

Expanding the Scope of Adenoviral Vectors by Utilizing Novel Tools for Recombination and Vector Rescue
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells, Viruses, and Bacterial Strains
2.2. Construction of Viral Bacmids
2.3. Rescue of Recombinant Adenovirus
2.4. Time Course Analysis
2.5. Sequencing and Data Analysis
2.6. Software
3. Results
3.1. Screening of Mastadenovirus Types Usable for CTR
- Human adenovirus species A, type 12;
- Human adenovirus species B1, type 3;
- Human adenovirus species B2, type 35;
- Human adenovirus species C, type 2;
- Human adenovirus species F, type 41;
- Simian adenovirus species E, type 25.
3.2. HFR Allows for the Reliable Assembly of Novel Vectors
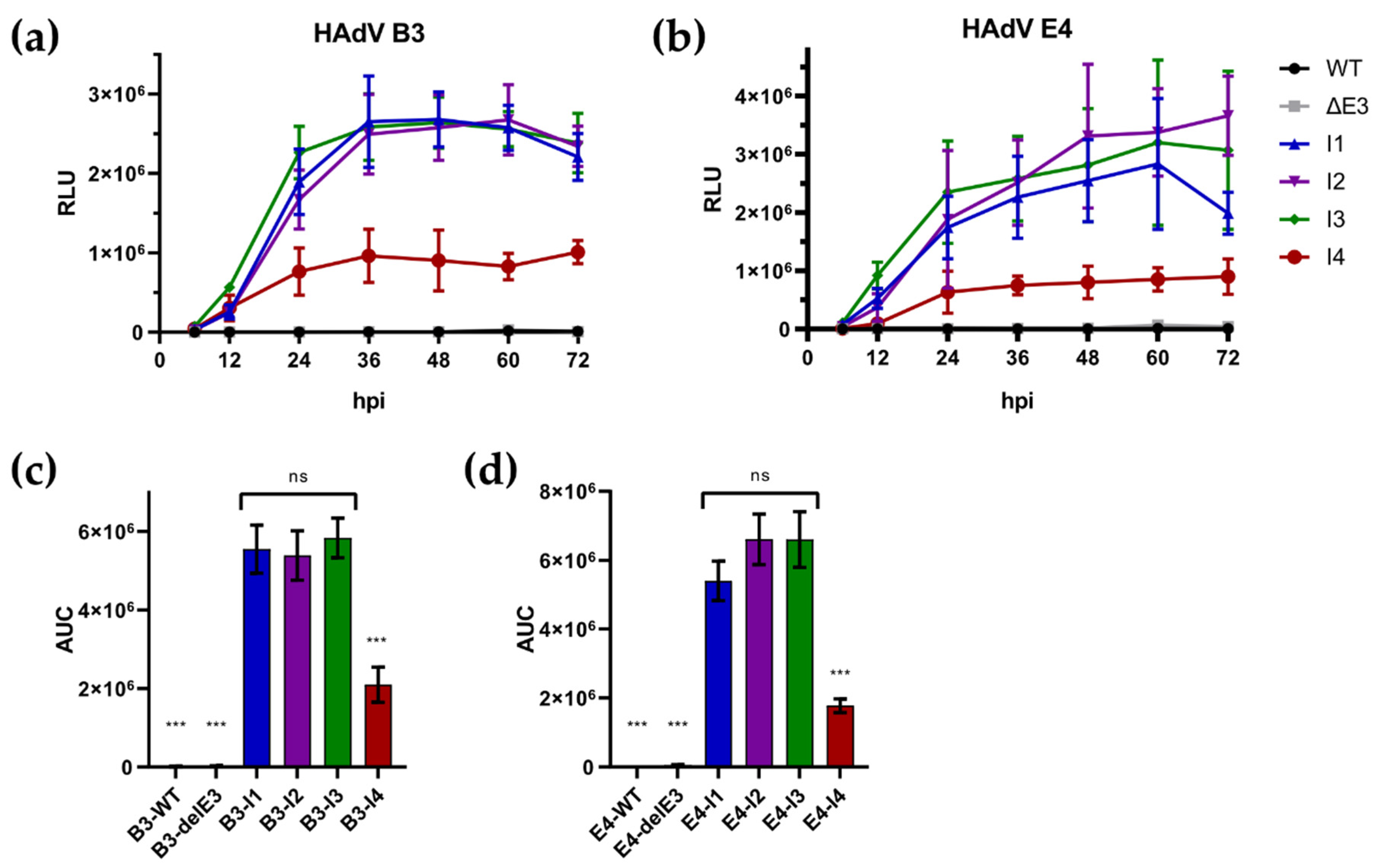
3.3. Insertion Site Directly Affects Transgene Expression but Does Not Affect Viral Replication
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Construction of CTR-Proficient Bacmids Containing Wild-Type Adenoviral Genomic Sequences
Appendix A.2. HFR Cloning of Vector Variants
Appendix A.2.1. Kanamycin Intermediates for Inserting Transgenes into HAdV-B3 and HAdV-E4

Appendix A.2.2. Transfer Vectors and Bacmid Assembly for Transgenic HAdV-B3 and HAdV-E4
References
- Echavarría, M. Adenoviruses in Immunocompromised Hosts. Clin. Microbiol. Rev. 2008, 21, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bergelson, J.M. Adenovirus Receptors. J. Virol. 2005, 79, 12125–12131. [Google Scholar] [CrossRef] [PubMed]
- Windheim, M.; Hilgendorf, A.; Burgert, H.G. Immune Evasion by Adenovirus E3 Proteins: Exploitation of Intracellular Trafficking Pathways. Curr. Top. Microbiol. Immunol. 2004, 273, 29–85. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Anselmo, A.C.; Mitragotri, S. Viral Vector-based Gene Therapies in the Clinic. Bioeng. Transl. Med. 2022, 7, e10258. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.; Subramanian, G.; Silayeva, L.; Newkirk, I.; Doctor, D.; Chawla, K.; Chattopadhyay, S.; Chandra, D.; Chilukuri, N.; Betapudi, V. Gene Therapy Leaves a Vicious Cycle. Front. Oncol. 2019, 9, 297. [Google Scholar] [CrossRef] [PubMed]
- Chartier, C.; Degryse, E.; Gantzer, M.; Dieterle, A.; Pavirani, A.; Mehtali, M. Efficient Generation of Recombinant Adenovirus Vectors by Homologous Recombination in Escherichia Coli. J. Virol. 1996, 70, 4805–4810. [Google Scholar] [CrossRef] [PubMed]
- Mach, N.; Gao, J.; Schaffarczyk, L.; Janz, S.; Ehrke-Schulz, E.; Dittmar, T.; Ehrhardt, A.; Zhang, W. Spectrum-Wide Exploration of Human Adenoviruses for Breast Cancer Therapy. Cancers 2020, 12, 1403. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chen, Y.; Feng, Y.; Li, J.; Kang, X.; Zhang, S.; Li, Y.; Zhao, Z.; Yang, W.; Zhao, L.; et al. Generation and Characterization of a Replication-Competent Human Adenovirus Type 55 Encoding EGFP. Viruses 2023, 15, 1192. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, O.; Diaconu, I.; Cerullo, V.; Pesonen, S.K.; Kanerva, A.; Joensuu, T.; Kairemo, K.; Laasonen, L.; Partanen, K.; Kangasniemi, L.; et al. Ad3-HTERT-E1A, a Fully Serotype 3 Oncolytic Adenovirus, in Patients with Chemotherapy Refractory Cancer. Mol. Ther. 2012, 20, 1821–1830. [Google Scholar] [CrossRef]
- Zafar, S.; Basnet, S.; Launonen, I.; Quixabeira, D.C.A.; Santos, J.; Hemminki, O.; Malmstedt, M.; Cervera-Carrascon, V.; Aronen, P.; Kalliokoski, R.; et al. Oncolytic Adenovirus Type 3 Coding for CD40L Facilitates Dendritic Cell Therapy of Prostate Cancer in Humanized Mice and Patient Samples. Hum. Gene Ther. 2021, 32, 192–202. [Google Scholar] [CrossRef]
- Becker, J.; Fakhiri, J.; Grimm, D. Fantastic AAV Gene Therapy Vectors and How to Find Them— Random Diversification, Rational Design and Machine Learning. Pathogens 2022, 11, 756. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Fedotova, A.; Jaki, L.; Sallard, E.; Erhardt, A.; Fuchs, J.; Ruzsics, Z. Combining CRISPR/Cas Mediated Terminal Resolution with a Novel Genetic Workflow to Achieve High-Diversity Adenoviral Libraries. Mol. Ther.-Methods Clin. Dev. 2024, 32, 101241. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Yan, Y.; Zhang, J.; Zhao, S.; Feng, L.; Ou, J.; Cao, N.; Li, M.; Zhao, W.; Wan, C.; et al. Rapid Construction of a Replication-Competent Infectious Clone of Human Adenovirus Type 14 by Gibson Assembly. Viruses 2018, 10, 568. [Google Scholar] [CrossRef] [PubMed]
- Riedl, A.; Fischer, J.; Burgert, H.-G.; Ruzsics, Z. Rescue of Recombinant Adenoviruses by CRISPR/Cas-Mediated in Vivo Terminal Resolution. Front. Microbiol. 2022, 13, 495. [Google Scholar] [CrossRef] [PubMed]
- Datsenko, K.A.; Wanner, B.L. One-Step Inactivation of Chromosomal Genes in Escherichia Coli K-12 Using PCR Products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic Assembly of DNA Molecules up to Several Hundred Kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Ruzsics, Z.; Wagner, M.; Osterlehner, A.; Cook, J.; Koszinowski, U.; Burgert, H.-G.H.-G. Transposon-Assisted Cloning and Traceless Mutagenesis of Adenoviruses: Development of a Novel Vector Based on Species D. J. Virol. 2006, 80, 8100–8113. [Google Scholar] [CrossRef] [PubMed]
- Lock, M.; Korn, M.; Wilson, J.; Sena-Esteves, M.; Gao, G. Measuring the Infectious Titer of Recombinant Adenovirus Using Tissue Culture Infection Dose 50% (TCID 50 ) End-Point Dilution and Quantitative Polymerase Chain Reaction (QPCR). Cold Spring Harb. Protoc. 2019, 2019, pdb.prot095562. [Google Scholar] [CrossRef]
- Ruzsics, Z.; Hoffmann, K.; Riedl, A.; Krawczyk, A.; Widera, M.; Sertznig, H.; Schipper, L.; Kapper-Falcone, V.; Debreczeny, M.; Ernst, W.; et al. A Novel, Broad-Acting Peptide Inhibitor of Double-Stranded DNA Virus Gene Expression and Replication. Front. Microbiol. 2020, 11, 601555. [Google Scholar] [CrossRef]
- Weigang, S.; Fuchs, J.; Zimmer, G.; Schnepf, D.; Kern, L.; Beer, J.; Luxenburger, H.; Ankerhold, J.; Falcone, V.; Kemming, J.; et al. Within-Host Evolution of SARS-CoV-2 in an Immunosuppressed COVID-19 Patient as a Source of Immune Escape Variants. Nat. Commun. 2021, 12, 6405. [Google Scholar] [CrossRef]
- Biomatters developement, team. Geneious Prime, version 2019.2.3; Biomatters Ltd.: Auckland, New Zealand, 2019. [Google Scholar]
- Afgan, E.; Nekrutenko, A.; Grüning, B.A.; Blankenberg, D.; Goecks, J.; Schatz, M.C.; Ostrovsky, A.E.; Mahmoud, A.; Lonie, A.J.; Syme, A.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2022 Update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef]
- Uusi-Kerttula, H.; Hulin-Curtis, S.; Davies, J.; Parker, A.L. Oncolytic Adenovirus: Strategies and Insights for Vector Design and Immuno-Oncolytic Applications. Viruses 2015, 7, 5987–6020. [Google Scholar] [CrossRef]
- Wang, H.; Li, Z.-Y.; Liu, Y.; Persson, J.; Beyer, I.; Möller, T.; Koyuncu, D.; Drescher, M.R.; Strauss, R.; Zhang, X.-B.; et al. Desmoglein 2 Is a Receptor for Adenovirus Serotypes 3, 7, 11 and 14. Nat. Med. 2011, 17, 96–104. [Google Scholar] [CrossRef]
- Matsuda, K.; Migueles, S.A.; Huang, J.; Bolkhovitinov, L.; Stuccio, S.; Griesman, T.; Pullano, A.A.; Kang, B.H.; Ishida, E.; Zimmerman, M.; et al. A Replication-Competent Adenovirus-Vectored Influenza Vaccine Induces Durable Systemic and Mucosal Immunity. J. Clin. Investig. 2021, 131, e140794. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Huang, J.; Zhou, T.; Sheng, Z.; Kang, B.H.; Ishida, E.; Griesman, T.; Stuccio, S.; Bolkhovitinov, L.; Wohlbold, T.J.; et al. Prolonged Evolution of the Memory B Cell Response Induced by a Replicating Adenovirus-Influenza H5 Vaccine. Sci. Immunol. 2019, 4, eaau2710. [Google Scholar] [CrossRef]
- Kaufman, R.J.; Davies, M.V.; Pathak, V.K.; Hershey, J.W. The Phosphorylation State of Eucaryotic Initiation Factor 2 Alters Translational Efficiency of Specific MRNAs. Mol. Cell. Biol. 1989, 9, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kang, S.; Song, J.J.; Kim, J.-H. The Effectiveness of the Oncolytic Activity Induced by Ad5/F35 Adenoviral Vector Is Dependent on the Cumulative Cellular Conditions of Survival and Autophagy. Int. J. Oncol. 2013, 42, 1337–1348. [Google Scholar] [CrossRef]
- Burgert, H.-G.; Fischer, J.; Heinzinger, S.; England, H.; Elsing, A.; Steinle, A.; Ruzsics, Z. Distinct down regulation by common E3 functions of HAdVs B-E. Unpublished manuscript, last modified April 10th 2024. Microsoft Word file.
- Syyam, A.; Nawaz, A.; Ijaz, A.; Sajjad, U.; Fazil, A.; Irfan, S.; Muzaffar, A.; Shahid, M.; Idrees, M.; Malik, K.; et al. Adenovirus Vector System: Construction, History and Therapeutic Applications. Biotechniques 2022, 73, 297–305. [Google Scholar] [CrossRef]
- Kovesdi, I.; Hedley, S.J. Adenoviral Producer Cells. Viruses 2010, 2, 1681–1703. [Google Scholar] [CrossRef] [PubMed]
- Thirion, C.; Lochmüller, H.; Ruzsics, Z.; Boelhauve, M.; König, C.; Thedieck, C.; Kutik, S.; Geiger, C.; Kochanek, S.; Volpers, C.; et al. Adenovirus Vectors Based on Human Adenovirus Type 19a Have High Potential for Human Muscle-Directed Gene Therapy. Hum. Gene Ther. 2006, 17, 193–205. [Google Scholar] [CrossRef]
- Sallard, E.; Schulte, L.; van den Boom, A.; Klimovitskii, A.; Knierer, J.; Hagedorn, C.; Knocks, M.; Zhang, W.; Kreppel, F.; Ehrhardt, A.; et al. Development of Oncolytic and Gene Therapy Vectors Based on Adenovirus Serotype 4 as an Alternative to Adenovirus Serotype 5. J. Gene Med. 2024, 26, e3576. [Google Scholar] [CrossRef] [PubMed]
- Sirena, D.; Ruzsics, Z.; Schaffner, W.; Greber, U.F.; Hemmi, S. The Nucleotide Sequence and a First Generation Gene Transfer Vector of Species B Human Adenovirus Serotype 3. Virology 2005, 343, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Abbink, P.; Lemckert, A.A.C.; Ewald, B.A.; Lynch, D.M.; Denholtz, M.; Smits, S.; Holterman, L.; Damen, I.; Vogels, R.; Thorner, A.R.; et al. Comparative Seroprevalence and Immunogenicity of Six Rare Serotype Recombinant Adenovirus Vaccine Vectors from Subgroups B and D. J. Virol. 2007, 81, 4654–4663. [Google Scholar] [CrossRef]
- Vogels, R.; Zuijdgeest, D.; van Rijnsoever, R.; Hartkoorn, E.; Damen, I.; de Béthune, M.-P.; Kostense, S.; Penders, G.; Helmus, N.; Koudstaal, W.; et al. Replication-Deficient Human Adenovirus Type 35 Vectors for Gene Transfer and Vaccination: Efficient Human Cell Infection and Bypass of Preexisting Adenovirus Immunity. J. Virol. 2003, 77, 8263–8271. [Google Scholar] [CrossRef]
- Holterman, L.; Vogels, R.; van der Vlugt, R.; Sieuwerts, M.; Grimbergen, J.; Kaspers, J.; Geelen, E.; van der Helm, E.; Lemckert, A.; Gillissen, G.; et al. Novel Replication-Incompetent Vector Derived from Adenovirus Type 11 (Ad11) for Vaccination and Gene Therapy: Low Seroprevalence and Non-Cross-Reactivity with Ad5. J. Virol. 2004, 78, 13207–13215. [Google Scholar] [CrossRef]
- Abrahamsen, K.; Kong, H.L.; Mastrangeli, A.; Brough, D.; Lizonova, A.; Crystal, R.G.; Falck-Pedersen, E. Construction of an Adenovirus Type 7a E1A- Vector. J. Virol. 1997, 71, 8946–8951. [Google Scholar] [CrossRef]
- Fallaux, F.J.; Bout, A.; van der Velde, I.; van den Wollenberg, D.J.M.; Hehir, K.M.; Keegan, J.; Auger, C.; Cramer, S.J.; van Ormondt, H.; van der Eb, A.J.; et al. New Helper Cells and Matched Early Region 1-Deleted Adenovirus Vectors Prevent Generation of Replication-Competent Adenoviruses. Hum. Gene Ther. 1998, 9, 1909–1917. [Google Scholar] [CrossRef]
- Fallaux, F.J.; Kranenburg, O.; Cramer, S.J.; Houweling, A.; van Ormondt, H.; Hoeben, R.C.; van der Eb, A.J. Characterization of 911: A New Helper Cell Line for the Titration and Propagation of Early Region 1-Deleted Adenoviral Vectors. Hum. Gene Ther. 1996, 7, 215–222. [Google Scholar] [CrossRef]
- Hatfield, L.; Hearing, P. Redundant Elements in the Adenovirus Type 5 Inverted Terminal Repeat Promote Bidirectional Transcriptionin Vitro and Are Important for Virus Growthin Vivo. Virology 1991, 184, 265–276. [Google Scholar] [CrossRef]
- Charman, M.; Herrmann, C.; Weitzman, M.D. Viral and Cellular Interactions during Adenovirus DNA Replication. FEBS Lett. 2019, 593, 3531–3550. [Google Scholar] [CrossRef]
- Danthinne, X.; Imperiale, M.J. Production of First Generation Adenovirus Vectors: A Review. Gene Ther. 2000, 7, 1707–1714. [Google Scholar] [CrossRef]
- Bett, A.J.; Prevec, L.; Graham, F.L. Packaging Capacity and Stability of Human Adenovirus Type 5 Vectors. J. Virol. 1993, 67, 5911–5921. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Alemany, R.; Yamamoto, M.; Curiel, D.T. The Presence of the Adenovirus E3 Region Improves the Oncolytic Potency of Conditionally Replicative Adenoviruses. Clin. Cancer Res. 2002, 8, 3348–3359. [Google Scholar] [PubMed]
- Wen, S.; Driscoll, R.M.; Schneider, D.B.; Dichek, D.A. Inclusion of the E3 Region in an Adenoviral Vector Decreases Inflammation and Neointima Formation After Arterial Gene Transfer. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Greer, A.E.; Hearing, P.; Ketner, G. The Adenovirus E4 11 k Protein Binds and Relocalizes the Cytoplasmic P-Body Component Ddx6 to Aggresomes. Virology 2011, 417, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Yondola, M.A.; Hearing, P. The Adenovirus E4 ORF3 Protein Binds and Reorganizes the TRIM Family Member Transcriptional Intermediary Factor 1 Alpha. J. Virol. 2007, 81, 4264–4271. [Google Scholar] [CrossRef] [PubMed]
- Vink, E.; Zheng, Y.; Yeasmin, R.; Stamminger, T.; Krug, L.; Hearing, P. Impact of Adenovirus E4-ORF3 Oligomerization and Protein Localization on Cellular Gene Expression. Viruses 2015, 7, 2428–2449. [Google Scholar] [CrossRef]
- Reichel, R.; Neill, S.D.; Kovesdi, I.; Simon, M.C.; Raychaudhuri, P.; Nevins, J.R. The Adenovirus E4 Gene, in Addition to the E1A Gene, Is Important for Trans-Activation of E2 Transcription and for E2F Activation. J. Virol. 1989, 63, 3643–3650. [Google Scholar] [CrossRef]
- Sandler, A.B.; Ketner, G. Adenovirus Early Region 4 Is Essential for Normal Stability of Late Nuclear RNAs. J. Virol. 1989, 63, 624–630. [Google Scholar] [CrossRef]
- Zhang, W.; Fu, J.; Liu, J.; Wang, H.; Schiwon, M.; Janz, S.; Schaffarczyk, L.; von der Goltz, L.; Ehrke-Schulz, E.; Dörner, J.; et al. An Engineered Virus Library as a Resource for the Spectrum-Wide Exploration of Virus and Vector Diversity. Cell Rep. 2017, 19, 1698–1709. [Google Scholar] [CrossRef]
- Graham, F.L.L. Covalently Closed Circles of Human Adenovirus DNA Are Infectious. EMBO J. 1984, 3, 2917–2922. [Google Scholar] [CrossRef] [PubMed]
- Berman, C.M.; Papa, L.J.; Hendel, S.J.; Moore, C.L.; Suen, P.H.; Weickhardt, A.F.; Doan, N.-D.; Kumar, C.M.; Uil, T.G.; Butty, V.L.; et al. An Adaptable Platform for Directed Evolution in Human Cells. J. Am. Chem. Soc. 2018, 140, 18093–18103. [Google Scholar] [CrossRef] [PubMed]
- Uil, T.G.; Vellinga, J.; de Vrij, J.; van den Hengel, S.K.; Rabelink, M.J.W.E.; Cramer, S.J.; Eekels, J.J.M.; Ariyurek, Y.; van Galen, M.; Hoeben, R.C. Directed Adenovirus Evolution Using Engineered Mutator Viral Polymerases. Nucleic Acids Res. 2011, 39, e30. [Google Scholar] [CrossRef]
- Kuhn, I.; Harden, P.; Bauzon, M.; Chartier, C.; Nye, J.; Thorne, S.; Reid, T.; Ni, S.; Lieber, A.; Fisher, K.; et al. Directed Evolution Generates a Novel Oncolytic Virus for the Treatment of Colon Cancer. PLoS ONE 2008, 3, e2409. [Google Scholar] [CrossRef]
- Myers, N.D.; Skorohodova, K.V.; Gounder, A.P.; Smith, J.G. Directed Evolution of Mutator Adenoviruses Resistant to Antibody Neutralization. J. Virol. 2013, 87, 6047–6050. [Google Scholar] [CrossRef]
- New England BioLabs NEBuilder® HiFi DNA Assembly—Benefits Over GeneArt Gibson Assembly® and In-Fusion® Snap Assembly. Available online: https://www.neb.com/en/applications/cloning-and-synthetic-biology/dna-assembly-and-cloning/nebuilder-hifi-dna-assembly/nebuilder-hifi-dna-assembly---benefits-over-neb-gibson-assembly (accessed on 26 October 2023).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Source |
|---|---|
| HAdV-A12 | Prof. Dr. Anja Erhardt, University Witten-Herdecke |
| HAdV-B3 | Prof. Dr. Anja Erhardt, University Witten-Herdecke |
| HAdV-B35 | Prof. Dr. Albert Heim, German Adenovirus Reference Laboratory, Hannover |
| HAdV-C2 | ATCC VR-846 |
| HAdV-E4 | ATCC-1572 |
| HAdV-F41 | ATCC VR-930 |
| SAdV-E25 | ATCC VR-594 |
| Human | Simian | |||||||
|---|---|---|---|---|---|---|---|---|
| Species | A | B1 | B2 | C | E | F | E | |
| Type | 12 | 3 | 35 | 2 | 5 | 4 | 41 | 25 |
| Ad-BAC | - | 3 | - | - | 7 | 2 | - | 4 |
| Ad-BAC-RC | 3 | 2 | 9 | 8 | 2 | 1 | 4 | 2 |
| Correct clones * | 30% | 50% | 90% | 80% | 90% | 30% | 40% | 60% |
| Human | Simian | |||||||
|---|---|---|---|---|---|---|---|---|
| Species | A | B1 | B2 | C | E | F | E | |
| Type | 12 | 3 | 35 | 2 | 5 | 4 | 41 | 25 |
| HeLa | ND | ++ | ++ | ND | ++ | ++ | − | ND |
| A549 | ++ | ++ | +++ | ++ | ++ | ++ | + | + |
| 293A | +++ | − | +++ | +++ | +++ | ++ | + | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, J.; Fedotova, A.; Bühler, C.; Darriba, L.; Schreiner, S.; Ruzsics, Z. Expanding the Scope of Adenoviral Vectors by Utilizing Novel Tools for Recombination and Vector Rescue. Viruses 2024, 16, 658. https://doi.org/10.3390/v16050658
Fischer J, Fedotova A, Bühler C, Darriba L, Schreiner S, Ruzsics Z. Expanding the Scope of Adenoviral Vectors by Utilizing Novel Tools for Recombination and Vector Rescue. Viruses. 2024; 16(5):658. https://doi.org/10.3390/v16050658
Chicago/Turabian StyleFischer, Julian, Ariana Fedotova, Clara Bühler, Laura Darriba, Sabrina Schreiner, and Zsolt Ruzsics. 2024. "Expanding the Scope of Adenoviral Vectors by Utilizing Novel Tools for Recombination and Vector Rescue" Viruses 16, no. 5: 658. https://doi.org/10.3390/v16050658
APA StyleFischer, J., Fedotova, A., Bühler, C., Darriba, L., Schreiner, S., & Ruzsics, Z. (2024). Expanding the Scope of Adenoviral Vectors by Utilizing Novel Tools for Recombination and Vector Rescue. Viruses, 16(5), 658. https://doi.org/10.3390/v16050658