
Developmental Dynamics of the Gut Virome in Tibetan Pigs at High Altitude: A Metagenomic Perspective across Age Groups

 , , , and
, , , and 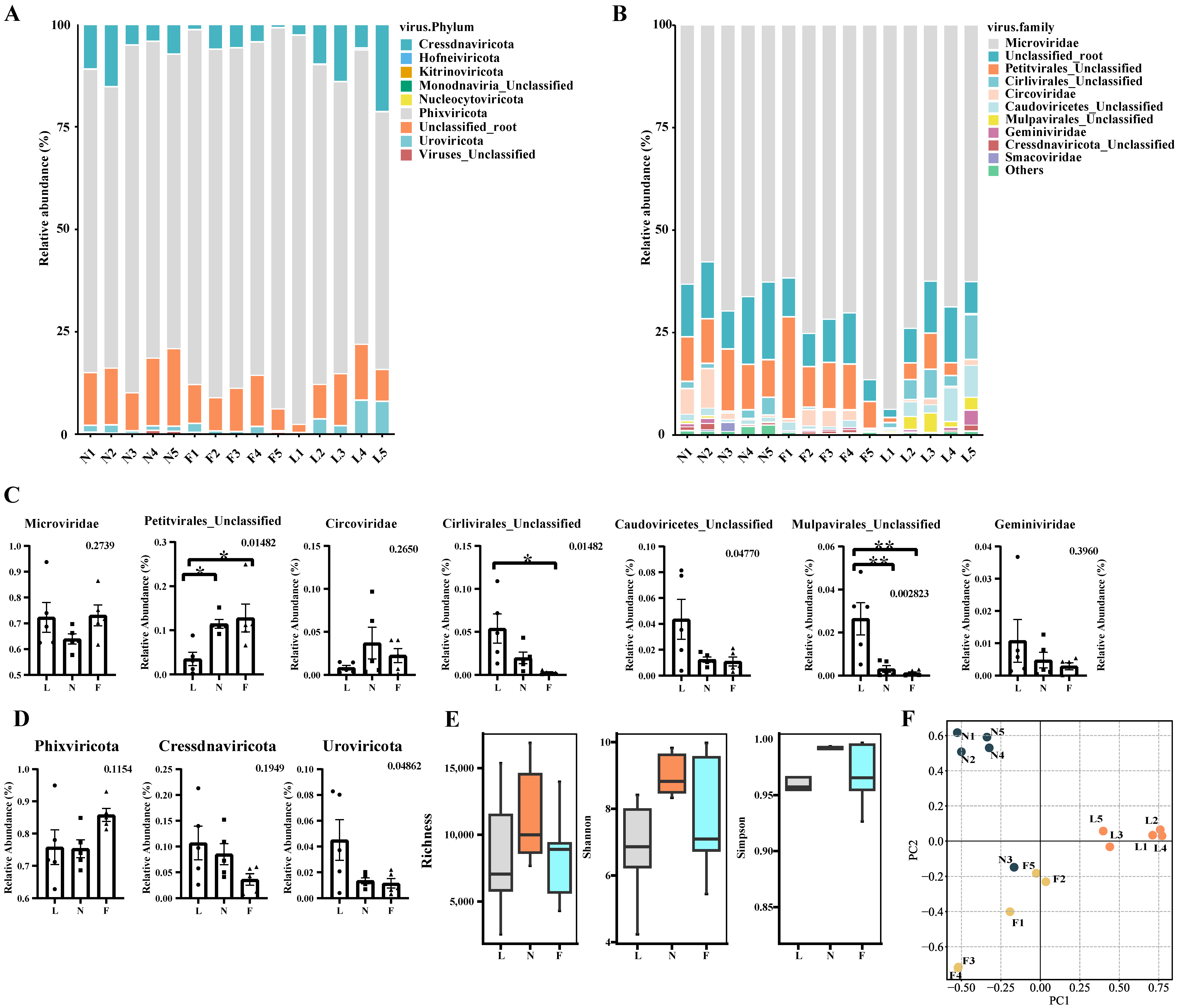{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Management and Sample Collection
2.2. Metagenomes Collection and Sequencing
2.3. Metagenomic Assembly
2.4. Virus Operational Taxonomic Unit (vOTU) Clustering and Annotation
2.5. Fluctuation Trend of Variables and Function Identification of vOTU
2.6. The Identification and Analysis of Viral Auxiliary Metabolic Genes (vAMGs)
2.7. Methods of Lysogenic and Lytic Lifestyle Identification
2.8. Statistics Analysis
3. Results
3.1. Sequencing Data and Quality Control
3.2. Assemble of Metagenomic Data and vOTUs Construction
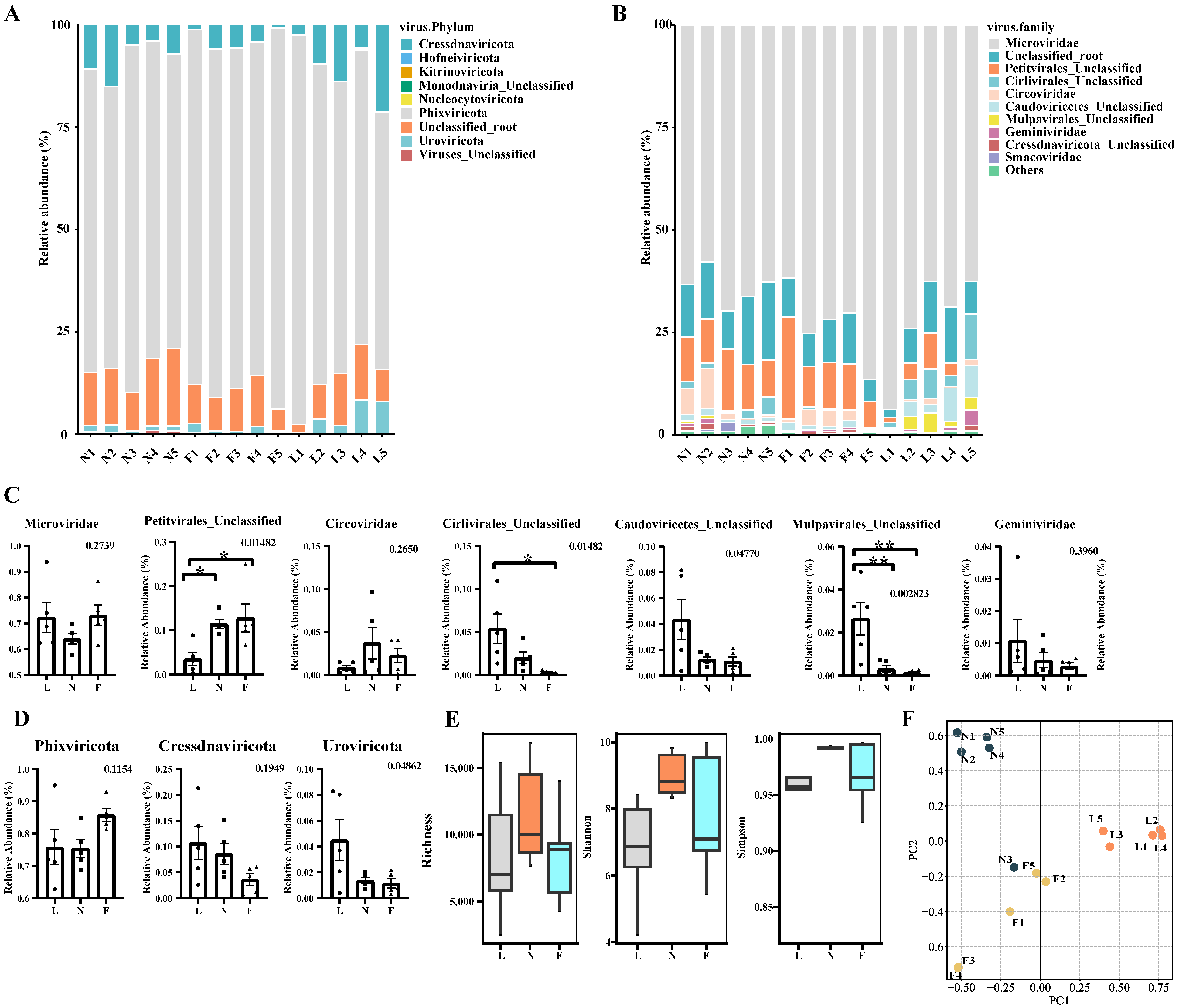
3.3. Taxonomy Composition of Tibetan Pig Gut Virome
3.4. Analysis of Diversity Differences between Samples Based on vOTUs
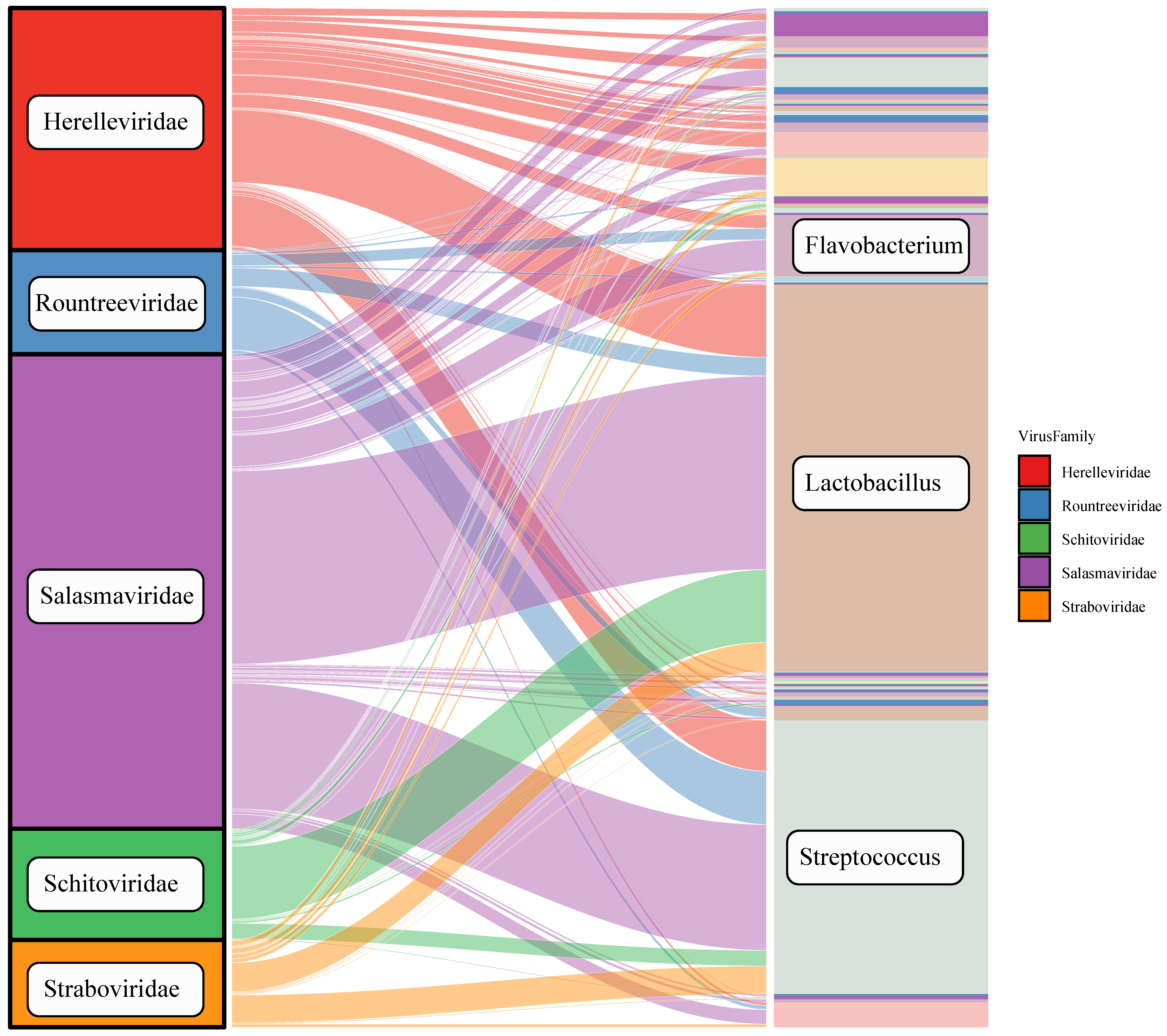
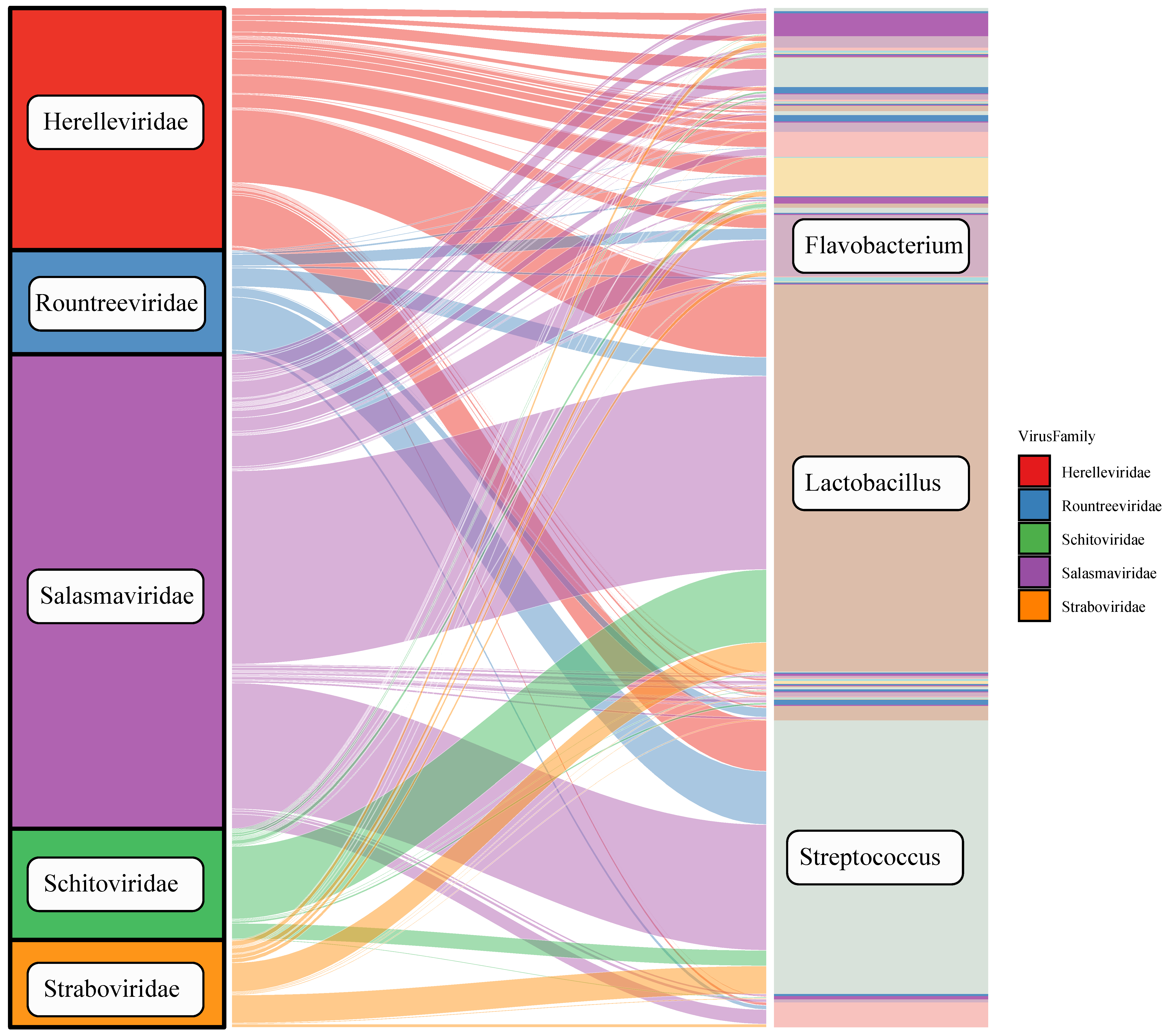
3.5. Host Prediction of Tibetan Pig Gut Virome
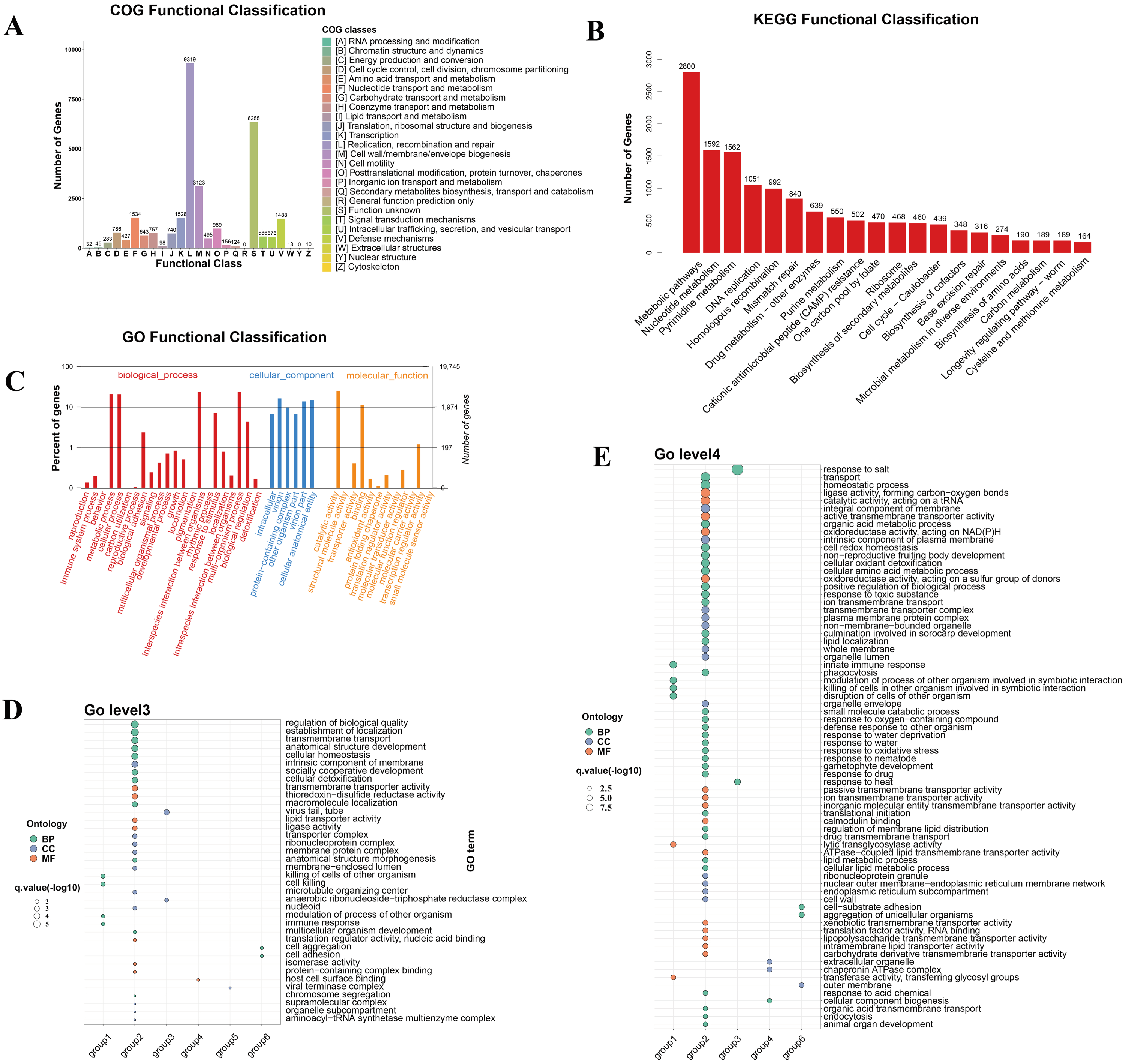
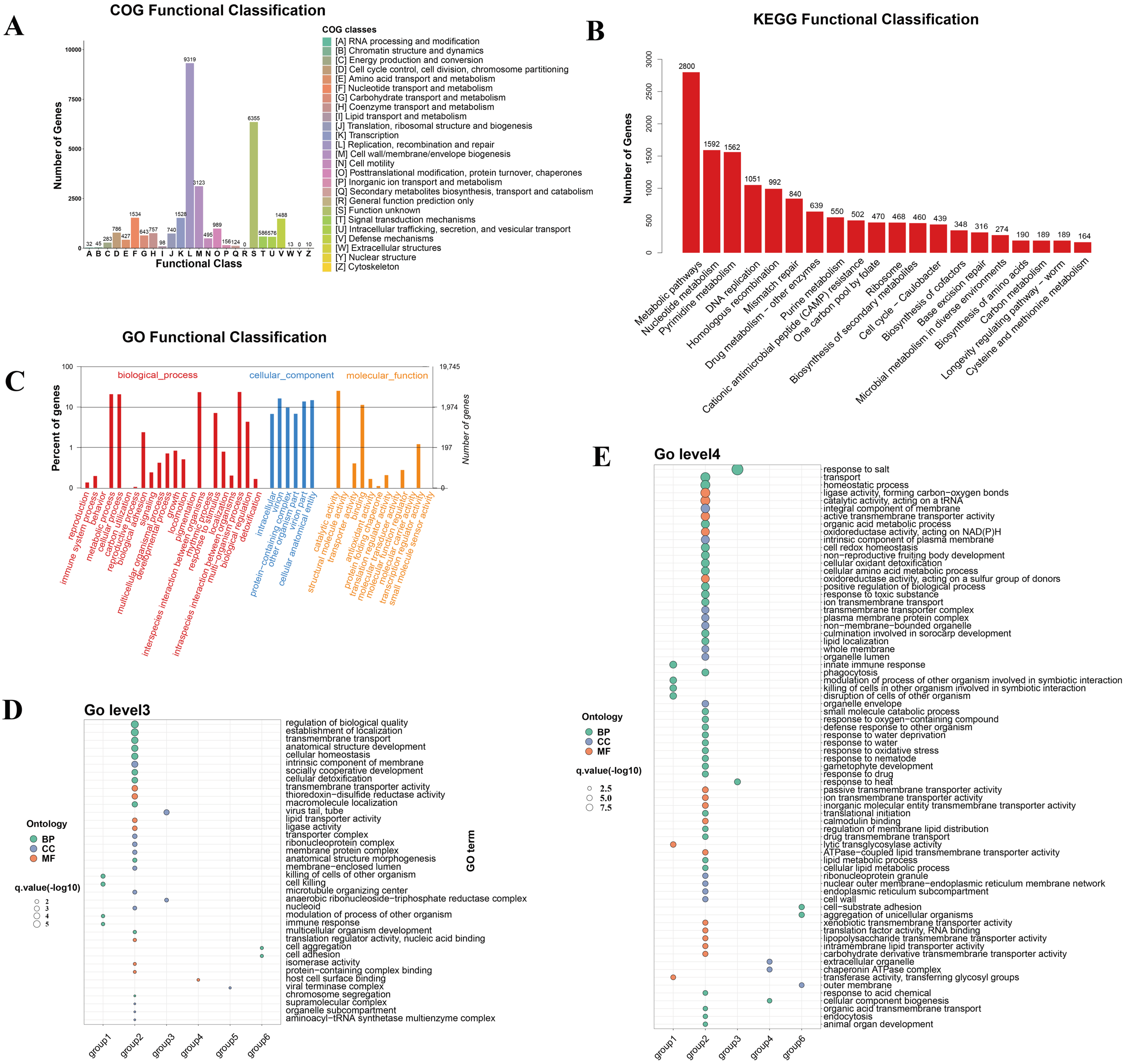
3.6. Functional Annotation and Functional Enrichment Analysis of vOTUs
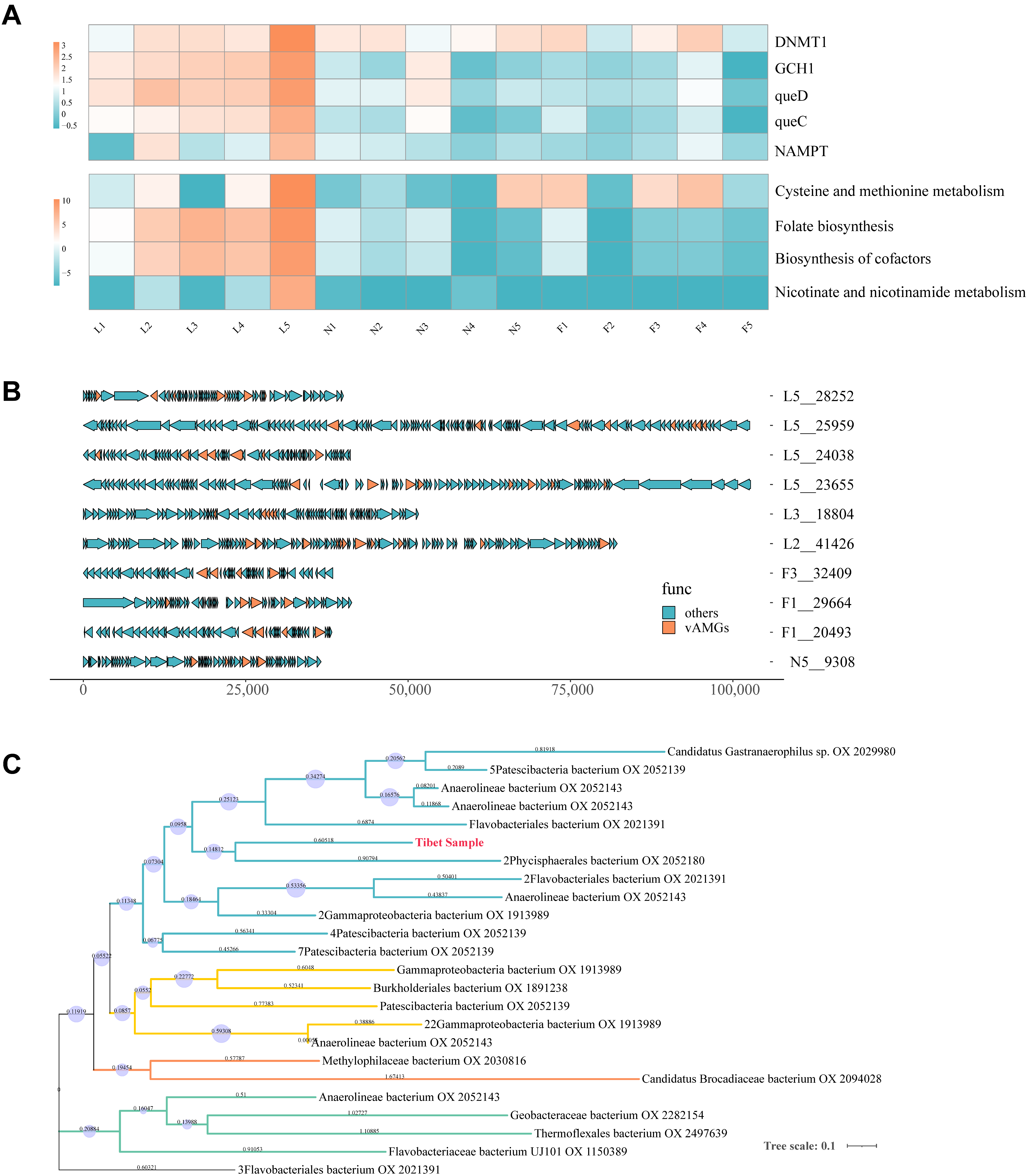
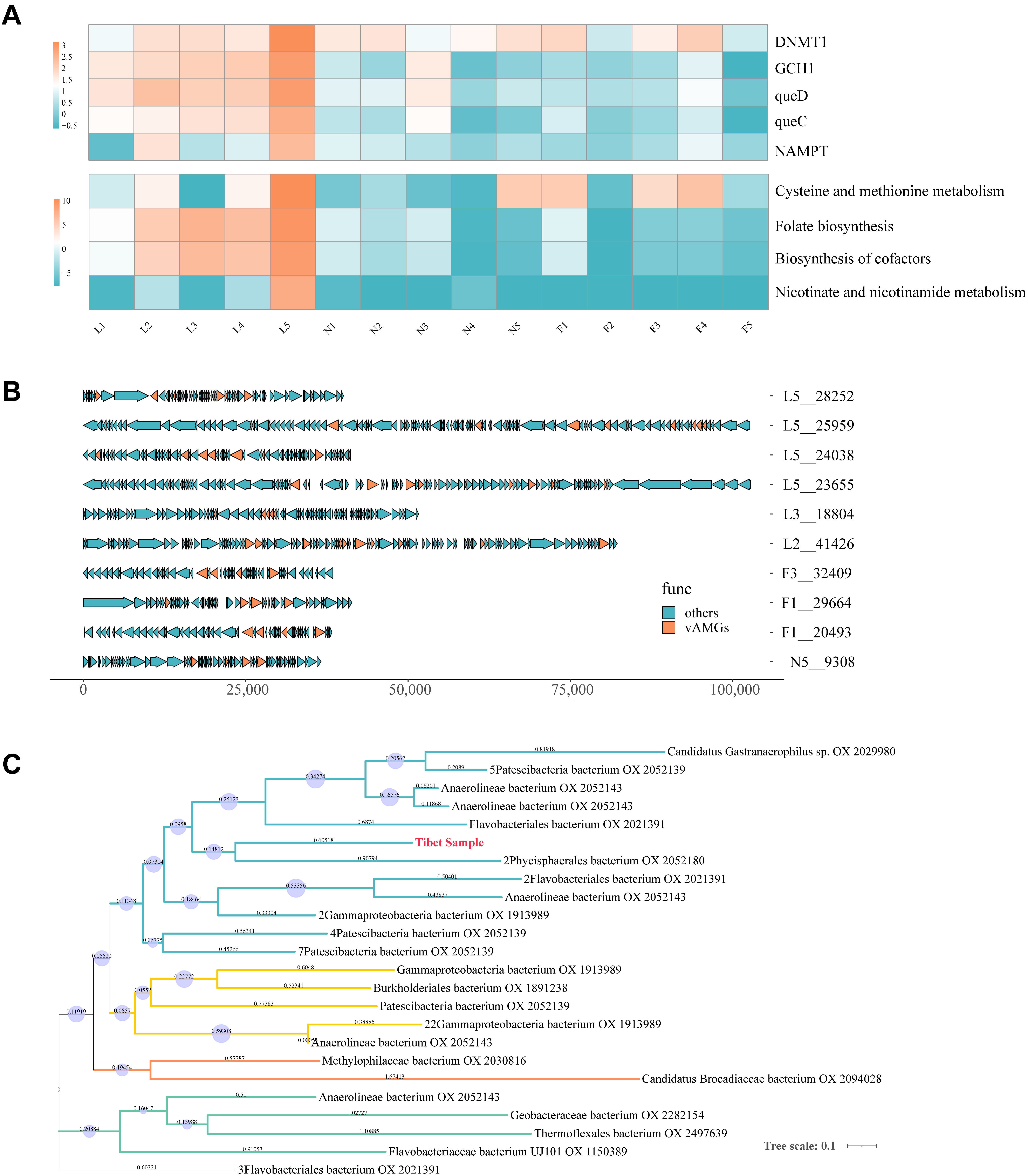
3.7. Auxiliary Metabolic Genes Identification from Tibetan Pig Gut Virome
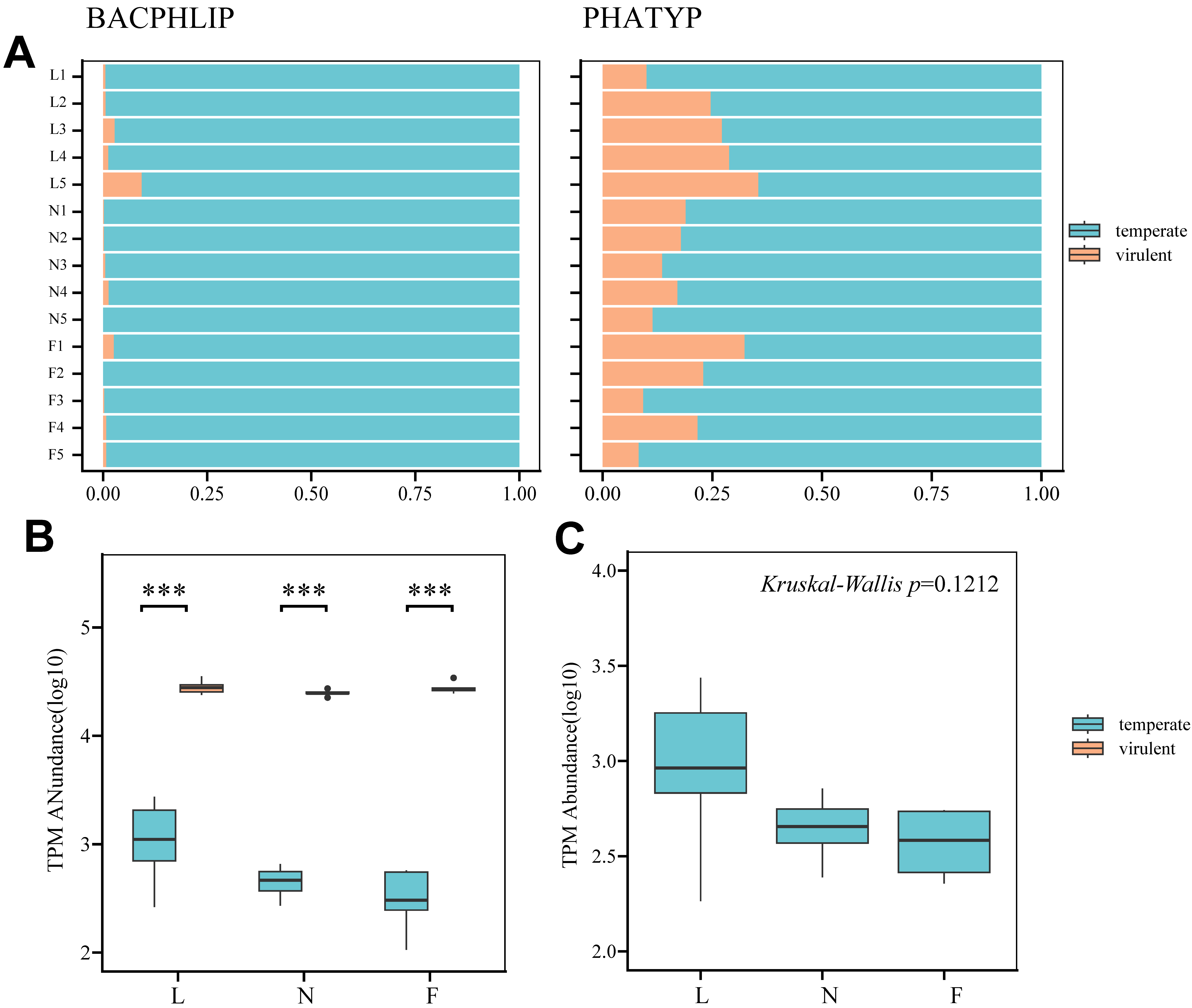
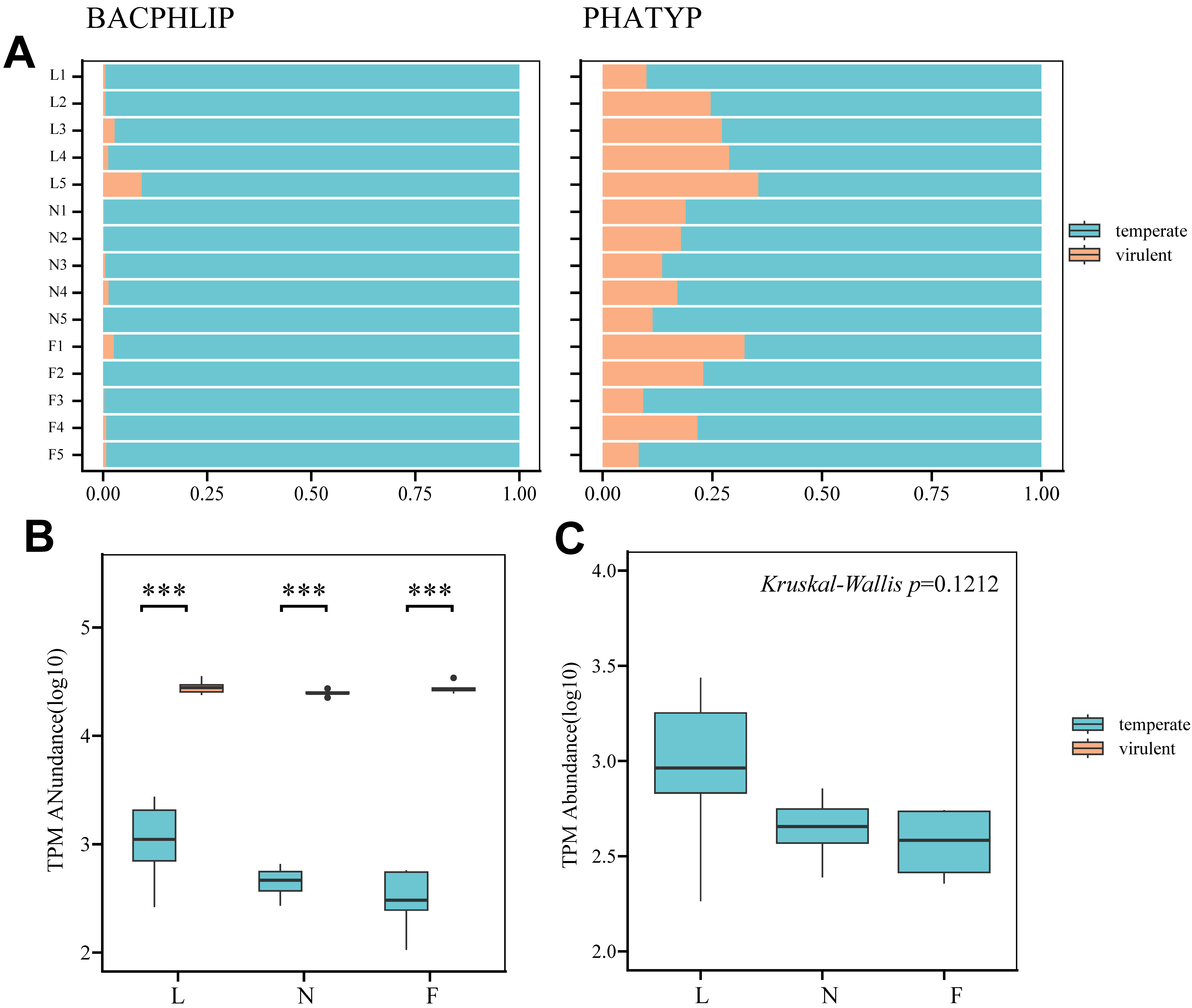
3.8. Phage Lifestyle Identification Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ma, Y.F.; Han, X.M.; Huang, C.P.; Zhong, L.; Adeola, A.C.; Irwin, D.M.; Xie, H.B.; Zhang, Y.P. Population Genomics Analysis Revealed Origin and High-altitude Adaptation of Tibetan Pigs. Sci. Rep. 2019, 9, 11463. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Yang, L.; Zhang, T.; Zhuang, D.; Wu, Q.; Yu, J.; Tian, C.; Zhang, Z. Gut microbiome signatures of extreme environment adaption in Tibetan pig. NPJ Biofilms Microbiomes 2023, 9, 27. [Google Scholar] [CrossRef]
- Zhou, S.; Luo, R.; Gong, G.; Wang, Y.; Gesang, Z.; Wang, K.; Xu, Z.; Suolang, S. Characterization of Metagenome-Assembled Genomes and Carbohydrate-Degrading Genes in the Gut Microbiota of Tibetan Pig. Front. Microbiol. 2020, 11, 595066. [Google Scholar] [CrossRef]
- Ai, H.; Yang, B.; Li, J.; Xie, X.; Chen, H.; Ren, J. Population history and genomic signatures for high-altitude adaptation in Tibetan pigs. BMC Genom. 2014, 15, 834. [Google Scholar] [CrossRef]
- Gregory, A.C.; Zablocki, O.; Zayed, A.A.; Howell, A.; Bolduc, B.; Sullivan, M.B. The Gut Virome Database Reveals Age-Dependent Patterns of Virome Diversity in the Human Gut. Cell Host Microbe 2020, 28, 724–740.e728. [Google Scholar] [CrossRef]
- Cao, Z.; Sugimura, N.; Burgermeister, E.; Ebert, M.P.; Zuo, T.; Lan, P. The gut virome: A new microbiome component in health and disease. EBioMedicine 2022, 81, 104113. [Google Scholar] [CrossRef]
- Shang, P.; Wei, M.; Duan, M.; Yan, F.; Chamba, Y. Healthy Gut Microbiome Composition Enhances Disease Resistance and Fat Deposition in Tibetan Pigs. Front. Microbiol. 2022, 13, 965292. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Zhang, S.; Xu, H.; Kong, F.; Yu, X.; Wang, P.; Yang, M.; Li, D.; Zhang, M.; Ni, Q.; et al. Gut microbiota of Tibetans and Tibetan pigs varies between high and low altitude environments. Microbiol. Res. 2020, 235, 126447. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tsai, T.; Deng, F.; Wei, X.; Chai, J.; Knapp, J.; Apple, J.; Maxwell, C.V.; Lee, J.A.; Li, Y.; et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 2019, 7, 109. [Google Scholar] [CrossRef]
- Xia, B.; Wu, W.; Zhang, L.; Wen, X.; Xie, J.; Zhang, H. Gut microbiota mediates the effects of inulin on enhancing sulfomucin production and mucosal barrier function in a pig model. Food Funct. 2021, 12, 10967–10982. [Google Scholar] [CrossRef]
- Frese, S.A.; Parker, K.; Calvert, C.C.; Mills, D.A. Diet shapes the gut microbiome of pigs during nursing and weaning. Microbiome 2015, 3, 28. [Google Scholar] [CrossRef] [PubMed]
- Folgueiras-Gonzalez, A.; van den Braak, R.; Deijs, M.; Kuller, W.; Sietsma, S.; Thuring, V.; van der Hoek, L.; de Groof, A. Dynamics of the Enteric Virome in a Swine Herd Affected by Non-PCV2/PRRSV Postweaning Wasting Syndrome. Viruses 2021, 13, 2538. [Google Scholar] [CrossRef] [PubMed]
- Smolak, D.; Salamunova, S.; Jackova, A.; Harsanyova, M.; Budis, J.; Szemes, T.; Vilcek, S. Analysis of RNA virome in rectal swabs of healthy and diarrheic pigs of different age. Comp. Immunol. Microbiol. Infect. Dis. 2022, 90–91, 101892. [Google Scholar] [CrossRef]
- Sachsenroder, J.; Twardziok, S.O.; Scheuch, M.; Johne, R. The general composition of the faecal virome of pigs depends on age, but not on feeding with a probiotic bacterium. PLoS ONE 2014, 9, e88888. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Zou, H.; Li, J.; Wei, H. Landscapes of Enteric Virome Signatures in Early-Weaned Piglets. Microbiol. Spectr. 2022, 10, e01698-22. [Google Scholar] [CrossRef] [PubMed]
- Puente, H.; Arguello, H.; Cortey, M.; Gomez-Garcia, M.; Mencia-Ares, O.; Perez-Perez, L.; Diaz, I.; Carvajal, A. Detection and genetic characterization of enteric viruses in diarrhoea outbreaks from swine farms in Spain. Porc. Health Manag. 2023, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Johansen, J.; Atarashi, K.; Arai, Y.; Hirose, N.; Sorensen, S.J.; Vatanen, T.; Knip, M.; Honda, K.; Xavier, R.J.; Rasmussen, S.; et al. Centenarians have a diverse gut virome with the potential to modulate metabolism and promote healthy lifespan. Nat. Microbiol. 2023, 8, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Erez, Z.; Steinberger-Levy, I.; Shamir, M.; Doron, S.; Stokar-Avihail, A.; Peleg, Y.; Melamed, S.; Leavitt, A.; Savidor, A.; Albeck, S.; et al. Communication between viruses guides lysis-lysogeny decisions. Nature 2017, 541, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.; Yi, Y.; Wang, J.; Hao, Y.; Zhang, M.; Wang, S.; Meng, C.; Zhang, Y.; Jing, H.; Wang, Y.; et al. Diversity and distribution of viruses inhabiting the deepest ocean on Earth. ISME J. 2021, 15, 3094–3110. [Google Scholar] [CrossRef]
- Kim, M.S.; Bae, J.W. Lysogeny is prevalent and widely distributed in the murine gut microbiota. ISME J. 2018, 12, 1127–1141. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Turkington, C.J.; Hill, C. Mutualistic interplay between bacteriophages and bacteria in the human gut. Nat. Rev. Microbiol. 2022, 20, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Maaroufi, H.; Levesque, R.C. Glycoside hydrolase family 32 is present in Bacillus subtilis phages. Virol. J. 2015, 12, 157. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.L.A.; Andrade, M.E.R.; Trindade, L.M.; Leocadio, P.C.L.; Alvarez-Leite, J.I.; Dos Reis, D.C.; Cassali, G.D.; Souza, E.M.E.L.S.; Dos Santos Martins, F.; Fernandes, S.O.A.; et al. Prophylactic and therapeutic supplementation using fructo-oligosaccharide improves the intestinal homeostasis after mucositis induced by 5-fluorouracil. Biomed. Pharmacother. 2021, 133, 111012. [Google Scholar] [CrossRef] [PubMed]
- Mangalea, M.R.; Paez-Espino, D.; Kieft, K.; Chatterjee, A.; Chriswell, M.E.; Seifert, J.A.; Feser, M.L.; Demoruelle, M.K.; Sakatos, A.; Anantharaman, K.; et al. Individuals at risk for rheumatoid arthritis harbor differential intestinal bacteriophage communities with distinct metabolic potential. Cell Host Microbe 2021, 29, 726–739.e5. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Wilks, C.; Antonescu, V.; Charles, R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 2019, 35, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Camargo, A.P.; Nayfach, S.; Chen, I.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Ritter, S.J.; Reddy, T.B.K.; Mukherjee, S.; Schulz, F.; et al. IMG/VR v4: An expanded database of uncultivated virus genomes within a framework of extensive functional, taxonomic, and ecological metadata. Nucleic Acids Res. 2023, 51, D733–D743. [Google Scholar] [CrossRef]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitua, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Camargo, A.P.; Roux, S.; Schulz, F.; Babinski, M.; Xu, Y.; Hu, B.; Chain, P.S.G.; Nayfach, S.; Kyrpides, N.C. Identification of mobile genetic elements with geNomad. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef]
- Shang, J.; Peng, C.; Liao, H.; Tang, X.; Sun, Y. PhaBOX: A web server for identifying and characterizing phage contigs in metagenomic data. Bioinform. Adv. 2023, 3, vbad101. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; LoCascio, P.F.; Hauser, L.J.; Uberbacher, E.C. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 2012, 28, 2223–2230. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Jang, K.; Park, K.; Kim, H. Real-time measurement of fibers using an HY-differential mobility analyzer with an optical particle counter (KOFAM). PLoS ONE 2017, 12, e0182119. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Famiglietti, M.L.; Estreicher, A.; Breuza, L.; Poux, S.; Redaschi, N.; Xenarios, I.; Bridge, A.; UniProt, C. An enhanced workflow for variant interpretation in UniProtKB/Swiss-Prot improves consistency and reuse in ClinVar. Database 2019, 2019, baz040. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernandez-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, M.; Borton, M.A.; McGivern, B.B.; Zayed, A.A.; La Rosa, S.L.; Solden, L.M.; Liu, P.; Narrowe, A.B.; Rodriguez-Ramos, J.; Bolduc, B.; et al. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res. 2020, 48, 8883–8900. [Google Scholar] [CrossRef]
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021, 39, 578–585. [Google Scholar] [CrossRef]
- Qin, J.; Ji, B.; Ma, Y.; Liu, X.; Wang, T.; Liu, G.; Li, B.; Wang, G.; Gao, P. Diversity and potential function of pig gut DNA viruses. Heliyon 2023, 9, e14020. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Pan, D.; Wei, G.; Pi, W.; Zhang, C.; Wang, J.H.; Peng, Y.; Zhang, L.; Wang, Y.; Hubert, C.R.J.; et al. Deep sea sediments associated with cold seeps are a subsurface reservoir of viral diversity. ISME J. 2021, 15, 2366–2378. [Google Scholar] [CrossRef] [PubMed]
- Ter Horst, A.M.; Santos-Medellin, C.; Sorensen, J.W.; Zinke, L.A.; Wilson, R.M.; Johnston, E.R.; Trubl, G.; Pett-Ridge, J.; Blazewicz, S.J.; Hanson, P.J.; et al. Correction to: Minnesota peat virome reveal terrestrial and aquatic niche partitioning for local and global viral populations. Microbiome 2022, 10, 17. [Google Scholar] [CrossRef]
- Hockenberry, A.J.; Wilke, C.O. BACPHLIP: Predicting bacteriophage lifestyle from conserved protein domains. PeerJ 2021, 9, e11396. [Google Scholar] [CrossRef]
- Shang, J.; Tang, X.; Sun, Y. PhaTYP: Predicting the lifestyle for bacteriophages using BERT. Brief. Bioinform. 2023, 24, bbac487. [Google Scholar] [CrossRef]
- Niu, L.; Zhao, S.; Chen, Y.; Li, Y.; Zou, G.; Tao, Y.; Zhang, W.; Wang, L.; Zhang, H. Diversity and potential functional characteristics of phage communities colonizing microplastic biofilms. Environ. Res. 2023, 219, 115103. [Google Scholar] [CrossRef]
- Zhu, W.; Yang, J.; Lu, S.; Jin, D.; Pu, J.; Wu, S.; Luo, X.L.; Liu, L.; Li, Z.; Xu, J. RNA virus diversity in birds and small mammals from Qinghai–Tibet Plateau of China. Front. Microbiol. 2022, 13, 780651. [Google Scholar] [CrossRef]
- Pan, J.; Ji, L.; Wu, H.; Wang, X.; Wang, Y.; Wu, Y.; Yang, S.; Shen, Q.; Liu, Y.; Zhang, W.; et al. Metagenomic analysis of herbivorous mammalian viral communities in the Northwest Plateau. BMC Genom. 2023, 24, 568. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Hu, C.; Zhou, Q.; Yang, D.; Wang, L.; Zhang, B. Viral communities associated with porcine diarrhoeal disease and genetic characterization of a bufavirus in Tibetan pigs in China. Arch. Virol. 2021, 166, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, J.; Ma, L.; Liu, X.; Wei, H.; Xiao, Y.; Tao, S. Metagenomic Sequencing Identified Specific Bacteriophage Signature Discriminating between Healthy and Diarrheal Neonatal Piglets. Nutrients 2023, 15, 1616. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Ravet, V.; Pereira, O.; Sullivan, M.B. Genomic characteristics and environmental distributions of the uncultivated Far-T4 phages. Front. Microbiol. 2015, 6, 199. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Gong, W.; Yan, X.; Zhao, Z.; Yang, L.; Tan, Z.; Xu, L.; Zhu, A.; Zhang, J.; Rao, J.; et al. Viral Metagenome-Based Precision Surveillance of Pig Population at Large Scale Reveals Viromic Signatures of Sample Types and Influence of Farming Management on Pig Virome. mSystems 2021, 6, e00420-21. [Google Scholar] [CrossRef]
- Neu, U.; Mainou, B.A. Virus interactions with bacteria: Partners in the infectious dance. PLoS Pathog. 2020, 16, e1008234. [Google Scholar] [CrossRef] [PubMed]
- Veldsman, W.P.; Yang, C.; Zhang, Z.; Huang, Y.; Chowdhury, D.; Zhang, L. Structural and Functional Disparities within the Human Gut Virome in Terms of Genome Topology and Representative Genome Selection. Viruses 2024, 16, 134. [Google Scholar] [CrossRef]
- Huang, L.; Wu, X.; Guo, S.; Lv, Y.; Zhou, P.; Huang, G.; Duan, Z.; Sun, W. Metagenomic-based characterization of the gut virome in patients with polycystic ovary syndrome. Front. Microbiol. 2022, 13, 951782. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Ju, F. Potential Auxiliary Metabolic Capabilities and Activities Reveal Biochemical Impacts of Viruses in Municipal Wastewater Treatment Plants. Environ. Sci. Technol. 2023, 57, 5485–5498. [Google Scholar] [CrossRef]
- El Yacoubi, B.; Bailly, M.; de Crecy-Lagard, V. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 2012, 46, 69–95. [Google Scholar] [CrossRef]
- Sabri, M.; Hauser, R.; Ouellette, M.; Liu, J.; Dehbi, M.; Moeck, G.; Garcia, E.; Titz, B.; Uetz, P.; Moineau, S. Genome annotation and intraviral interactome for the Streptococcus pneumoniae virulent phage Dp-1. J. Bacteriol. 2011, 193, 551–562. [Google Scholar] [CrossRef]
- Lallès, J.-P.; Bosi, P.; Smidt, H.; Stokes, C.R. Weaning—A challenge to gut physiologists. Livest. Sci. 2007, 108, 82–93. [Google Scholar] [CrossRef]
- Hurwitz, B.L.; U’Ren, J.M. Viral metabolic reprogramming in marine ecosystems. Curr. Opin. Microbiol. 2016, 31, 161–168. [Google Scholar] [CrossRef]
- Sharon, I.; Alperovitch, A.; Rohwer, F.; Haynes, M.; Glaser, F.; Atamna-Ismaeel, N.; Pinter, R.Y.; Partensky, F.; Koonin, E.V.; Wolf, Y.I.; et al. Photosystem I gene cassettes are present in marine virus genomes. Nature 2009, 461, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Smith, F.; Clark, J.E.; Overman, B.L.; Tozel, C.C.; Huang, J.H.; Rivier, J.E.; Blikslager, A.T.; Moeser, A.J. Early weaning stress impairs development of mucosal barrier function in the porcine intestine. Am. J. Physiol.Gastrointest. Liver Physiol. 2010, 298, G352–G363. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, S.G.; Reyes, A.; Maurice, C.F. Bacteriophages playing nice: Lysogenic bacteriophage replication stable in the human gut microbiota. iScience 2023, 26, 106007. [Google Scholar] [CrossRef]
- Abedon, S.T. Commentary: Communication between Viruses Guides Lysis-Lysogeny Decisions. Front. Microbiol. 2017, 8, 983. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, T.; Yu, M.; Chen, Y.L.; Jin, M. The Life Cycle Transitions of Temperate Phages: Regulating Factors and Potential Ecological Implications. Viruses 2022, 14, 1904. [Google Scholar] [CrossRef]
- Matos, R.C.; Leulier, F. Lactobacilli-Host mutualism: “learning on the fly”. Microb. Cell Fact. 2014, 13 (Suppl. S1), S6. [Google Scholar] [CrossRef]
- Qi, R.; Qiu, X.; Du, L.; Wang, J.; Wang, Q.; Huang, J.; Liu, Z. Changes of Gut Microbiota and Its Correlation With Short Chain Fatty Acids and Bioamine in Piglets at the Early Growth Stage. Front. Vet. Sci. 2020, 7, 617259. [Google Scholar] [CrossRef]
- Li, G.; Cortez, M.H.; Dushoff, J.; Weitz, J.S. When to be temperate: On the fitness benefits of lysis vs. lysogeny. Virus Evol. 2020, 6, veaa042. [Google Scholar] [CrossRef]
- Wahl, L.M.; Betti, M.I.; Dick, D.W.; Pattenden, T.; Puccini, A.J. Evolutionary stability of the lysis-lysogeny decision: Why be virulent? Evolution 2019, 73, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Stewart, F.M.; Levin, B.R. The population biology of bacterial viruses: Why be temperate. Theor. Popul. Biol. 1984, 26, 93–117. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Qi, M.; Shang, P.; Zhang, H.; Nawaz, S.; Ghaffar, A.; Wu, Q.; Dong, H. Characterization, estimation of virulence and drug resistance of diarrheagenic escherichia coli (DEC) isolated from Tibetan pigs. Microb. Pathog. 2023, 177, 106046. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Cao, Z.; Shang, P.; Zhang, H.; Hussain, R.; Mehmood, K.; Chang, Z.; Wu, Q.; Dong, H. Comparative analysis of fecal microbiota composition diversity in Tibetan piglets suffering from diarrheagenic Escherichia coli (DEC). Microb. Pathog. 2023, 158, 105106. [Google Scholar] [CrossRef]
- Vahedi, A.; Soltan Dallal, M.M.; Douraghi, M.; Nikkhahi, F.; Rajabi, Z.; Yousefi, M.; Mousavi, M. Isolation and identification of specific bacteriophage against enteropathogenic Escherichia coli (EPEC) and in vitro and in vivo characterization of bacteriophage. FEMS Microbiol. Lett. 2018, 365, fny136. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, R.; Guan, A.; Ma, B.; Gao, Y.; Peng, Y.; He, Y.; Xu, Q.; Li, K.; Zhong, Y.; Luo, R.; et al. Developmental Dynamics of the Gut Virome in Tibetan Pigs at High Altitude: A Metagenomic Perspective across Age Groups. Viruses 2024, 16, 606. https://doi.org/10.3390/v16040606
Luo R, Guan A, Ma B, Gao Y, Peng Y, He Y, Xu Q, Li K, Zhong Y, Luo R, et al. Developmental Dynamics of the Gut Virome in Tibetan Pigs at High Altitude: A Metagenomic Perspective across Age Groups. Viruses. 2024; 16(4):606. https://doi.org/10.3390/v16040606
Chicago/Turabian StyleLuo, Runbo, Aohan Guan, Bin Ma, Yuan Gao, Yuna Peng, Yanling He, Qianshuai Xu, Kexin Li, Yanan Zhong, Rui Luo, and et al. 2024. "Developmental Dynamics of the Gut Virome in Tibetan Pigs at High Altitude: A Metagenomic Perspective across Age Groups" Viruses 16, no. 4: 606. https://doi.org/10.3390/v16040606
APA StyleLuo, R., Guan, A., Ma, B., Gao, Y., Peng, Y., He, Y., Xu, Q., Li, K., Zhong, Y., Luo, R., Cao, R., Jin, H., Lin, Y., & Shang, P. (2024). Developmental Dynamics of the Gut Virome in Tibetan Pigs at High Altitude: A Metagenomic Perspective across Age Groups. Viruses, 16(4), 606. https://doi.org/10.3390/v16040606