
Influenza Virus Genomic Surveillance, Arizona, USA, 2023–2024
 ,
,  ,
, 
 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Sample Processing and Testing
2.3. Next Generation Sequencing
2.4. Sequencing Analysis
2.5. Phylogenetic Analysis
2.6. Protein Structure Modeling
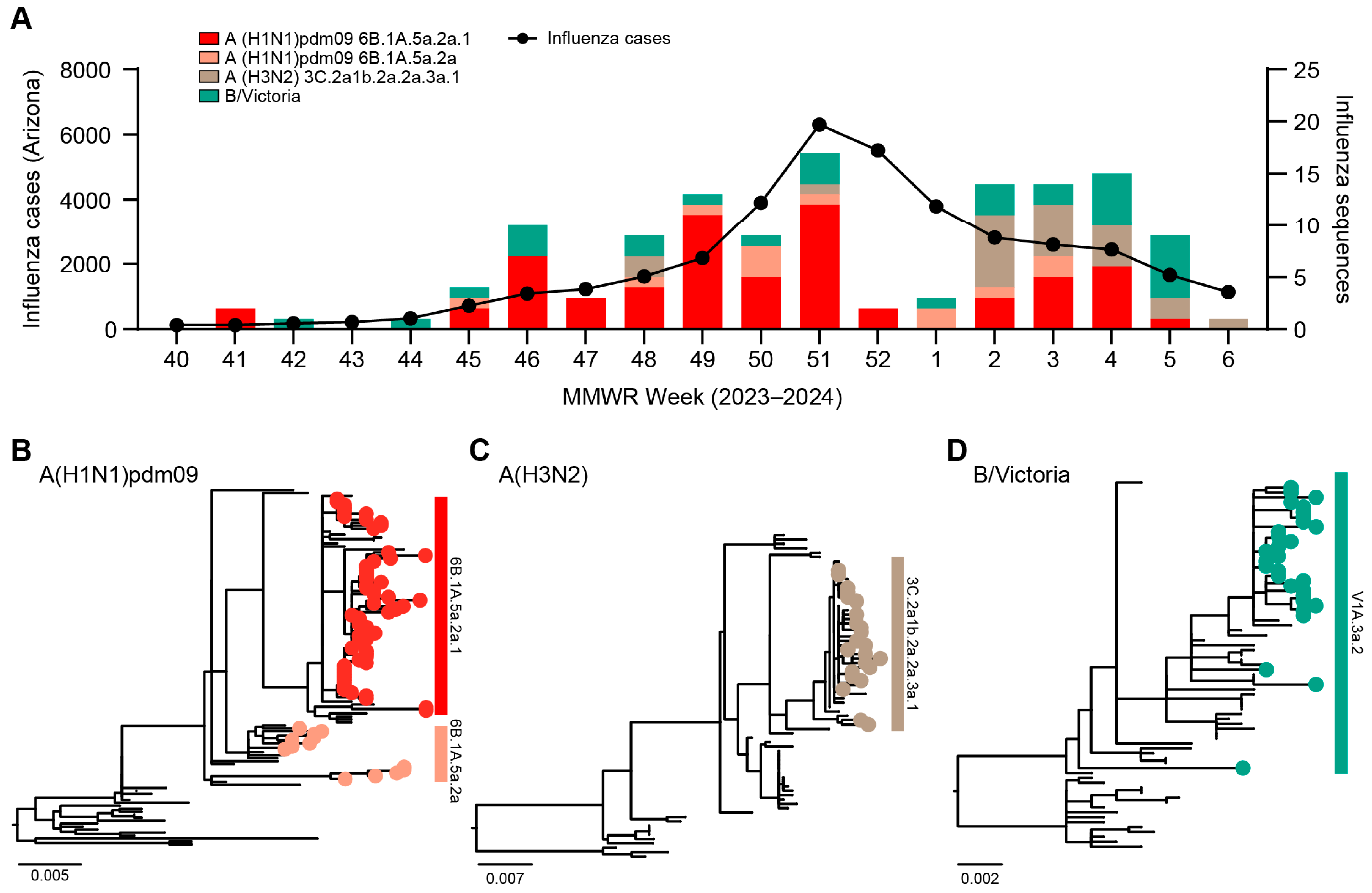
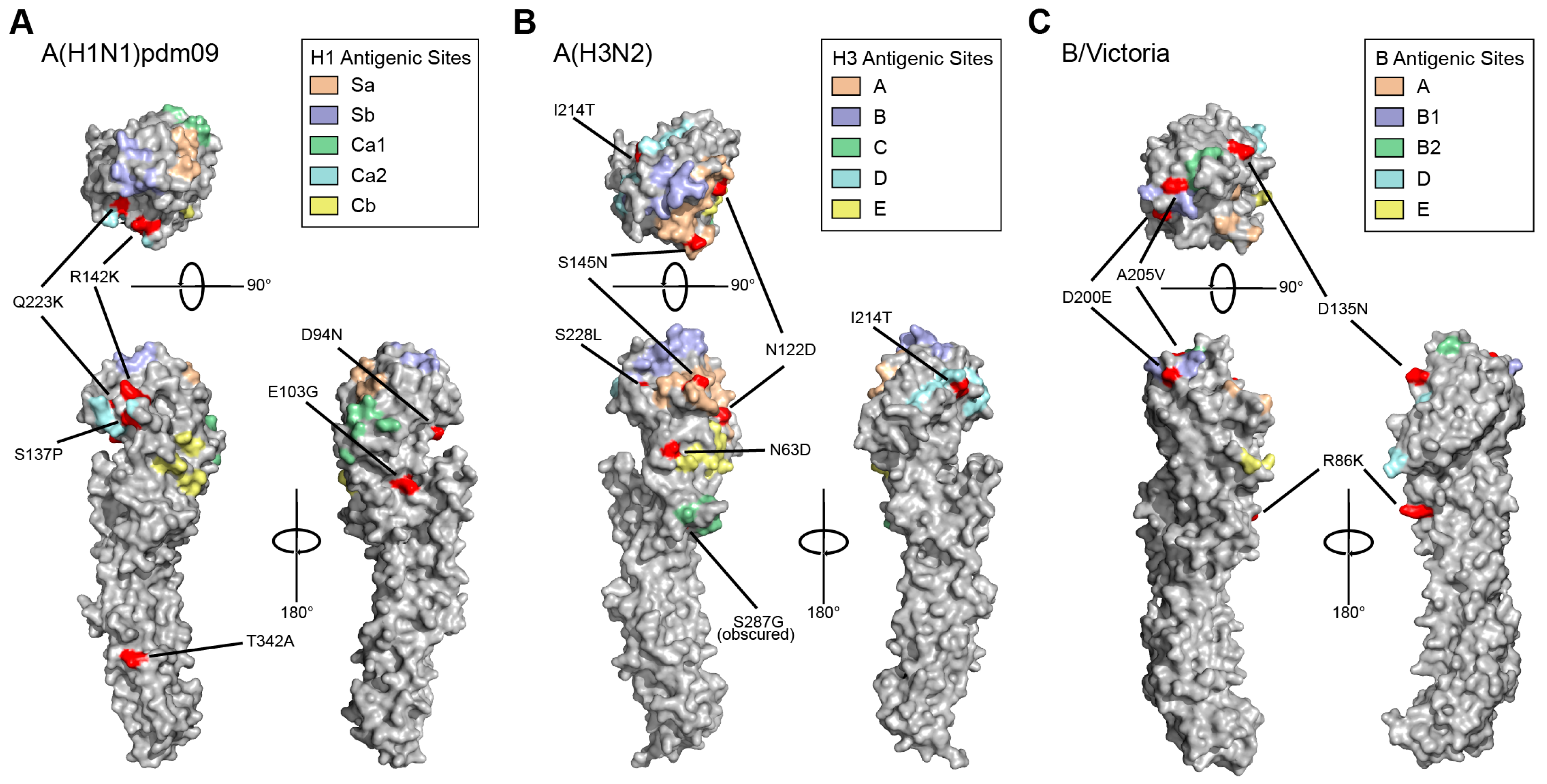
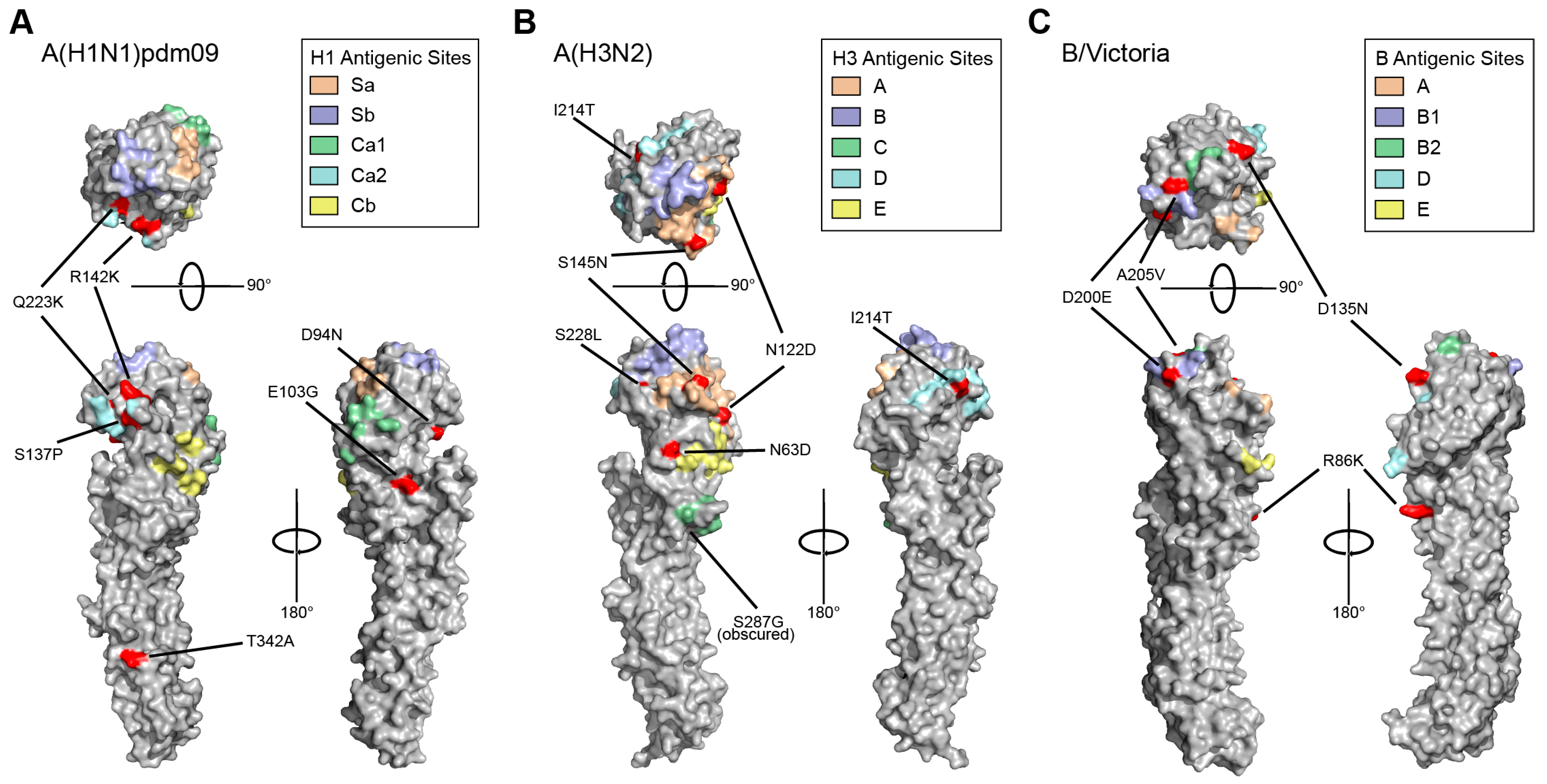
3. Results
3.1. Genomic Surveillance 2023–2024
3.2. Phylogenetic Analysis of Influenza Viruses
3.3. HA and NA Mutations Identified in Circulating 2023–2024 Influenza Viruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Collaborators, G.B.D.I. Mortality, morbidity, and hospitalisations due to influenza lower respiratory tract infections, 2017: An analysis for the Global Burden of Disease Study 2017. Lancet Respir. Med. 2019, 7, 69–89. [Google Scholar] [CrossRef]
- Garjani, A.; Chegini, A.M.; Salehi, M.; Tabibzadeh, A.; Yousefi, P.; Razizadeh, M.H.; Esghaei, M.; Esghaei, M.; Rohban, M.H. Forecasting influenza hemagglutinin mutations through the lens of anomaly detection. Sci. Rep. 2023, 13, 14944. [Google Scholar] [CrossRef] [PubMed]
- Han, A.X.; de Jong, S.P.J.; Russell, C.A. Co-evolution of immunity and seasonal influenza viruses. Nat. Rev. Microbiol. 2023, 21, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Huddleston, J.; Barnes, J.R.; Rowe, T.; Xu, X.; Kondor, R.; Wentworth, D.E.; Whittaker, L.; Ermetal, B.; Daniels, R.S.; McCauley, J.W.; et al. Integrating genotypes and phenotypes improves long-term forecasts of seasonal influenza A/H3N2 evolution. Elife 2020, 9, e60067. [Google Scholar] [CrossRef] [PubMed]
- Rolfes, M.A.; Foppa, I.M.; Garg, S.; Flannery, B.; Brammer, L.; Singleton, J.A.; Burns, E.; Jernigan, D.; Olsen, S.J.; Bresee, J.; et al. Annual estimates of the burden of seasonal influenza in the United States: A tool for strengthening influenza surveillance and preparedness. Influenza Other Respir. Viruses 2018, 12, 132–137. [Google Scholar] [CrossRef] [PubMed]
- WHO. Global Influenza Surveillance and Response System (GISRS); World Health Organization: Geneva, Switzerland, 2024.
- Mitchell, P.K.; Cronk, B.D.; Voorhees, I.E.H.; Rothenheber, D.; Anderson, R.R.; Chan, T.H.; Wasik, B.R.; Dubovi, E.J.; Parrish, C.R.; Goodman, L.B. Method comparison of targeted influenza A virus typing and whole-genome sequencing from respiratory specimens of companion animals. J. Vet. Diagn. Investig. 2021, 33, 191–201. [Google Scholar] [CrossRef]
- CDC. Influenza Virus Genome Sequencing and Genetic Characterization; Center for Disease Control and Prevention: Atlanta, GA, USA, 2024.
- Smith, M.F.; Holland, S.C.; Lee, M.B.; Hu, J.C.; Pham, N.C.; Sullins, R.A.; Holland, L.A.; Mu, T.; Thomas, A.W.; Fitch, R.; et al. Baseline Sequencing Surveillance of Public Clinical Testing, Hospitals, and Community Wastewater Reveals Rapid Emergence of SARS-CoV-2 Omicron Variant of Concern in Arizona, USA. mBio 2023, 14, e0310122. [Google Scholar] [CrossRef]
- CDC. Influenza Activity in the United States during the 2022–23 Season and Composition of the 2023–24 Influenza Vaccine. Available online: https://www.cdc.gov/flu/spotlights/2023-2024/22-23-summary-technical-report.htm (accessed on 23 February 2024).
- Zhou, B.; Wentworth, D.E. Influenza A virus molecular virology techniques. Methods Mol. Biol. 2012, 865, 175–192. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F. Trim Galore. Available online: https://github.com/FelixKrueger/TrimGalore (accessed on 23 February 2024).
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Burke, D.F.; Smith, D.J. A recommended numbering scheme for influenza A HA subtypes. PLoS ONE 2014, 9, e112302. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- A*STAR Bioinformatics Institute (BII). FluSurver Tool. 2024. Available online: https://flusurver.bii.a-star.edu.sg/ (accessed on 23 February 2024).
- Stray, S.J.; Pittman, L.B. Subtype- and antigenic site-specific differences in biophysical influences on evolution of influenza virus hemagglutinin. Virol. J. 2012, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Holland, L.A.; Holland, S.C.; Smith, M.F.; Leonard, V.R.; Murugan, V.; Nordstrom, L.; Mulrow, M.; Salgado, R.; White, M.; Lim, E.S. Genomic Sequencing Surveillance to Identify Respiratory Syncytial Virus Mutations, Arizona, USA. Emerg. Infect. Dis. 2023, 29, 2380–2382. [Google Scholar] [CrossRef] [PubMed]
- Grohskopf, L.A.; Blanton, L.H.; Ferdinands, J.M.; Chung, J.R.; Broder, K.R.; Talbot, H.K.; Morgan, R.L.; Fry, A.M. Prevention and Control of Seasonal Influenza with Vaccines: Recommendations of the Advisory Committee on Immunization Practices—United States, 2022–23 Influenza Season. MMWR Recomm. Rep. 2022, 71, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.G.; Pierse, N.; Sue Huang, Q.; Prasad, N.; Duque, J.; Claire Newbern, E.; Baker, M.G.; Turner, N.; McArthur, C.; On behalf of SHIVERS Investigation Team. Influenza vaccine effectiveness in preventing influenza-associated intensive care admissions and attenuating severe disease among adults in New Zealand 2012–2015. Vaccine 2018, 36, 5916–5925. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Symptoms | Yes | No | Do Not Know/Unknown |
|---|---|---|---|
| Fever/feverishness | 84.3% | 11.4% | 4.3% |
| Chills | 77.1% | 21.4% | 1.4% |
| Sore throat | 77.1% | 21.4% | 1.4% |
| Difficulty breathing/shortness of breath | 60.0% | 40.0% | 0.0% |
| Runny nose or nasal congestion | 88.6% | 11.4% | 0.0% |
| Decreased/complete loss of sense of taste or smell | 22.1% | 75.0% | 2.9% |
| Headache | 80.0% | 20.0% | 0.0% |
| Wheezing | 37.9% | 60.7% | 1.4% |
| Nausea/vomiting | 47.9% | 51.4% | 0.7% |
| Diarrhea | 20.0% | 79.3% | 0.7% |
| Muscle pain or body aches | 77.1% | 22.1% | 0.7% |
| Fatigue | 89.3% | 10.0% | 0.7% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maqsood, R.; Smith, M.F.; Holland, L.A.; Sullins, R.A.; Holland, S.C.; Tan, M.; Hernandez Barrera, G.M.; Thomas, A.W.; Islas, M.; Kramer, J.L.; et al. Influenza Virus Genomic Surveillance, Arizona, USA, 2023–2024. Viruses 2024, 16, 692. https://doi.org/10.3390/v16050692
Maqsood R, Smith MF, Holland LA, Sullins RA, Holland SC, Tan M, Hernandez Barrera GM, Thomas AW, Islas M, Kramer JL, et al. Influenza Virus Genomic Surveillance, Arizona, USA, 2023–2024. Viruses. 2024; 16(5):692. https://doi.org/10.3390/v16050692
Chicago/Turabian StyleMaqsood, Rabia, Matthew F. Smith, LaRinda A. Holland, Regan A. Sullins, Steven C. Holland, Michelle Tan, Gabrielle M. Hernandez Barrera, Alexis W. Thomas, Mario Islas, Joanna L. Kramer, and et al. 2024. "Influenza Virus Genomic Surveillance, Arizona, USA, 2023–2024" Viruses 16, no. 5: 692. https://doi.org/10.3390/v16050692
APA StyleMaqsood, R., Smith, M. F., Holland, L. A., Sullins, R. A., Holland, S. C., Tan, M., Hernandez Barrera, G. M., Thomas, A. W., Islas, M., Kramer, J. L., Nordstrom, L., Mulrow, M., White, M., Murugan, V., & Lim, E. S. (2024). Influenza Virus Genomic Surveillance, Arizona, USA, 2023–2024. Viruses, 16(5), 692. https://doi.org/10.3390/v16050692