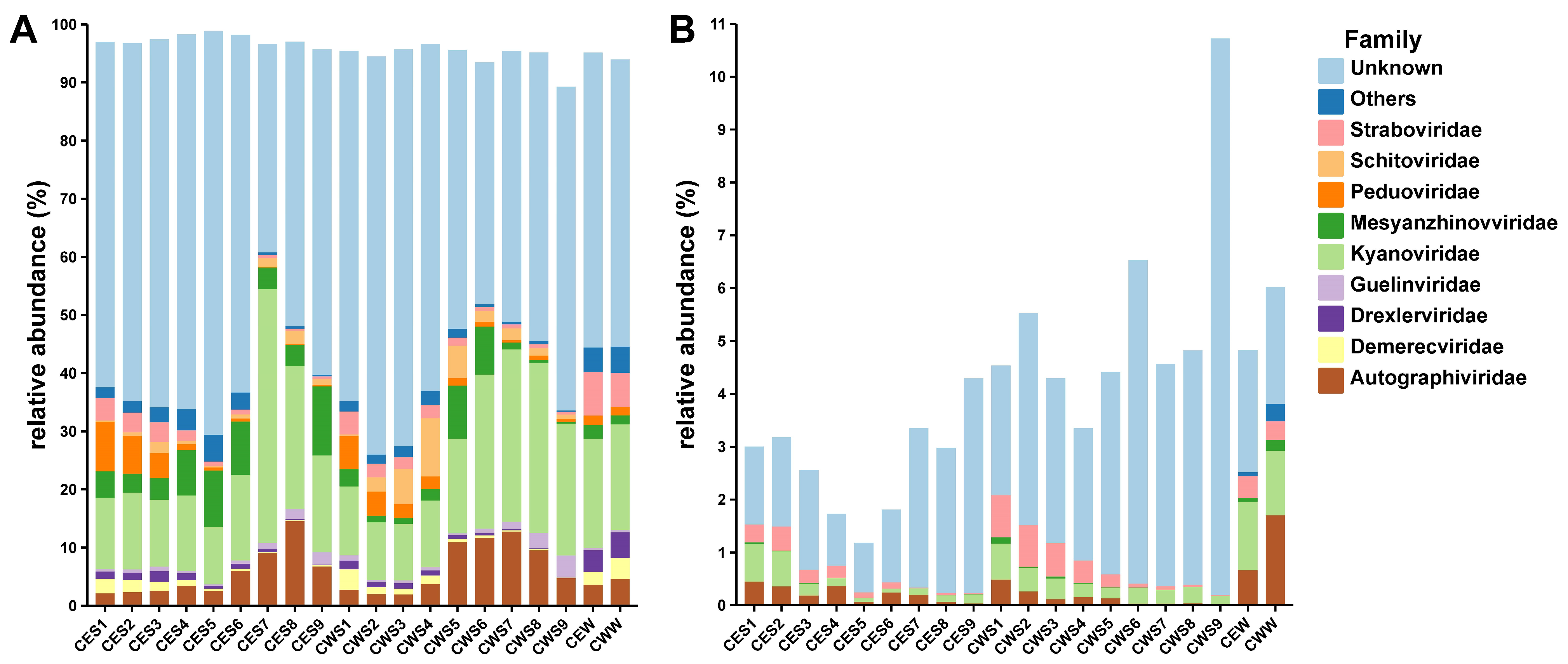
3.2. Community Composition of Viruses in Chaohu Lake
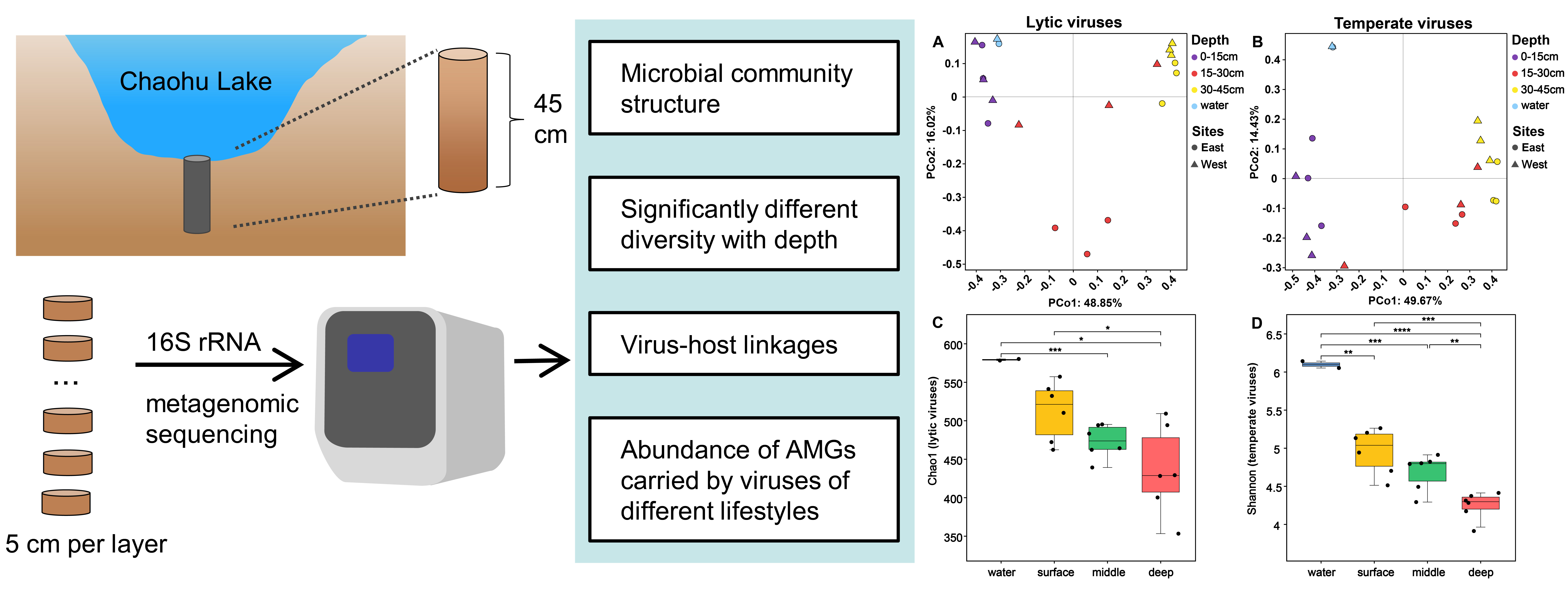
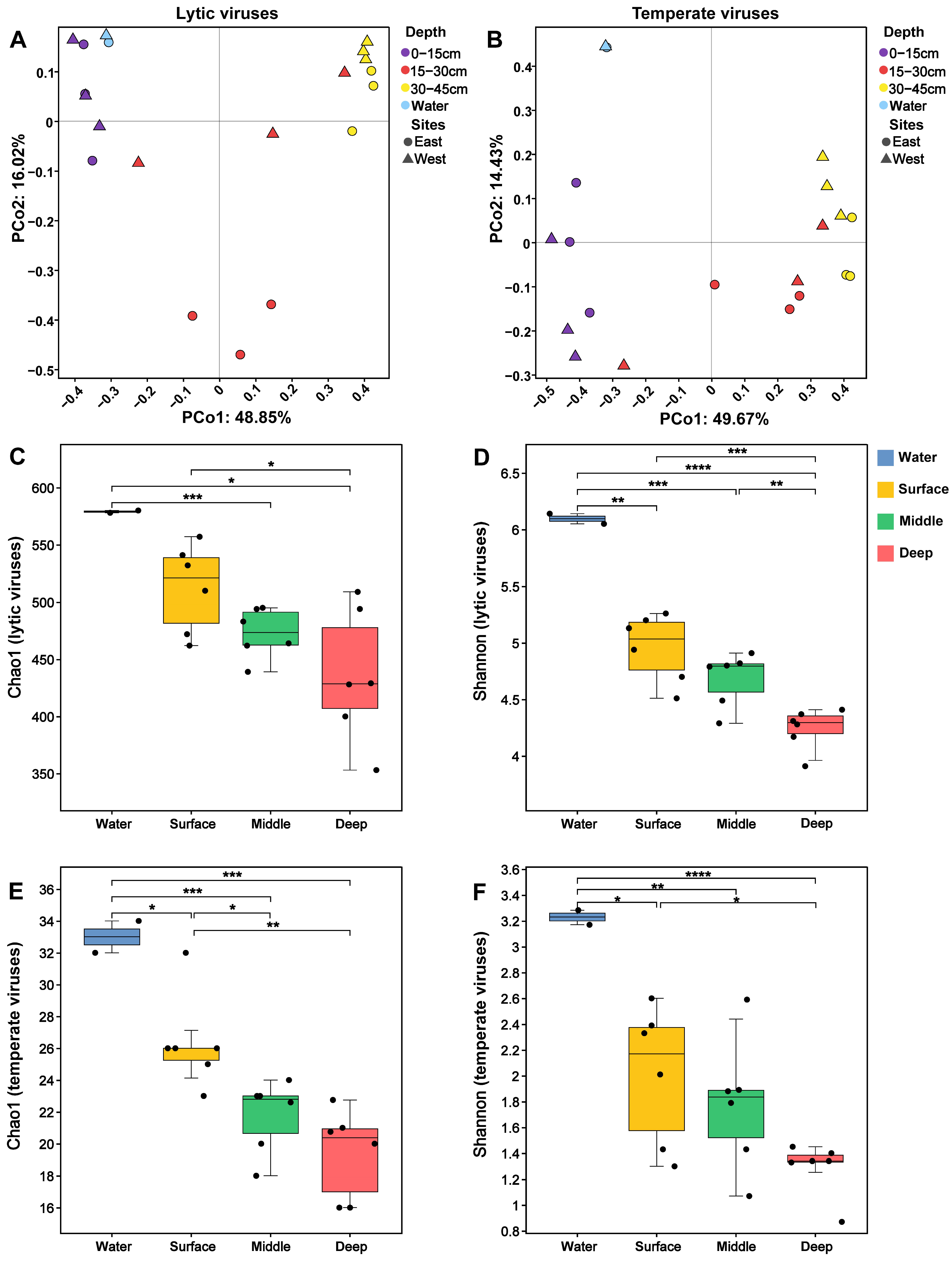
3.3. Diversity of Chaohu Viruses
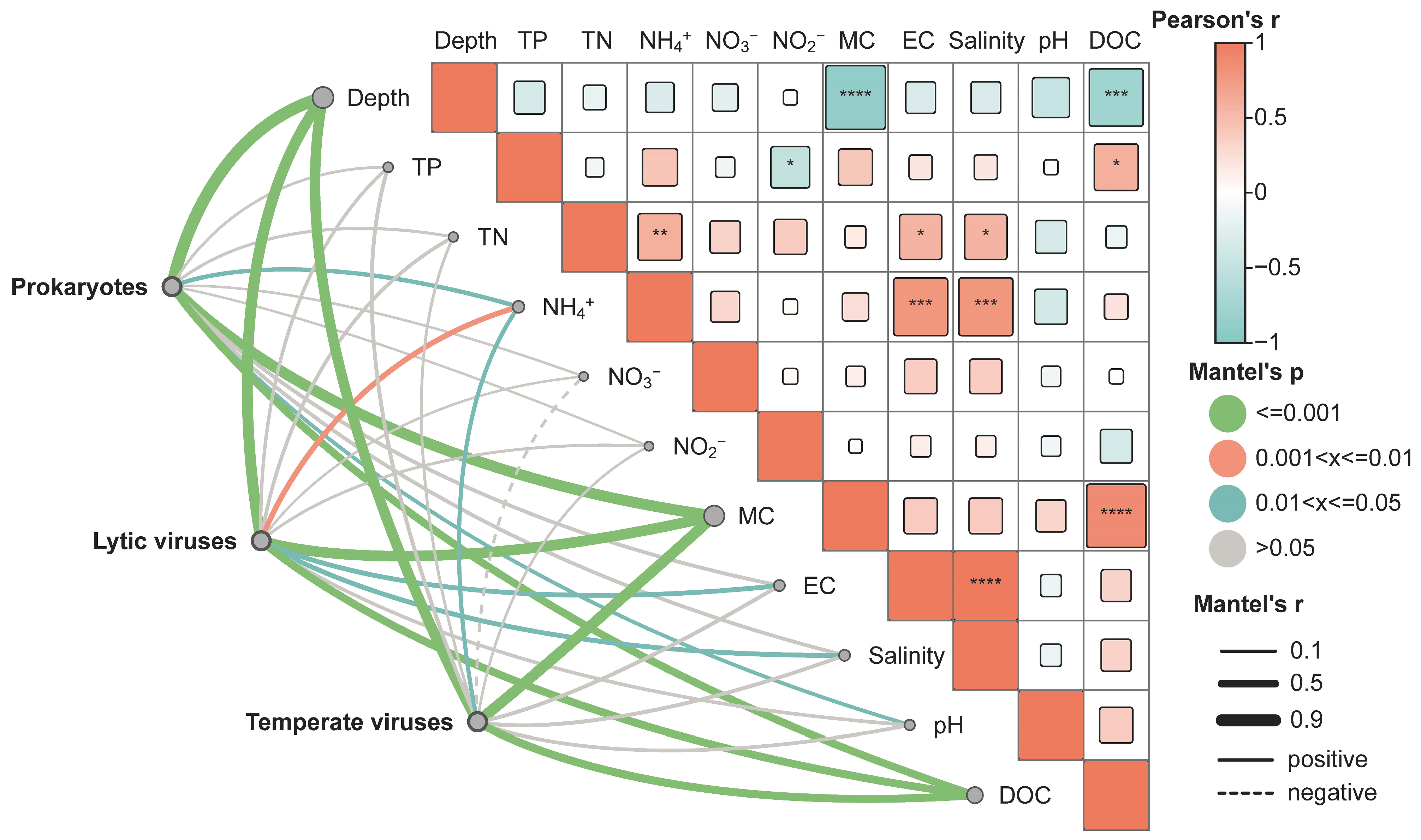
3.4. The Driving Factors of Virus Community Construction in Chaohu Lake
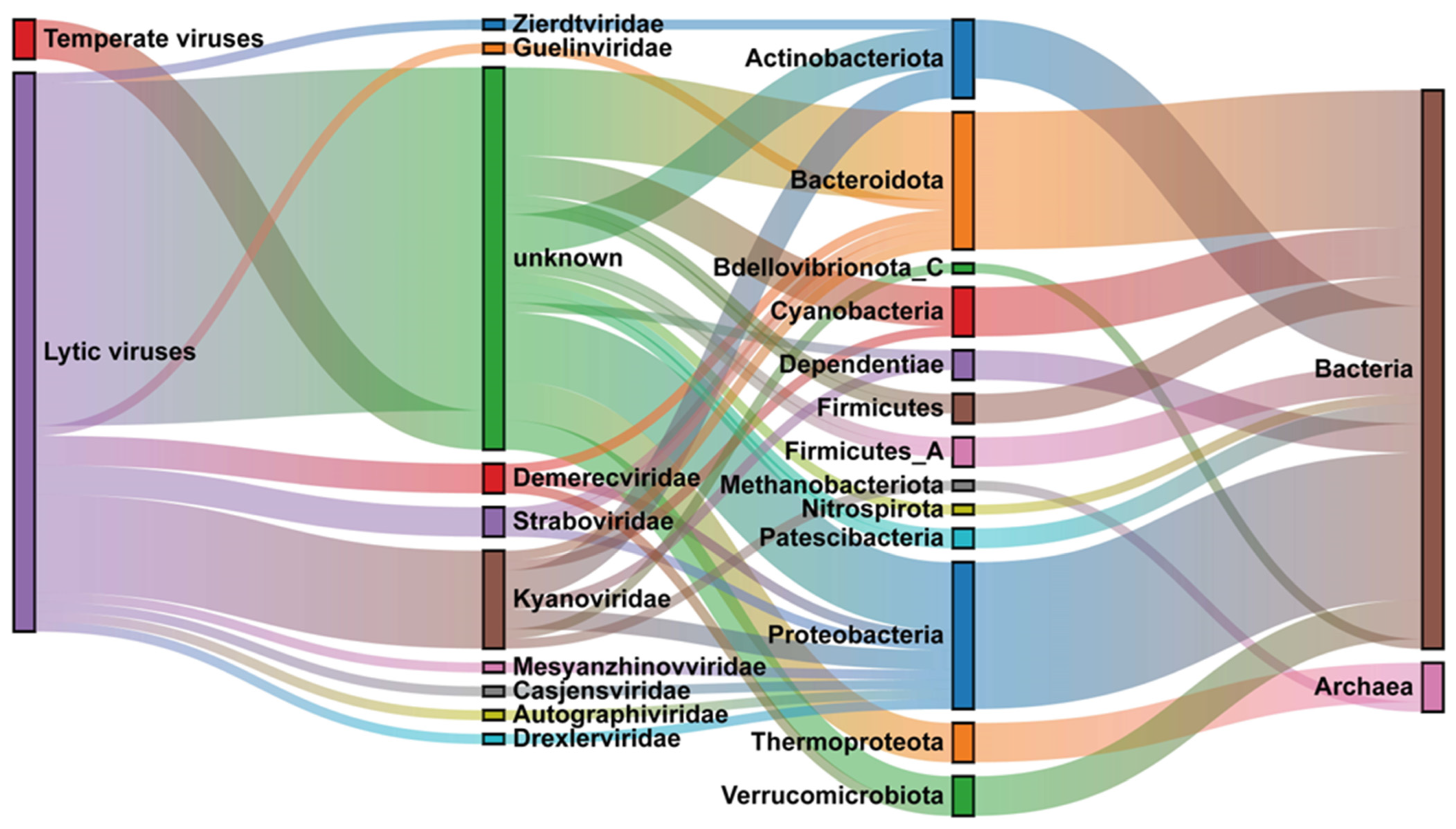
3.5. Host Prediction of Chaohu Viruses
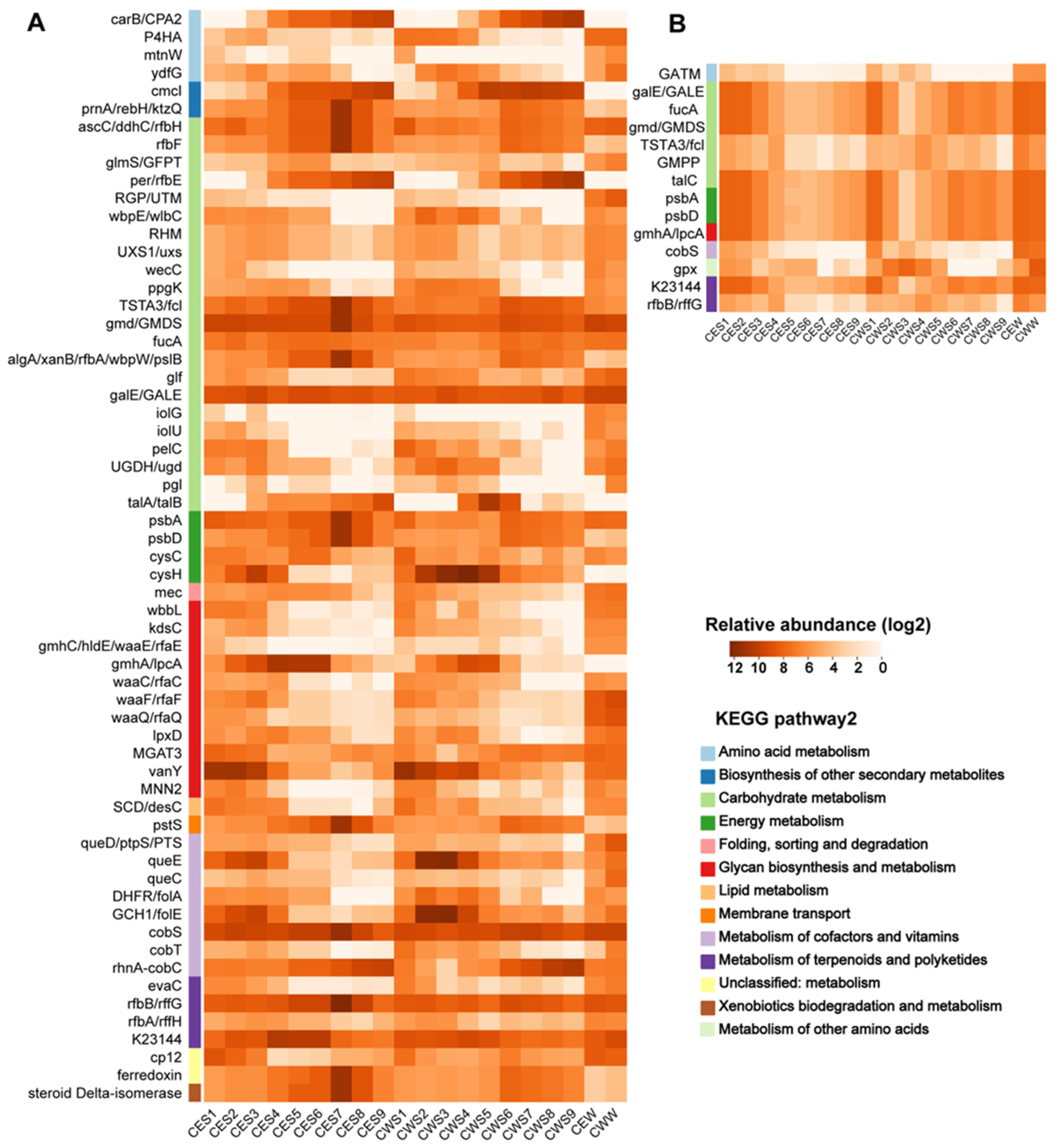
3.6. Composition of AMGs for Different Lifestyle Viruses
4. Discussion
4.1. Linkages between the Chaohu Viruses and Other Ecosystems around the Globe
4.2. Driving Mechanism of Virus Community Construction in Chaohu Lake
4.3. Relationship between Viruses and Hosts
4.4. Differences in AMGs of Viruses with Different Lifestyles
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jansson, J.K.; Wu, R. Soil Viral Diversity, Ecology and Climate Change. Nat. Rev. Microbiol. 2023, 21, 296–311. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine Viruses—Major Players in the Global Ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Ignacio-Espinoza, J.C.; Roux, S.; Doulcier, G.; Acinas, S.G.; Alberti, A.; Chaffron, S.; Cruaud, C.; de Vargas, C.; Gasol, J.M.; et al. Patterns and Ecological Drivers of Ocean Viral Communities. Science 2015, 348, 1261498. [Google Scholar] [CrossRef]
- Cai, L.; Jørgensen, B.B.; Suttle, C.A.; He, M.; Cragg, B.A.; Jiao, N.; Zhang, R. Active and Diverse Viruses Persist in the Deep Sub-Seafloor Sediments over Thousands of Years. ISME J. 2019, 13, 1857–1864. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.; Yi, Y.; Wang, J.; Hao, Y.; Zhang, M.; Wang, S.; Meng, C.; Zhang, Y.; Jing, H.; Wang, Y.; et al. Diversity and Distribution of Viruses Inhabiting the Deepest Ocean on Earth. ISME J. 2021, 15, 3094–3110. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.-Q.; Wang, P.; Li, J.-L.; Ahmad, M.; Duan, L.; Yin, L.-Z.; Deng, Q.-Q.; Fang, B.-Z.; Li, S.-H.; Li, W.-J. Viral Community-Wide Auxiliary Metabolic Genes Differ by Lifestyles, Habitats, and Hosts. Microbiome 2022, 10, 190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Du, X.-P.; Zeng, Y.-H.; Zhu, J.-M.; Zhang, S.-J.; Cai, Z.-H.; Zhou, J. The Communities and Functional Profiles of Virioplankton along a Salinity Gradient in a Subtropical Estuary. Sci. Total Environ. 2021, 759, 143499. [Google Scholar] [CrossRef]
- Che, R.; Bai, M.; Xiao, W.; Zhang, S.; Wang, Y.; Cui, X. Nutrient Levels and Prokaryotes Affect Viral Communities in Plateau Lakes. Sci. Total Environ. 2022, 839, 156033. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, F.H.; Cabello-Yeves, P.J.; Gonzalez-Serrano, R.; Rosselli, R.; López-Pérez, M.; Zemskaya, T.I.; Zakharenko, A.S.; Ivanov, V.G.; Rodriguez-Valera, F. New Viral Biogeochemical Roles Revealed through Metagenomic Analysis of Lake Baikal. Microbiome 2020, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Elbehery, A.H.A.; Deng, L. Insights into the Global Freshwater Virome. Front. Microbiol. 2022, 13, 953500. [Google Scholar] [CrossRef]
- Prado, T.; Brandão, M.L.; Fumian, T.M.; Freitas, L.; Chame, M.; Leomil, L.; Magalhães, M.G.P.; Degrave, W.M.S.; Leite, J.P.G.; Miagostovich, M.P. Virome Analysis in Lakes of the South Shetland Islands, Antarctica—2020. Sci. Total Environ. 2022, 852, 158537. [Google Scholar] [CrossRef] [PubMed]
- Kallmeyer, J.; Pockalny, R.; Adhikari, R.R.; Smith, D.C.; D’Hondt, S. Global Distribution of Microbial Abundance and Biomass in Subseafloor Sediment. Proc. Natl. Acad. Sci. USA 2012, 109, 16213–16216. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, X.; Wang, M.; Qiao, Y.; Zheng, Y.; Zhang, X.-H. Bacterial and Archaeal Communities in Sediments of the North Chinese Marginal Seas. Microb. Ecol. 2015, 70, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Bonnain, C.; Malki, K.; Sawaya, N.A. Phage Puppet Masters of the Marine Microbial Realm. Nat. Microbiol. 2018, 3, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Paul, J.H. Prophages in Marine Bacteria: Dangerous Molecular Time Bombs or the Key to Survival in the Seas? ISME J. 2008, 2, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Segall, A.; Steward, G.; Seguritan, V.; Breitbart, M.; Wolven, F.; Farooq Azam, F. The Complete Genomic Sequence of the Marine Phage Roseophage SIO1 Shares Homology with Nonmarine Phages. Limnol. Oceanogr. 2000, 45, 408–418. [Google Scholar] [CrossRef]
- Mann, N.H.; Cook, A.; Millard, A.; Bailey, S.; Clokie, M. Bacterial Photosynthesis Genes in a Virus. Nature 2003, 424, 741. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Li, X.; Jiang, L.; Gong, L.; Geslin, C.; Shao, Z. Virus Diversity and Interactions with Hosts in Deep-Sea Hydrothermal Vents. Microbiome 2022, 10, 235. [Google Scholar] [CrossRef] [PubMed]
- Dalcin Martins, P.; Danczak, R.E.; Roux, S.; Frank, J.; Borton, M.A.; Wolfe, R.A.; Burris, M.N.; Wilkins, M.J. Viral and Metabolic Controls on High Rates of Microbial Sulfur and Carbon Cycling in Wetland Ecosystems. Microbiome 2018, 6, 138. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Guo, X.; Zhang, R.; Qu, W.; Gao, B.; Zeng, R. Diversities and Potential Biogeochemical Impacts of Mangrove Soil Viruses. Microbiome 2019, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, M.J.; Buchan, A. Lysogeny in the Oceans: Lessons from Cultivated Model Systems and a Reanalysis of Its Prevalence. Environ. Microbiol. 2020, 22, 4919–4933. [Google Scholar] [CrossRef] [PubMed]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in Nature: Mechanisms, Impact and Ecology of Temperate Phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kim, Y.; Ma, Q.; Hong, S.H.; Pokusaeva, K.; Sturino, J.M.; Wood, T.K. Cryptic Prophages Help Bacteria Cope with Adverse Environments. Nat. Commun. 2010, 1, 147. [Google Scholar] [CrossRef] [PubMed]
- Sekizuka, T.; Yamamoto, A.; Komiya, T.; Kenri, T.; Takeuchi, F.; Shibayama, K.; Takahashi, M.; Kuroda, M.; Iwaki, M. Corynebacterium ulcerans 0102 Carries the Gene Encoding Diphtheria Toxin on a Prophage Different from the C. diphtheriae NCTC 13129 Prophage. BMC Microbiol. 2012, 12, 72. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhou, X.; Lu, X.; Xu, Y.; Wei, Z.; Ruan, A. Grain Size Distribution Drives Microbial Communities Vertically Assemble in Nascent Lake Sediments. Environ. Res. 2023, 227, 115828. [Google Scholar] [CrossRef]
- Zhou, X.; Lennon, J.T.; Lu, X.; Ruan, A. Anthropogenic Activities Mediate Stratification and Stability of Microbial Communities in Freshwater Sediments. Microbiome 2023, 11, 191. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.-F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1.0: A Fast and Scalable Metagenome Assembler Driven by Advanced Methodologies and Community Practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Uritskiy, G.V.; DiRuggiero, J.; Taylor, J. MetaWRAP—A Flexible Pipeline for Genome-Resolved Metagenomic Data Analysis. Microbiome 2018, 6, 158. [Google Scholar] [CrossRef]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A Tool for Fast and Accurate Genomic Comparisons That Enables Improved Genome Recovery from Metagenomes through de-Replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk v2: Memory Friendly Classification with the Genome Taxonomy Database. Bioinformatics 2022, 38, 5315–5316. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A Multi-Classifier, Expert-Guided Approach to Detect Diverse DNA and RNA Viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Song, K.; Deng, C.; Ahlgren, N.A.; Fuhrman, J.A.; Li, Y.; Xie, X.; Poplin, R.; Sun, F. Identifying Viruses from Metagenomic Data Using Deep Learning. Quant. Biol. Beijing China 2020, 8, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV Assesses the Quality and Completeness of Metagenome-Assembled Viral Genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated Recovery, Annotation and Curation of Microbial Viruses, and Evaluation of Viral Community Function from Genomic Sequences. Microbiome 2020, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.-Z.; Yuan, W.-G.; Shang, J.; Shi, Y.-H.; Yang, L.-L.; Liu, M.; Zhu, P.; Jin, T.; Sun, Y.; Yuan, L.-H. Virus Classification for Viral Genomic Fragments Using PhaGCN2. Brief. Bioinform. 2023, 24, bbac505. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Zayed, A.A.; Conceição-Neto, N.; Temperton, B.; Bolduc, B.; Alberti, A.; Ardyna, M.; Arkhipova, K.; Carmichael, M.; Cruaud, C.; et al. Marine DNA Viral Macro- and Microdiversity from Pole to Pole. Cell 2019, 177, 1109–1123.e14. [Google Scholar] [CrossRef]
- Li, Z.; Pan, D.; Wei, G.; Pi, W.; Zhang, C.; Wang, J.-H.; Peng, Y.; Zhang, L.; Wang, Y.; Hubert, C.R.J.; et al. Deep Sea Sediments Associated with Cold Seeps Are a Subsurface Reservoir of Viral Diversity. ISME J. 2021, 15, 2366–2378. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN Analysis of Metagenomic Data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic Assignment of Uncultivated Prokaryotic Virus Genomes Is Enabled by Gene-Sharing Networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Emerson, J.B.; Roux, S.; Brum, J.R.; Bolduc, B.; Woodcroft, B.J.; Jang, H.B.; Singleton, C.M.; Solden, L.M.; Naas, A.E.; Boyd, J.A.; et al. Host-Linked Soil Viral Ecology along a Permafrost Thaw Gradient. Nat. Microbiol. 2018, 3, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Camargo, A.P.; Coutinho, F.H.; Dabdoub, S.M.; Dutilh, B.E.; Nayfach, S.; Tritt, A. iPHoP: An Integrated Machine Learning Framework to Maximize Host Prediction for Metagenome-Derived Viruses of Archaea and Bacteria. PLOS Biol. 2023, 21, e3002083. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.-A.; Hugenholtz, P. GTDB: An Ongoing Census of Bacterial and Archaeal Diversity through a Phylogenetically Consistent, Rank Normalized and Complete Genome-Based Taxonomy. Nucleic Acids Res. 2022, 50, D785–D794. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-M.A.; Chu, K.; Palaniappan, K.; Ratner, A.; Huang, J.; Huntemann, M.; Hajek, P.; Ritter, S.; Varghese, N.; Seshadri, R.; et al. The IMG/M Data Management and Analysis System v.6.0: New Tools and Advanced Capabilities. Nucleic Acids Res. 2021, 49, D751–D763. [Google Scholar] [CrossRef]
- Nayfach, S.; Roux, S.; Seshadri, R.; Udwary, D.; Varghese, N.; Schulz, F.; Wu, D.; Paez-Espino, D.; Chen, I.-M.; Huntemann, M.; et al. A Genomic Catalog of Earth’s Microbiomes. Nat. Biotechnol. 2021, 39, 499–509. [Google Scholar] [CrossRef]
- Shaffer, M.; Borton, M.A.; McGivern, B.B.; Zayed, A.A.; La Rosa, S.L.; Solden, L.M.; Liu, P.; Narrowe, A.B.; Rodríguez-Ramos, J.; Bolduc, B.; et al. DRAM for Distilling Microbial Metabolism to Automate the Curation of Microbiome Function. Nucleic Acids Res. 2020, 48, 8883–8900. [Google Scholar] [CrossRef] [PubMed]
- ter Horst, A.M.; Santos-Medellín, C.; Sorensen, J.W.; Zinke, L.A.; Wilson, R.M.; Johnston, E.R.; Trubl, G.; Pett-Ridge, J.; Blazewicz, S.J.; Hanson, P.J.; et al. Minnesota Peat Viromes Reveal Terrestrial and Aquatic Niche Partitioning for Local and Global Viral Populations. Microbiome 2021, 9, 233. [Google Scholar] [CrossRef]
- Hurwitz, B.L.; U’Ren, J.M. Viral Metabolic Reprogramming in Marine Ecosystems. Curr. Opin. Microbiol. 2016, 31, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Helsley, K.R.; Brown, T.M.; Furlong, K.; Williamson, K.E. Applications and Limitations of Tea Extract as a Virucidal Agent to Assess the Role of Phage Predation in Soils. Biol. Fertil. Soils 2014, 50, 263–274. [Google Scholar] [CrossRef]
- Hwang, Y.; Roux, S.; Coclet, C.; Krause, S.J.E.; Girguis, P.R. Viruses Interact with Hosts That Span Distantly Related Microbial Domains in Dense Hydrothermal Mats. Nat. Microbiol. 2023, 8, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s Virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Correa, A.M.S.; Howard-Varona, C.; Coy, S.R.; Buchan, A.; Sullivan, M.B.; Weitz, J.S. Revisiting the Rules of Life for Viruses of Microorganisms. Nat. Rev. Microbiol. 2021, 19, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Zhang, R.; He, Y.; Feng, X.; Jiao, N. Metagenomic Analysis of Virioplankton of the Subtropical Jiulong River Estuary, China. Viruses 2016, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- ter Horst, A.M.; Fudyma, J.D.; Sones, J.L.; Emerson, J.B. Dispersal, Habitat Filtering, and Eco-Evolutionary Dynamics as Drivers of Local and Global Wetland Viral Biogeography. ISME J. Multidiscip. J. Microb. Ecol. 2023, 17, 2079–2089. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Shan, B.; Zhang, H.; Mao, Z. Heavy Metal Sources and Associated Risk in Response to Agricultural Intensification in the Estuarine Sediments of Chaohu Lake Valley, East China. J. Hazard. Mater. 2010, 176, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Combi, T.; Taniguchi, S.; Figueira, R.C.L.; de Mahiques, M.M.; Martins, C.C. Spatial Distribution and Historical Input of Polychlorinated Biphenyls (PCBs) and Organochlorine Pesticides (OCPs) in Sediments from a Subtropical Estuary (Guaratuba Bay, SW Atlantic). Mar. Pollut. Bull. 2013, 70, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Woodcroft, B.J.; Singleton, C.M.; Boyd, J.A.; Evans, P.N.; Emerson, J.B.; Zayed, A.A.F.; Hoelzle, R.D.; Lamberton, T.O.; McCalley, C.K.; Hodgkins, S.B.; et al. Genome-Centric View of Carbon Processing in Thawing Permafrost. Nature 2018, 560, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Karner, M.B.; DeLong, E.F.; Karl, D.M. Archaeal Dominance in the Mesopelagic Zone of the Pacific Ocean. Nature 2001, 409, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Buckley, D.H.; Graber, J.R.; Schmidt, T.M. Phylogenetic Analysis of Nonthermophilic Members of the Kingdom Crenarchaeota and Their Diversity and Abundance in Soils. Appl. Environ. Microbiol. 1998, 64, 4333–4339. [Google Scholar] [CrossRef] [PubMed]
- Casamayor, E.O.; Schäfer, H.; Bañeras, L.; Pedrós-Alió, C.; Muyzer, G. Identification of and Spatio-Temporal Differences between Microbial Assemblages from Two Neighboring Sulfurous Lakes: Comparison by Microscopy and Denaturing Gradient Gel Electrophoresis. Appl. Environ. Microbiol. 2000, 66, 499–508. [Google Scholar] [CrossRef]
- Stahl, D.A.; de la Torre, J.R. Physiology and Diversity of Ammonia-Oxidizing Archaea. Annu. Rev. Microbiol. 2012, 66, 83–101. [Google Scholar] [CrossRef] [PubMed]
- Danovaro, R.; Dell’Anno, A.; Corinaldesi, C.; Rastelli, E.; Cavicchioli, R.; Krupovic, M.; Noble, R.T.; Nunoura, T.; Prangishvili, D. Virus-Mediated Archaeal Hecatomb in the Deep Seafloor. Sci. Adv. 2016, 2, e1600492. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, T.; Goordial, J.; Lindsay, M.R.; McGonigle, J.; Booker, A.; Moser, D.; Stepanauskus, R.; Orcutt, B.N. Replicated Life-History Patterns and Subsurface Origins of the Bacterial Sister Phyla Nitrospirota and Nitrospinota. ISME J. 2023, 17, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Thompson, L.; Suttle, C.; Sullivan, M. Exploring the Vast Diversity of Marine Viruses. Oceanography 2007, 20, 135–139. [Google Scholar] [CrossRef]
- Moura de Sousa, J.A.; Pfeifer, E.; Touchon, M.; Rocha, E.P.C. Causes and Consequences of Bacteriophage Diversification via Genetic Exchanges across Lifestyles and Bacterial Taxa. Mol. Biol. Evol. 2021, 38, 2497–2512. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.; Clokie, M.R.J.; Millard, A.; Mann, N.H. Cyanophage Infection and Photoinhibition in Marine Cyanobacteria. Res. Microbiol. 2004, 155, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Hevroni, G.; Enav, H.; Rohwer, F.; Béjà, O. Diversity of Viral Photosystem-I psaA Genes. ISME J. 2015, 9, 1892–1898. [Google Scholar] [CrossRef] [PubMed]
- Focardi, A.; Ostrowski, M.; Goossen, K.; Brown, M.V.; Paulsen, I. Investigating the Diversity of Marine Bacteriophage in Contrasting Water Masses Associated with the East Australian Current (EAC) System. Viruses 2020, 12, 317. [Google Scholar] [CrossRef]
- Thompson, L.R.; Zeng, Q.; Kelly, L.; Huang, K.H.; Singer, A.U.; Stubbe, J.; Chisholm, S.W. Phage Auxiliary Metabolic Genes and the Redirection of Cyanobacterial Host Carbon Metabolism. Proc. Natl. Acad. Sci. USA 2011, 108, E757–E764. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, A.E.; Howard-Varona, C.; Needham, D.M.; John, S.G.; Worden, A.Z.; Sullivan, M.B.; Waldbauer, J.R.; Coleman, M.L. Metabolic and Biogeochemical Consequences of Viral Infection in Aquatic Ecosystems. Nat. Rev. Microbiol. 2020, 18, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Warwick-Dugdale, J.; Buchholz, H.H.; Allen, M.J.; Temperton, B. Host-Hijacking and Planktonic Piracy: How Phages Command the Microbial High Seas. Virol. J. 2019, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Guo, C.; Wang, M.; Wang, M.; You, S.; Liu, Y.; Zhang, X.; Liu, H.; Jiang, Y.; Shao, H.; et al. Isolation and Complete Genome Sequence of a Novel Cyanophage, S-B05, Infecting an Estuarine Synechococcus Strain: Insights into Environmental Adaptation. Arch. Virol. 2020, 165, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, Y.; Yan, S.; Chen, X.; Xie, S. Viral Community Structure and Functional Potential Vary with Lifestyle and Altitude in Soils of Mt. Everest. Environ. Int. 2023, 178, 108055. [Google Scholar] [CrossRef]
- Pratama, A.A.; Bolduc, B.; Zayed, A.A.; Zhong, Z.-P.; Guo, J.; Vik, D.R.; Gazitúa, M.C.; Wainaina, J.M.; Roux, S.; Sullivan, M.B. Expanding Standards in Viromics: In Silico Evaluation of dsDNA Viral Genome Identification, Classification, and Auxiliary Metabolic Gene Curation. PeerJ 2021, 9, e11447. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, Y.; Gao, Z.; Wu, S.; Ruan, A. Community Structure, Drivers, and Potential Functions of Different Lifestyle Viruses in Chaohu Lake. Viruses 2024, 16, 590. https://doi.org/10.3390/v16040590
Zheng Y, Gao Z, Wu S, Ruan A. Community Structure, Drivers, and Potential Functions of Different Lifestyle Viruses in Chaohu Lake. Viruses. 2024; 16(4):590. https://doi.org/10.3390/v16040590
Chicago/Turabian StyleZheng, Yu, Zihao Gao, Shuai Wu, and Aidong Ruan. 2024. "Community Structure, Drivers, and Potential Functions of Different Lifestyle Viruses in Chaohu Lake" Viruses 16, no. 4: 590. https://doi.org/10.3390/v16040590