
Whole-Genome Sequencing and Genetic Diversity of Human Respiratory Syncytial Virus in Patients with Influenza-like Illness in Sicily (Italy) from 2017 to 2023
 , , , , ,
, , , , ,  , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Case Definition
2.2. Phylogenetic Analysis
2.3. Analysis of Deduced Amino Acid Sequences and Mutations
2.4. Selection Pressure Prediction
2.5. Statistical Analysis
2.6. Ethical Review
3. Results
3.1. Demographic Characteristics of Study Population
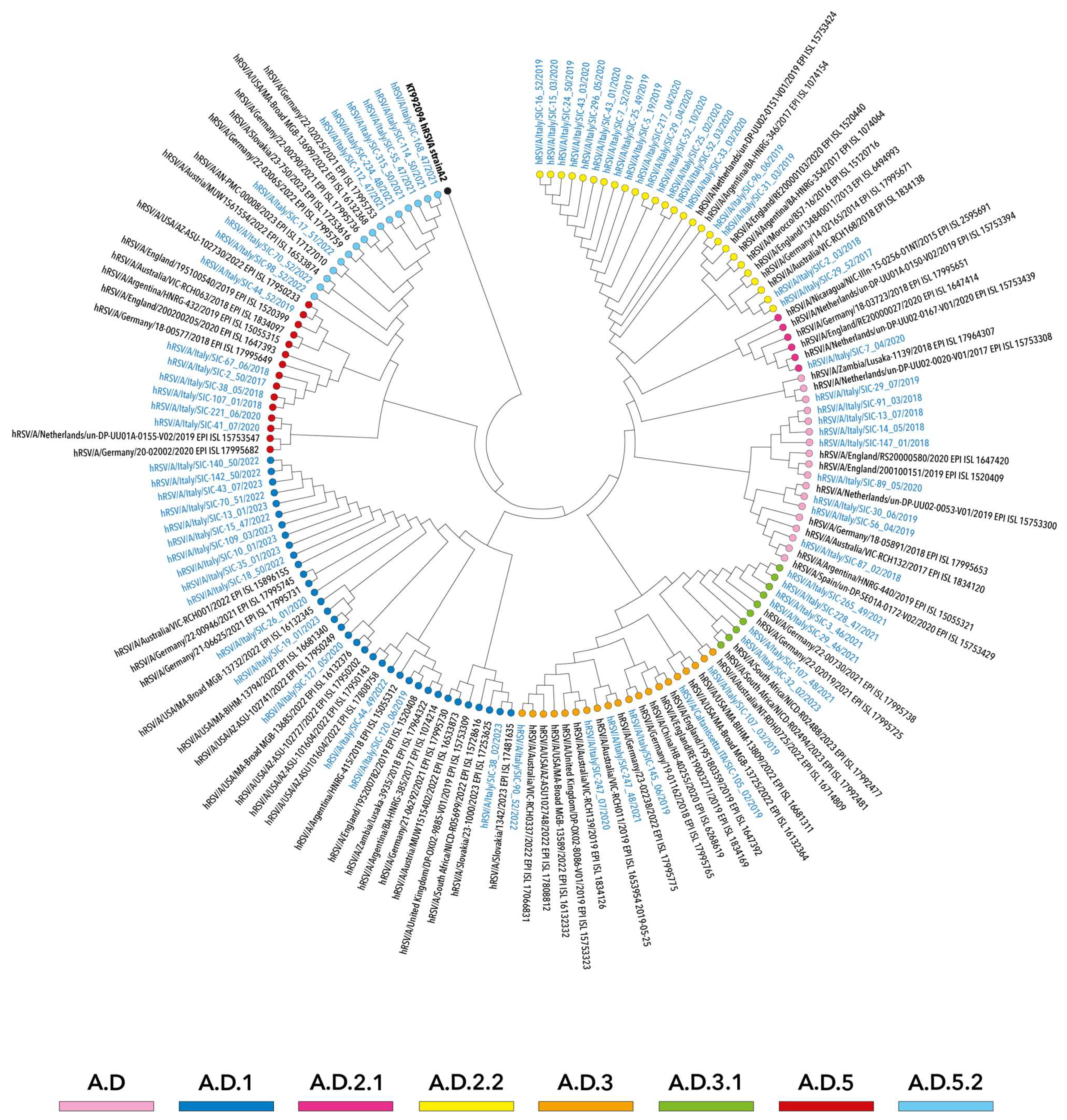
3.2. Phylogenetic Analysis of hRSV Genomes
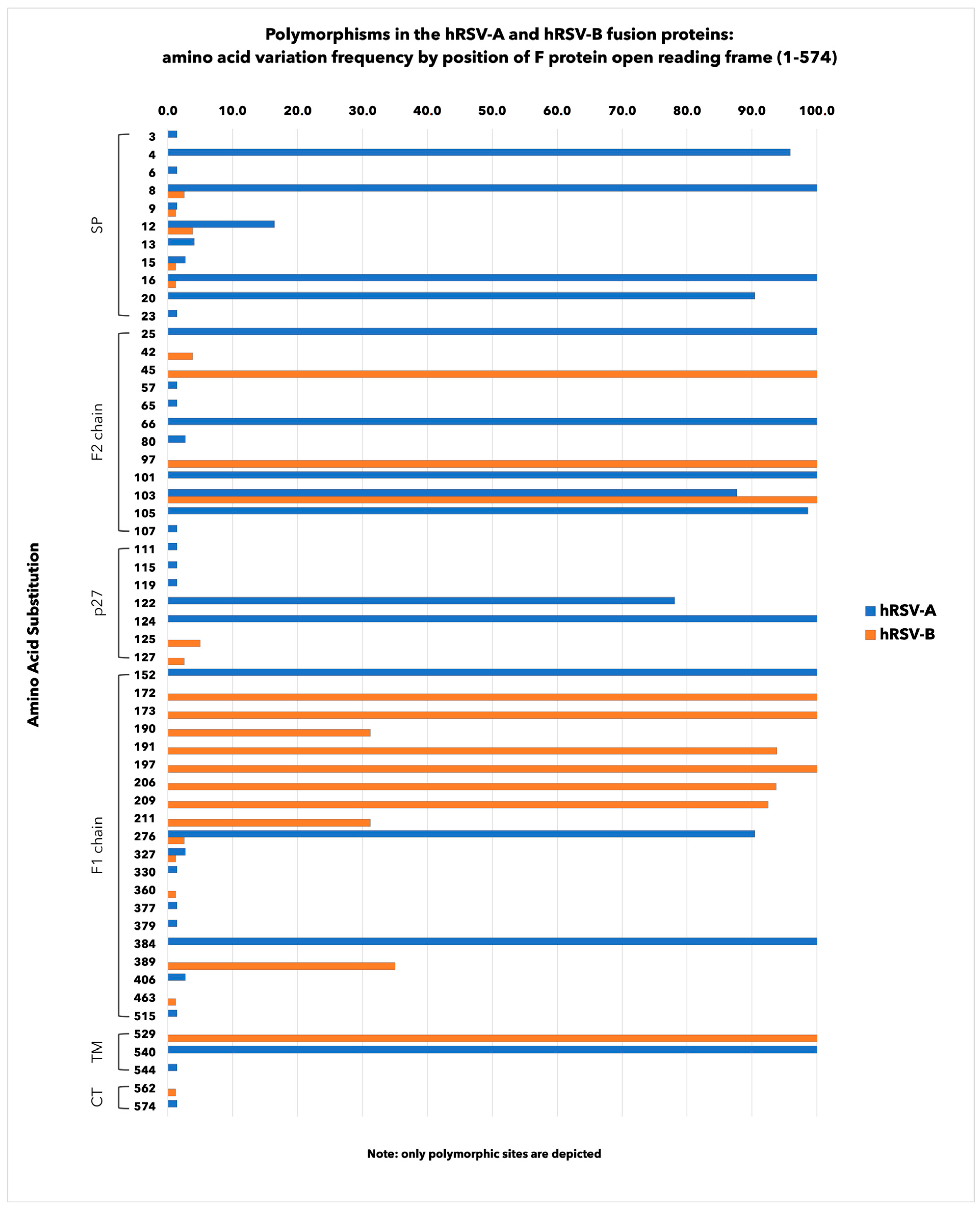
3.3. Amino Acid Polymorphisms in hRSV F Surface Proteins
3.4. Determination of the Selection Pressure on hRSV F Gene
3.5. Analysis of NS1, NS2, N, P, SH, M2-1, M2-2 and L Proteins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Y.; Wang, X.; Blau, D.M.; Caballero, M.T.; Feikin, D.R.; Gill, C.J.; Madhi, S.A.; Omer, S.B.; Simões, E.A.F.; Campbell, H.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in children younger than 5 years in 2019: A systematic analysis. Lancet 2022, 399, 2047–2064. [Google Scholar] [CrossRef] [PubMed]
- Díez-Gandía, E.; Gómez-Álvarez, C.; López-Lacort, M.; Muñoz-Quiles, C.; Úbeda-Sansano, I.; Díez-Domingo, J.; Orrico-Sánchez, A.; Study Collaborators. The impact of childhood RSV infection on children’s and parents’ quality of life: A prospective multicenter study in Spain. BMC Infect. Dis. 2021, 21, 924. [Google Scholar] [CrossRef] [PubMed]
- Tramuto, F.; Maida, C.M.; Di Naro, D.; Randazzo, G.; Vitale, F.; Restivo, V.; Costantino, C.; Amodio, E.; Casuccio, A.; Graziano, G.; et al. Respiratory Syncytial Virus: New Challenges for Molecular Epidemiology Surveillance and Vaccination Strategy in Patients with ILI/SARI. Vaccines 2021, 9, 1334. [Google Scholar] [CrossRef]
- Hao, Y.; Cheng, L.; Lu, D. Epidemiological Study of Respiratory Virus Infections among Hospitalized Children Aged 14 Years and Younger during COVID-19 Pandemic in Wuhan, China, 2018–2022. J. Glob. Infect. Dis. 2023, 15, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Van-Tam, J.S.; O’Leary, M.; Martin, E.T.; Heijnen, E.; Callendret, B.; Fleischhackl, R.; Comeaux, C.; Tran, T.M.P.; Weber, K. Burden of respiratory syncytial virus infection in older and high-risk adults: A systematic review and meta-analysis of the evidence from developed countries. Eur. Respir. Rev. 2022, 31, 220105. [Google Scholar] [CrossRef] [PubMed]
- Weycker, D.; Averin, A.; Houde, L.; Ottino, K.; Shea, K.; Sato, R.; Gessner, B.D.; Yacisin, K.; Curcio, D.; Begier, E.; et al. Rates of Lower Respiratory Tract Illness in US Adults by Age and Comorbidity Profile. Infect. Dis. Ther. 2024, 13, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Savic, M.; Penders, Y.; Shi, T.; Branche, A.; Pirçon, J.Y. Respiratory syncytial virus disease burden in adults aged 60 years and older in high-income countries: A systematic literature review and meta-analysis. Influenza Other Respir. Viruses 2023, 17, e13031. [Google Scholar] [CrossRef] [PubMed]
- Correa, R.A.; Arancibia, F.; De Ávila Kfouri, R.; Chebabo, A.; García, G.; Gutiérrez Robledo, L.M.; Lopardo, G.; Nemerovsky, J.; Pérez, C.M.; Rendon, A.; et al. Understanding the Burden of Respiratory Syncytial Virus in Older Adults in Latin America: An Expert Perspective on Knowledge Gaps. Pulm. Ther. 2024, 10, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Avadhanula, V.; Agustinho, D.P.; Menon, V.K.; Chemaly, R.F.; Shah, D.P.; Qin, X.; Surathu, A.; Doddapaneni, H.; Muzny, D.M.; Metcalf, G.A.; et al. Inter and intra-host diversity of RSV in hematopoietic stem cell transplant adults with normal and delayed viral clearance. Virus Evol. 2023, 10, ead086. [Google Scholar] [CrossRef]
- Mejias, A.; Rodríguez-Fernández, R.; Oliva, S.; Peeples, M.E.; Ramilo, O. The journey to a respiratory syncytial virus vaccine. Ann. Allergy Asthma Immunol. 2020, 125, 36–46. [Google Scholar] [CrossRef]
- Johnson, P.R.; Spriggs, M.K.; Olmsted, R.A.; Collins, P.L. The G glycoprotein of human respiratory syncytial viruses of subgroups A and B: Extensive sequence divergence between antigenically related proteins. Proc. Natl. Acad. Sci. USA 1987, 84, 5625–5629. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ng, T.F.F.; Castro, C.J.; Marine, R.L.; Magaña, L.C.; Esona, M.; Peret, T.C.T.; Thornburg, N.J. Next-generation sequencing of human respiratory syncytial virus subgroups A and B genomes. J. Virol. Methods 2022, 299, 114335. [Google Scholar] [CrossRef] [PubMed]
- Tabor, D.E.; Fernandes, F.; Langedijk, A.C.; Wilkins, D.; Lebbink, R.J.; Tovchigrechko, A.; Ruzin, A.; Kragten-Tabatabaie, L.; Jin, H.; Esser, M.T.; et al. Global Molecular Epidemiology of Respiratory Syncytial Virus from the 2017–2018 INFORM-RSV Study. J. Clin. Microbiol. 2020, 59, e01828-20. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Williams, T.C.; Viboud, C.; Campbell, H.; Chen, J.; Spiro, D.J. RSV genomic diversity and the development of a globally effective RSV intervention. Vaccine 2021, 39, 2811–2820. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Jiang, M.; Wang, F.; Qian, Y.; Song, Q.; Sun, Y.; Zhu, R.; Wang, F.; Qu, D.; Cao, L.; et al. Immune escaping of the novel genotypes of human respiratory syncytial virus based on gene sequence variation. Front. Immunol. 2023, 10, 1084139. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.S.; Modjarrad, K.; McLellan, J.S. Novel antigens for RSV vaccines. Curr. Opin. Immunol. 2015, 35, 30–38. [Google Scholar] [CrossRef]
- Rezende, W.; Ye, X.; Angelo, L.S.; Carisey, A.F.; Avadhanula, V.; Piedra, P.A. The Efficiency of p27 Cleavage during In Vitro Respiratory Syncytial Virus (RSV) Infection Is Cell Line and RSV Subtype Dependent. J. Virol. 2023, 97, e0025423. [Google Scholar] [CrossRef]
- Sun, M.; Lai, H.; Na, F.; Li, S.; Qiu, X.; Tian, J.; Zhang, Z.; Ge, L. Monoclonal Antibody for the Prevention of Respiratory Syncytial Virus in Infants and Children: A Systematic Review and Network Meta-analysis. JAMA Netw. Open. 2023, 6, e230023. [Google Scholar] [CrossRef]
- Ison, M.G.; Papi, A.; Athan, E.; Feldman, R.G.; Langley, J.M.; Lee, D.G.; Leroux-Roels, I.; Martinon-Torres, F.; Schwarz, T.F.; van Zyl-Smit, R.N.; et al. Efficacy and safety of respiratory syncytial virus prefusion F protein vaccine (RSVPreF3 OA) in older adults over 2 RSV seasons. Clin. Infect. Dis. 2024, 22, ciae010. [Google Scholar] [CrossRef]
- Schaerlaekens, S.; Jacobs, L.; Stobbelaar, K.; Cos, P.; Delputte, P. All Eyes on the Prefusion-Stabilized F Construct, but Are We Missing the Potential of Alternative Targets for Respiratory Syncytial Virus Vaccine Design? Vaccines 2024, 12, 97. [Google Scholar] [CrossRef]
- European Medicine Agency (EMA). Abrysvo-Respiratory Syncytial Virus Vaccine (Bivalent, Recombinant); EMA: Amsterdam, The Netherlands; Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/abrysvo (accessed on 15 April 2024).
- European Medicine Agency (EMA). Arexvy-Recombinant Respiratory Syncytial Virus Pre-Fusion F Protein, Adjuvanted with AS01E; EMA: Amsterdam, The Netherlands; Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/arexvy (accessed on 15 April 2024).
- Wilson, E.; Goswami, J.; Baqui, A.H.; Doreski, P.A.; Perez-Marc, G.; Zaman, K.; Monroy, J.; Duncan, C.J.A.; Ujiie, M.; Rämet, M.; et al. Efficacy and Safety of an mRNA-Based RSV PreF Vaccine in Older Adults. N. Engl. J. Med. 2023, 389, 2233–2244. [Google Scholar] [CrossRef] [PubMed]
- Schepens, B.; Sedeyn, K.; Vande Ginste, L.; De Baets, S.; Schotsaert, M.; Roose, K.; Houspie, L.; Van Ranst, M.; Gilbert, B.; van Rooijen, N.; et al. Protection and mechanism of action of a novel human respiratory syncytial virus vaccine candidate based on the extracellular domain of small hydrophobic protein. EMBO Mol. Med. 2014, 6, 1436–1454. [Google Scholar] [CrossRef] [PubMed]
- Langley, J.M.; MacDonald, L.D.; Weir, G.M.; MacKinnon-Cameron, D.; Ye, L.; McNeil, S.; Schepens, B.; Saelens, X.; Stanford, M.M.; Halperin, S.A. A Respiratory Syncytial Virus Vaccine Based on the Small Hydrophobic Protein Ectodomain Presented With a Novel Lipid-Based Formulation Is Highly Immunogenic and Safe in Adults: A First-in-Humans Study. J. Infect. Dis. 2018, 218, 378–387. [Google Scholar] [CrossRef]
- Samy, N.; Reichhardt, D.; Schmidt, D.; Chen, L.M.; Silbernagl, G.; Vidojkovic, S.; Meyer, T.P.; Jordan, E.; Adams, T.; Weidenthaler, H.; et al. Safety and immunogenicity of novel modified vaccinia Ankara-vectored RSV vaccine: A randomized phase I clinical trial. Vaccine 2020, 38, 2608–2619. [Google Scholar] [CrossRef] [PubMed]
- Endt, K.; Wollmann, Y.; Haug, J.; Bernig, C.; Feigl, M.; Heiseke, A.; Kalla, M.; Hochrein, H.; Suter, M.; Chaplin, P.; et al. A recombinant. MVA-based RSV vaccine induces T-cell and antibody responses that cooperate in the protection against RSV infection. Front. Immunol. 2022, 13, 841471. [Google Scholar] [CrossRef] [PubMed]
- Díez-Domingo, J.; Sáez-Llorens, X.; Rodriguez-Weber, M.A.; Epalza, C.; Chatterjee, A.; Chiu, C.H.; Lin, C.Y.; Berry, A.A.; Martinón-Torres, F.; Baquero-Artigao, F.; et al. Safety and Immunogenicity of a ChAd155-Vectored Respiratory Syncytial Virus (RSV) Vaccine in Healthy RSV-Seropositive Children 12–23 Months of Age. J. Infect. Dis. 2023, 227, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Llorens, X.; Norero, X.; Mussi-Pinhata, M.M.; Luciani, K.; de la Cueva, I.S.; Díez-Domingo, J.; Lopez-Medina, E.; Epalza, C.; Brzostek, J.; Szymański, H.; et al. Safety and immunogenicity of a ChAd155-vectored respiratory syncytial virus vaccine in infants 6–7 months of age: A phase 1/2 randomized trial. J. Infect. Dis. 2024, 229, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Blunck, B.N.; Aideyan, L.; Ye, X.; Avadhanula, V.; Ferlic-Stark, L.; Zechiedrich, L.; Gilbert, B.E.; Piedra, P.A. Antibody responses of healthy adults to the p27 peptide of respiratory syncytial virus fusion protein. Vaccine 2022, 40, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, Y.; Klenow, L.; Coyle, E.M.; Tang, J.; Ravichandran, S.; Golding, H.; Khurana, S. Protective antigenic sites identified in respiratory syncytial virus fusion protein reveals importance of p27 domain. EMBO Mol. Med. 2022, 14, e13847. [Google Scholar] [CrossRef]
- Mas, V.; Nair, H.; Campbell, H.; Melero, J.A.; Williams, T.C. Antigenic and sequence variability of the human respiratory syncytial virus F glycoprotein compared to related viruses in a comprehensive dataset. Vaccine 2018, 36, 6660–6673. [Google Scholar] [CrossRef]
- Hause, A.M.; Henke, D.M.; Avadhanula, V.; Shaw, C.A.; Tapia, L.I.; Piedra, P.A. Sequence variability of the respiratory syncytial virus (RSV) fusion gene among contemporary and historical genotypes of RSV/A and RSV/B. PLoS ONE 2017, 12, e0175792. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lu, B.; Tabor, D.E.; Tovchigrechko, A.; Wilkins, D.; Jin, H.; Madhi, S.A.; Soofie, N.; Esser, M.T.; Nunes, M.C. Characterization of human respiratory syncytial virus (RSV) isolated from HIV-exposed-uninfected and HIV-unexposed infants in South Africa during 2015–2017. Influenza Other Respir. Viruses 2020, 14, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Simões, E.A.F.; Forleo-Neto, E.; Geba, G.P.; Kamal, M.; Yang, F.; Cicirello, H.; Houghton, M.R.; Rideman, R.; Zhao, Q.; Benvin, S.L.; et al. Suptavumab for the Prevention of Medically Attended Respiratory Syncytial Virus Infection in Preterm Infants. Clin. Infect. Dis. 2021, 73, e4400–e4408. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Lu, B.; McTamney, P.; Palaszynski, S.; Diallo, S.; Ren, K.; Ulbrandt, N.D.; Kallewaard, N.; Wang, W.; Fernandes, F.; et al. Prevalence and Significance of Substitutions in the Fusion Protein of Respiratory Syncytial Virus Resulting in Neutralization Escape From Antibody MEDI8897. J. Infect. Dis. 2018, 218, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Li, Y.; Song, Q.; Wang, Z.; Li, M.; Zhang, Q.; Wang, Y.; Ye, W.; Li, Y. Mechanism of Cross-Resistance to Fusion Inhibitors Conferred by the K394R Mutation in Respiratory Syncytial Virus Fusion Protein. J. Virol. 2021, 95, e0120521. [Google Scholar] [CrossRef] [PubMed]
- Holland, L.A.; Holland, S.C.; Smith, M.F.; Leonard, V.R.; Murugan, V.; Nordstrom, L.; Mulrow, M.; Salgado, R.; White, M.; Lim, E.S. Genomic Sequencing Surveillance to Identify Respiratory Syncytial Virus Mutations, Arizona, USA. Emerg. Infect. Dis. 2023, 29, 2380–2382. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control. Influenza-LIKE ILLNESS DEFINITION. In Commission Implementing Decision (EU) 2018/945 of 22 June 2018. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32018D0945&from=EN#page=24 (accessed on 15 April 2024).
- Ministero della Salute. Direzione Generale della Prevenzione Sanitaria “Monitoraggio Dell’andamento Delle Forme Gravi e Complicate Di Malattia Da Virus Influenzali e/o da SARS-CoV-2 Confermate in Laboratorio, Stagione 2020–2021”—0042940-30/12/2020-DGPRE-DGPRE-P. Available online: https://www.trovanorme.salute.gov.it/norme/renderNormsanPdf?anno=2020&codLeg=78081&parte=1%20&serie=null (accessed on 15 April 2024).
- Istituto Superiore di Sanità. RespiVirNet, Sistema di Sorveglianza Integrata dei Virus Respiratori. Protocollo Operativo, Stagione 2023–2024. Available online: https://respivirnet.iss.it/pagine/Documenti.aspx (accessed on 15 April 2024).
- Jiang, X.W.; Huang, T.S.; Xie, L.; Chen, S.Z.; Wang, S.D.; Huang, Z.W.; Li, X.Y.; Ling, W.P. Development of a diagnostic assay by three-tube multiplex real-time PCR for simultaneous detection of nine microorganisms causing acute respiratory infections. Sci. Rep. 2022, 12, 13306. [Google Scholar] [CrossRef] [PubMed]
- Goya, S.; Galiano, M.; Nauwelaers, I.; Trento, A.; Openshaw, P.J.; Mistchenko, A.S.; Zambon, M.; Viegas, M. Toward unified molecular surveillance of RSV: A proposal for genotype definition. Influenza Other Respir. Viruses 2020, 14, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular Evolutionary Genetics Analysis (MEGA) for macOS. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef]
- Tramuto, F.; Maida, C.M.; Mazzucco, W.; Costantino, C.; Amodio, E.; Sferlazza, G.; Previti, A.; Immordino, P.; Vitale, F. Molecular Epidemiology and Genetic Diversity of Human Respiratory Syncytial Virus in Sicily during Pre- and Post-COVID-19 Surveillance Seasons. Pathogens 2023, 12, 1099. [Google Scholar] [CrossRef]
- Shi, T.; Denouel, A.; Tietjen, A.K.; Campbell, I.; Moran, E.; Li, X.; Campbell, H.; Demont, C.; Nyawanda, B.O.; Chu, H.Y.; et al. Global Disease Burden Estimates of Respiratory Syncytial Virus-Associated Acute Respiratory Infection in Older Adults in 2015: A Systematic Review and Meta-Analysis. J. Infect. Dis. 2020, 222, S577–S583. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Bergna, A.; Fabiano, V.; Ventura, C.D.; Fumagalli, G.; Mari, A.; Loiodice, M.; Zuccotti, G.V.; Zehender, G. Epidemiology and molecular analyses of respiratory syncytial virus in the 2021–2022 season in northern Italy. Front. Microbiol. 2024, 14, 1327239. [Google Scholar] [CrossRef] [PubMed]
- Rojo-Alba, S.; Martínez, Z.P.; González-Alba, J.M.; Boga, J.A.; Varela, C.O.; Álvarez, M.A.A.; Fonseca, C.P.; Clemente, M.M.G.; Rodriguez, J.G.; García, E.G.; et al. Respiratory syncytial virus incidence and typing in the last six seasons in the north of Spain (Asturias). Genetic characterization during the SARS-CoV-2 pandemic. J. Med. Virol. 2024, 96, e29499. [Google Scholar] [CrossRef] [PubMed]
- Eshaghi, A.; Duvvuri, V.R.; Lai, R.; Nadarajah, J.T.; Li, A.; Patel, S.N.; Low, D.E.; Gubbay, J.B. Genetic variability of human respiratory syncytial virus A strains circulating in Ontario: A novel genotype with a 72 nucleotide G gene duplication. PLoS ONE 2012, 7, e32807. [Google Scholar] [CrossRef] [PubMed]
- Dapat, I.C.; Shobugawa, Y.; Sano, Y.; Saito, R.; Sasaki, A.; Suzuki, Y.; Kumaki, A.; Zaraket, H.; Dapat, C.; Oguma, T.; et al. New genotypes within respiratory syncytial virus group B genotype BA in Niigata, Japan. J. Clin. Microbiol. 2010, 48, 3423–3427. [Google Scholar] [CrossRef] [PubMed]
- Park, P.H.; Huh, J.W.; Yun, H.J.; Lee, H.K.; Yoon, M.H.; Lee, S.; Ko, G. Molecular and clinical characterization of human respiratory syncytial virus in South Korea between 2009 and 2014. Epidemiol. Infect. 2017, 145, 3226–3242. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Wang, H.; Shi, J.; Cui, A.; Huang, Y.; Sun, L.; Xiang, X.; Ma, C.; Yu, P.; Yang, Z.; et al. Emergence of BA9 genotype of human respiratory syncytial virus subgroup B in China from 2006 to 2014. Sci. Rep. 2017, 7, 16765. [Google Scholar] [CrossRef] [PubMed]
- Sáez-López, E.; Cristóvão, P.; Costa, I.; Pechirra, P.; Conde, P.; Guiomar, R.; Portuguese Laboratory Network for the Diagnosis of Influenza Infection; Peres, M.J.; Viseu, R.; Lopes, P.; et al. Epidemiology and genetic variability of respiratory syncytial virus in Portugal, 2014–2018. J. Clin. Virol. 2019, 121, 104200. [Google Scholar] [CrossRef] [PubMed]
- Midulla, F.; Di Mattia, G.; Nenna, R.; Scagnolari, C.; Viscido, A.; Oliveto, G.; Petrarca, L.; Frassanito, A.; Arima, S.; Antonelli, G.; et al. Novel variants of respiratory syncytial virus a ON1 associated with increased clinical severity of bronchiolitis. J. Infect. Dis. 2020, 222, 102–110. [Google Scholar] [CrossRef]
- Korsun, N.; Angelova, S.; Trifonova, I.; Voleva, S.; Grigorova, I.; Tzotcheva, I.; Mileva, S.; Alexiev, I.; Perenovska, P. Predominance of ON1 and BA9 genotypes of respiratory syncytial virus (RSV) in Bulgaria, 2016–2018. J. Med. Virol. 2021, 93, 3401–3411. [Google Scholar] [CrossRef]
- Sun, Y.P.; Lei, S.Y.; Wang, Y.B.; Wang, Y.Z.; Qiang, H.S.; Yin, Y.F.; Jiang, Z.M.; Zhu, M.; Chen, X.L.; Ye, H.M.; et al. Molecular evolution of attachment glycoprotein (G) and fusion protein (F) genes of respiratory syncytial virus ON1 and BA9 strains in Xiamen, China. Microbiol. Spectr. 2022, 10, e0208321. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.; Moreno, G.K.; Petros, B.A.; Uddin, R.; Levine, Z.; Kotzen, B.; Messer, K.S.; Dobbins, S.T.; DeRuff, K.C.; Loreth, C.M.; et al. Viral Lineages in the 2022 RSV Surge in the United States. N. Engl. J. Med. 2023, 388, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Goya, S.; Sereewit, J.; Pfalmer, D.; Nguyen, T.V.; Bakhash, S.A.K.M.; Sobolik, E.B.; Greninger, A.L. Genomic Characterization of Respiratory Syncytial Virus during 2022–2023 Outbreak, Washington, USA. Emerg. Infect. Dis. 2023, 29, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Kasuya, F.; Mori, K.; Harada, S.; Kumagai, R.; Suzuki, A.; Amano, A.; Kosugi, T.; Hasegawa, M.; Nagashima, M.; Suzuki, J.; et al. Molecular and Epidemiological Analysis of Respiratory Syncytial Virus Detected in Tokyo, Japan in 2021 Season. Jpn. J. Infect. Dis. 2023, 76, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Köndgen, S.; Tolksdorf, K.; Dürrwald, R.; Schuler, E.; Biere, B.; Schweiger, B.; Goerlitz, L.; Haas, W.; Wolff, T.; et al. Atypical age distribution and high disease severity in children with RSV infections during two irregular epidemic seasons throughout the COVID-19 pandemic, Germany, 2021 to 2023. Eurosurveillance 2024, 29, 2300465. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, Y.; Yang, Y.; Dong, H.; Luo, Q.; Chen, Z.; Du, H.; Mei, G.; Wang, X.; Guan, Y.; et al. Molecular epidemiology and clinical characteristics of respiratory syncytial virus in hospitalized children during winter 2021–2022 in Bengbu, China. Front. Public Health 2024, 11, 1310293. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Cong, B.; Wei, X.; Wang, Y.; Kang, L.; Gong, C.; Huang, Q.; Wang, X.; Li, Y.; Huang, F. Characterising the changes in RSV epidemiology in Beijing, China during 2015–2023: Results from a prospective, multi-centre, hospital-based surveillance and serology study. Lancet Reg. Health West. Pac. 2024, 45, 101050. [Google Scholar] [CrossRef] [PubMed]
- Rochman, N.D.; Wolf, Y.I.; Koonin, E.V. Molecular adaptations during viral epidemics. EMBO Rep. 2022, 23, e55393. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Bhattacharya, M.; Lee, S.S. A Detailed Overview of Immune Escape, Antibody Escape, Partial Vaccine Escape of SARS-CoV-2 and Their Emerging Variants With Escape Mutations. Front. Immunol. 2022, 13, 801522. [Google Scholar] [CrossRef]
- Mazur, N.I.; Terstappen, J.; Baral, R.; Bardají, A.; Beutels, P.; Buchholz, U.J.; Cohen, C.; Crowe, J.E., Jr.; Cutland, C.L.; Eckert, L.; et al. Respiratory syncytial virus prevention within reach: The vaccine and monoclonal antibody landscape. Lancet Infect. Dis. 2023, 23, e2–e21. [Google Scholar] [CrossRef]
- Adhikari, B.; Hassan, F.; Harrison, C.J.; Dien Bard, J.; Dunn, J.; Kehl, S.; Selvarangan, R. A multi-center study to determine genetic variations in the fusion gene of respiratory syncytial virus (RSV) from children <2 years of age in the U.S. J. Clin. Virol. 2022, 154, 105223. [Google Scholar] [CrossRef]
- Adams, O.; Bonzel, L.; Kovacevic, A.; Mayatepek, E.; Hoehn, T.; Vogel, M. Palivizumab-resistant human respiratory syncytial virus infection in infancy. Clin. Infect. Dis. 2010, 51, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Papenburg, J.; Carbonneau, J.; Hamelin, M.È.; Isabel, S.; Bouhy, X.; Ohoumanne, N.; Déry, P.; Paes, B.A.; Corbeil, J.; Bergeron, M.G.; et al. Molecular evolution of respiratory syncytial virus fusion gene, Canada, 2006–2010. Emerg. Infect. Dis. 2012, 18, 120–124. [Google Scholar] [CrossRef]
- Zhu, Q.; Patel, N.K.; McAuliffe, J.M.; Zhu, W.; Wachter, L.; McCarthy, M.P.; Suzich, J.A. Natural polymorphisms and resistance-associated mutations in the fusion protein of respiratory syncytial virus (RSV): Effects on RSV susceptibility to palivizumab. J. Infect. Dis. 2012, 205, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Crowe, J.E.; Firestone, C.Y.; Crim, R.; Beeler, J.A.; Coelingh, K.L.; Barbas, C.F.; Burton, D.R.; Chanock, R.M.; Murphy, B.R. Monoclonal antibody-resistant mutants selected with a respiratory syncytial virus-neutralizing human antibody fab fragment (Fab 19) define a unique epitope on the fusion (F) glycoprotein. Virology 1998, 252, 373–375. [Google Scholar] [CrossRef]
- Zhu, Q.; McAuliffe, J.M.; Patel, N.K.; Palmer-Hill, F.J.; Yang, C.F.; Liang, B.; Su, L.; Zhu, W.; Wachter, L.; Wilson, S.; et al. Analysis of respiratory syncytial virus preclinical and clinical variants resistant to neutralization by monoclonal antibodies palivizumab and/or motavizumab. J. Infect. Dis. 2011, 203, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, F.P.; Sullender, W.M. Respiratory syncytial virus escape mutant derived in vitro resists palivizumab prophylaxis in cotton rats. Virology 2004, 318, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, E.; Chen, F.P.; Sullender, W.M. In vitro and in vivo fitness of respiratory syncytial virus monoclonal antibody escape mutants. J. Virol. 2006, 80, 11651–11657. [Google Scholar] [CrossRef]
- Fuentes, S.; Coyle, E.M.; Beeler, J.; Golding, H.; Khurana, S. Antigenic Fingerprinting following Primary RSV Infection in Young Children Identifies Novel Antigenic Sites and Reveals Unlinked Evolution of Human Antibody Repertoires to Fusion and Attachment Glycoproteins. PLoS Pathog. 2016, 12, e1005554. [Google Scholar] [CrossRef]
- Bin, L.; Liu, H.; Tabor, D.E.; Tovchigrechko, A.; Qi, Y.; Ruzin, A.; Esser, M.T.; Jin, H. Emergence of new antigenic epitopes in the glycoproteins of human respiratory syncytial virus collected from a US surveillance study, 2015–2017. Sci. Rep. 2019, 9, 3898. [Google Scholar] [CrossRef]
- Song, J.; Wang, H.; Ng, T.I.; Cui, A.; Zhu, S.; Huang, Y.; Sun, L.; Yang, Z.; Yu, D.; Yu, P.; et al. Sequence Analysis of the Fusion Protein Gene of Human Respiratory Syncytial Virus Circulating in China from 2003 to 2014. Sci. Rep. 2018, 8, 17618. [Google Scholar] [CrossRef] [PubMed]
- Shishir, T.A.; Saha, O.; Rajia, S.; Mondol, S.M.; Masum, M.H.U.; Rahaman, M.M.; Hossen, F.; Bahadur, N.M.; Ahmed, F.; Naser, I.B.; et al. Genome-wide study of globally distributed respiratory syncytial virus (RSV) strains implicates diversification utilizing phylodynamics and mutational analysis. Sci. Rep. 2023, 13, 13531. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wang, D.; Li, Y.; Wu, Z.; Liu, H.; Shi, Y.; Lu, X.; Liu, D. Prevalence, variation, and transmission patterns of human respiratory syncytial virus from pediatric patients in Hubei, China during 2020–2021. Virol. Sin. 2023, 38, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Ivancic-Jelecki, J.; Slovic, A.; Ljubin-Sternak, S.; Mlinarić Galinović, G.; Forcic, D. Variability analysis and inter-genotype comparison of human respiratory syncytial virus small hydrophobic gene. Virol. J. 2018, 15, 109. [Google Scholar] [CrossRef]
- Do, L.A.H.; Wilm, A.; van Doorn, H.R.; Lam, H.M.; Sim, S.; Sukumaran, R.; Tran, A.T.; Nguyen, B.H.; Tran, T.T.L.; Tran, Q.H.; et al. Direct whole-genome deep-sequencing of human respiratory syncytial virus A and B from Vietnamese children identifies distinct patterns of inter- and intra-host evolution. J. Gen. Virol. 2015, 96, 3470–3483. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographic Characteristic | Total | hRSV Positive | |||
|---|---|---|---|---|---|
| Overall | hRSV-A | hRSV-B | hRSV-A + B | ||
| Study population [n (%)] | 13,193 | 770 (5.8) | 355 (46.1) | 401 (52.1) | 14 (1.8) |
| Age group [years; n (%)] | |||||
| ≤11 months | 356 | 81 (22.7) | 36 (44.4) | 43 (53.1) | 2 (2.5) |
| 12–23 months | 588 | 135 (23.0) | 65 (48.1) | 66 (48.9) | 4 (3.0) |
| 2–4 | 1740 | 306 (17.6) | 157 (51.3) | 147 (48.0) | 2 (0.7) |
| 5–10 | 2000 | 108 (5.4) | 50 (46.3) | 52 (48.1) | 6 (5.6) |
| 11–18 | 954 | 22 (2.3) | 11 (50.0) | 11 (50.0) | 0 |
| 19–34 | 1012 | 20 (2.0) | 10 (50.0) | 10 (50.0) | 0 |
| 35–49 | 1518 | 11 (0.7) | 4 (36.4) | 7 (63.6) | 0 |
| 50–64 | 2147 | 38 (1.8) | 8 (21.0) | 30 (79.0) | 0 |
| ≥65 | 2878 | 49 (1.7) | 14 (28.6) | 35 (71.4) | 0 |
| Sex [n (%)] | |||||
| Female | 6576 | 381 (5.8) | 172 (45.1) | 203 (53.3) | 6 (1.6) |
| Male | 6617 | 389 (5.9) | 183 (47.0) | 198 (50.9) | 8 (2.1) |
| Healthcare setting [n (%)] | |||||
| Community based | 7313 | 713 (9.8) | 340 (47.7) | 359 (50.3) | 14 (2.0) |
| Hospital based | 5880 | 57 (1.0) | 15 (26.3) | 42 (73.7) | 0 |
| Surveillance season [n (%)] | |||||
| 2017–2018 | 2049 | 121 (5.9) | 53 (43.8) | 68 (56.2) | 0 |
| 2018–2019 | 2209 | 170 (7.7) | 36 (21.2) | 131 (77.1) | 3 (1.7) |
| 2019–2020 | 2320 | 216 (9.3) | 178 (82.4) | 35 (16.2) | 3 (1.4) |
| 2020–2021 | 4 | 1 (25.0) | 0 | 1 (100.0) | 0 |
| 2021–2022 | 5284 | 107 (2.0) | 30 (28.0) | 75 (70.1) | 2 (1.9) |
| 2022–2023 | 1327 | 155 (11.7) | 58 (37.4) | 91 (58.7) | 6 (3.9) |
| Total | hRSV-A | hRSV-B | |
|---|---|---|---|
| hRSV whole-genome sequences [n (%)] | 153 | 73 (47.7) | 80 (52.3) |
| Surveillance season | |||
| 2017–2018 | 22 | 11 | 11 |
| 2018–2019 | 28 | 9 | 19 |
| 2019–2020 | 32 | 23 | 9 |
| 2020–2021 | 1 | 0 | 1 |
| 2021–2022 | 31 | 12 | 19 |
| 2022–2023 | 39 | 18 | 21 |
| hRSV-A | hRSV-B | ||||
|---|---|---|---|---|---|
| Neutralizing Epitope | Position | AA Substitution | % | AA Substitution | % |
| Ø | 62–69 196–209 | K65R | 1.4 | S197N | 100.0 |
| K66E | 100.0 | I206M | 93.7 | ||
| Q209R | 92.5 | ||||
| I | 27–45 312–318 380–400 | V384I | 100.0 | R42K | 3.8 |
| F45L | 100.0 | ||||
| S389F/P | 35.0 | ||||
| II | 254–277 | N276S | 90.4 | S276N | 2.5 |
| III | 46–54 305–310 | ||||
| IV | 422–471 | E463D | 1.2 | ||
| V | 55–61 146–194 287–300 | I57V | 1.4 | S190N | 31.2 |
| V152I | 100.0 | K191R | 93.8 | ||
| Internal glycopeptide p27 | 100–136 | L111I | 1.4 | L125P/R | 5.0 |
| M115I | 1.4 | V127A | 2.5 | ||
| N119I | 1.4 | ||||
| A122T | 78.1 | ||||
| K124N | 100.0 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tramuto, F.; Maida, C.M.; Randazzo, G.; Guzzetta, V.; Santino, A.; Li Muli, R.; Costantino, C.; Graziano, G.; Amodio, E.; Mazzucco, W.; et al. Whole-Genome Sequencing and Genetic Diversity of Human Respiratory Syncytial Virus in Patients with Influenza-like Illness in Sicily (Italy) from 2017 to 2023. Viruses 2024, 16, 851. https://doi.org/10.3390/v16060851
Tramuto F, Maida CM, Randazzo G, Guzzetta V, Santino A, Li Muli R, Costantino C, Graziano G, Amodio E, Mazzucco W, et al. Whole-Genome Sequencing and Genetic Diversity of Human Respiratory Syncytial Virus in Patients with Influenza-like Illness in Sicily (Italy) from 2017 to 2023. Viruses. 2024; 16(6):851. https://doi.org/10.3390/v16060851
Chicago/Turabian StyleTramuto, Fabio, Carmelo Massimo Maida, Giulia Randazzo, Valeria Guzzetta, Arianna Santino, Rita Li Muli, Claudio Costantino, Giorgio Graziano, Emanuele Amodio, Walter Mazzucco, and et al. 2024. "Whole-Genome Sequencing and Genetic Diversity of Human Respiratory Syncytial Virus in Patients with Influenza-like Illness in Sicily (Italy) from 2017 to 2023" Viruses 16, no. 6: 851. https://doi.org/10.3390/v16060851