
Detection and Genetic Characterization of Bovine Torovirus in Uruguay
 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Viral RNA Extraction, Reverse Transcription, and Bovine Torovirus Screening by qPCR
2.3. Conventional PCR and Sequencing
2.4. Characterization of Three BToV Cases
2.5. Complete Genome
2.6. Sequence Analyses
2.7. Statistical Analysis
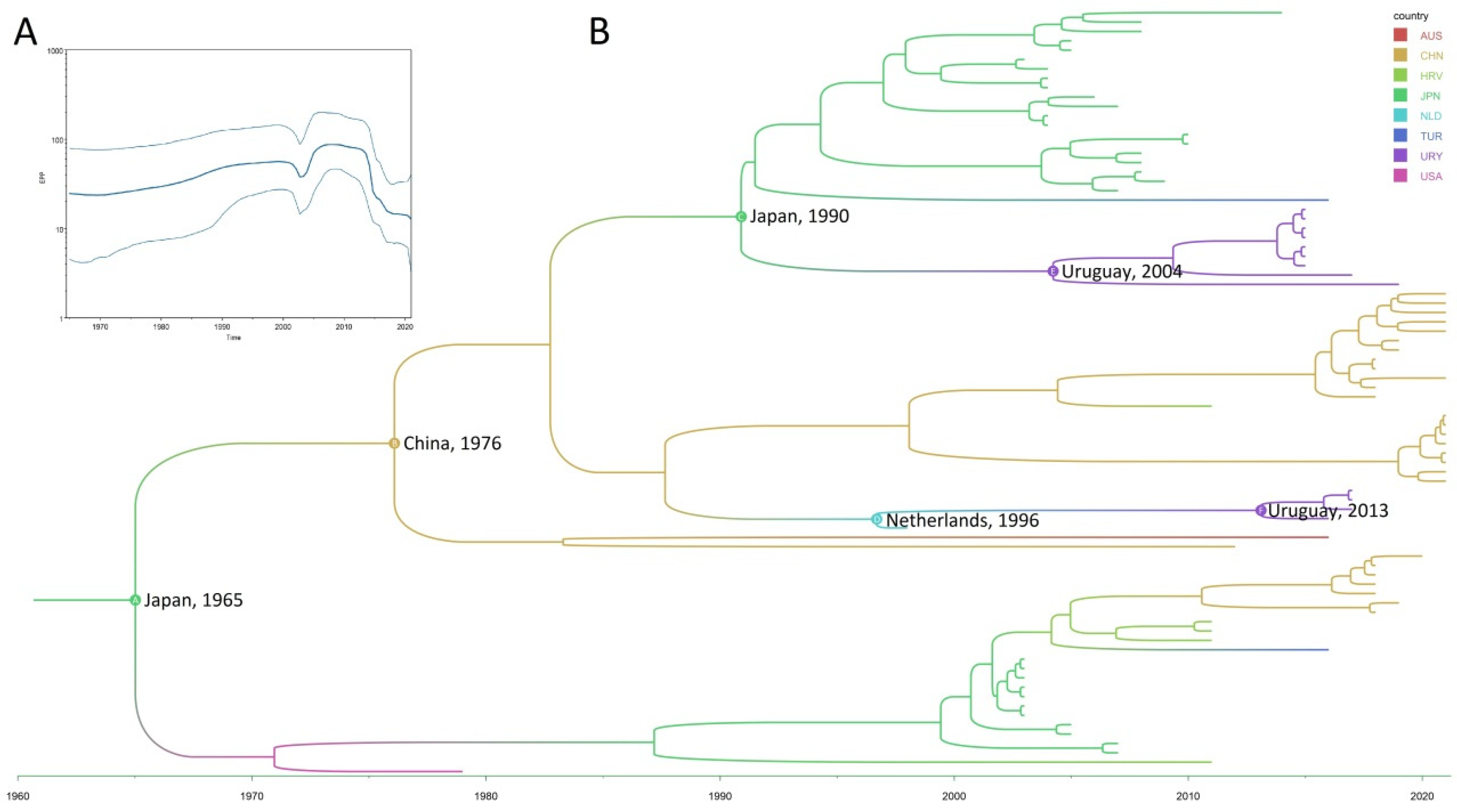
3. Results and Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woode, G.N.; Reed, D.E.; Runnels, P.L.; Herrig, M.A.; Hill, H.T. Studies with an unclassified virus isolated from diarrheic calves. Vet. Microbiol. 1982, 7, 221–240. [Google Scholar] [CrossRef] [PubMed]
- Horzinek, M.C.; Flewett, T.H.; Saif, L.J.; Spaan, W.J.; Weiss, M.; Woode, G.N. A new family of vertebrate viruses: Toroviridae. Intervirology 1987, 27, 17–24. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses (ICTV). Virus Taxonomy: 2022 Release (MSL #38). 2022. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 11 March 2024).
- Draker, R.; Roper, R.L.; Petric, M.; Tellier, R. The complete sequence of the bovine torovirus genome. Virus Res. 2006, 115, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Hoet, A.E.; Saif, L.J. Bovine torovirus (Breda virus) revisited. Anim. Health Res. Rev. 2004, 5, 157–171. [Google Scholar] [CrossRef]
- Aita, T.; Kuwabara, M.; Murayama, K.; Sasagawa, Y.; Yabe, S.; Higuchi, R.; Tamura, T.; Miyazaki, A.; Tsunemitsu, H. Characterization of epidemic diarrhea outbreaks associated with bovine torovirus in adult cows. Arch. Virol. 2012, 157, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Hoet, A.E.; Cho, K.O.; Chang, K.O.; Loerch, S.C.; Wittum, T.E.; Saif, L.J. Enteric and nasal shedding of bovine torovirus (Breda virus) in feedlot cattle. Am. J. Vet. Res. 2002, 63, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Holmes, K.V. Enteric infections with coronaviruses and toroviruses. Novartis Found. Symp. 2001, 238, 258–275. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, J.S.; Asano, K.M.; de Souza, S.P.; Brandão, P.E.; Richtzenhain, L.J. First detection and molecular diversity of Brazilian bovine torovirus (BToV) strains from young and adult cattle. Res. Vet. Sci. 2013, 95, 799–801. [Google Scholar] [CrossRef]
- USDA (United States Department of Agriculture). Livestock and Poultry: World Markets and Trade. Foreign Agricultural Service 9 April 2020. Available online: https://usda.library.cornell.edu/concern/publications/73666448x?locale=en (accessed on 20 May 2024).
- IDF (International Dairy Federation). The World Dairy Situation 2013. Bulletin of the International Dairy Federation 470/2013. 2013. Available online: https://shop.fil-idf.org/products/the-world-dairy-situation-2013-2 (accessed on 20 May 2024).
- Tsuchiaka, S.; Masuda, T.; Sugimura, S.; Kobayashi, S.; Komatsu, N.; Nagai, M.; Omatsu, T.; Furuya, T.; Oba, M.; Katayama, Y.; et al. Development of a novel detection system for microbes from bovine diarrhea by real-time PCR. J. Vet. Med. Sci. 2016, 78, 383–389. [Google Scholar] [CrossRef]
- Ito, T.; Okada, N.; Fukuyama, S. Epidemiological analysis of bovine torovirus in Japan. Virus Res. 2007, 126, 32–37. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Cho, Y.I.; Han, J.I.; Wang, C.; Cooper, V.; Schwartz, K.; Engelken, T.; Yoon, K.J. Case-control study of microbiological etiology associated with calf diarrhea. Vet. Microbiol. 2013, 166, 375–385. [Google Scholar] [CrossRef]
- Lojkić, I.; Krešić, N.; Šimić, I.; Bedeković, T. Detection and molecular characterisation of bovine corona and toroviruses from Croatian cattle. BMC Vet. Res. 2015, 11, 202. [Google Scholar] [CrossRef]
- Haschek, B.; Klein, D.; Benetka, V.; Herrera, C.; Sommerfeld-Stur, I.; Vilcek, S.; Moestl, K.; Baumgartner, W. Detection of bovine torovirus in neonatal calf diarrhoea in Lower Austria and Styria (Austria). J. Vet. Med. B Infect. Dis. Vet. Public Health 2006, 53, 160–165. [Google Scholar] [CrossRef]
- Kirisawa, R.; Takeyama, A.; Koiwa, M.; Iwai, H. Detection of bovine torovirus in fecal specimens of calves with diarrhea in Japan. J. Vet. Med. Sci. 2007, 69, 471–476. [Google Scholar] [CrossRef]
- Castells, M.; Giannitti, F.; Caffarena, R.D.; Casaux, M.L.; Schild, C.; Castells, D.; Riet-Correa, F.; Victoria, M.; Parreño, V.; Colina, R. Bovine coronavirus in Uruguay: Genetic diversity, risk factors and transboundary introductions from neighboring countries. Arch. Virol. 2019, 164, 2715–2724. [Google Scholar] [CrossRef] [PubMed]
- Caffarena, R.D.; Castells, M.; Schild, C.O.; Casaux, M.L.; Armendano, J.I.; Colina, R.; Giannitti, F. Determination of an RT-qPCR viral load cutoff point for the etiologic diagnosis of rotavirus A diarrhea in neonate dairy calves. Front. Vet. Sci. 2022, 9, 952197. [Google Scholar] [CrossRef]
- Castells, M.; Caffarena, R.D.; Casaux, M.L.; Schild, C.; Miño, S.; Castells, F.; Castells, D.; Victoria, M.; Riet-Correa, F.; Giannitti, F.; et al. Phylogenetic Analyses of Rotavirus A from Cattle in Uruguay Reveal the Circulation of Common and Uncommon Genotypes and Suggest Interspecies Transmission. Pathogens 2020, 9, 570. [Google Scholar] [CrossRef] [PubMed]
- Schild, C.O.; Caffarena, R.D.; Gil, A.; Sánchez, J.; Riet-Correa, F.; Giannitti, F. A survey of management practices that influence calf welfare and an estimation of the annual calf mortality risk in pastured dairy herds in Uruguay. J. Dairy. Sci. 2020, 103, 9418–9429. [Google Scholar] [CrossRef] [PubMed]
- Caffarena, R.D.; Casaux, M.L.; Schild, C.O.; Fraga, M.; Castells, M.; Colina, R.; Maya, L.; Corbellini, L.G.; Riet-Correa, F.; Giannitti, F. Causes of neonatal calf diarrhea and mortality in pasture-based dairy herds in Uruguay: A farm-matched case-control study. Braz. J. Microbiol. 2021, 52, 977–988. [Google Scholar] [CrossRef]
- Castells, M.; Bertoni, E.; Caffarena, R.D.; Casaux, M.L.; Schild, C.; Victoria, M.; Riet-Correa, F.; Giannitti, F.; Parreño, V.; Colina, R. Bovine Astrovirus Surveillance in Uruguay Reveals High Detection Rate of a Novel Mamastrovirus Species. Viruses 2019, 12, 32. [Google Scholar] [CrossRef]
- Castells, M.; Caffarena, R.D.; Casaux, M.L.; Schild, C.; Castells, F.; Castells, D.; Victoria, M.; Riet-Correa, F.; Giannitti, F.; Parreño, V.; et al. Detection, risk factors and molecular diversity of norovirus GIII in cattle in Uruguay. Infect. Genet. Evol. 2020, 86, 104613. [Google Scholar] [CrossRef]
- Casaux, M.L.; Neto, W.S.; Schild, C.O.; Costa, R.A.; Macías-Rioseco, M.; Caffarena, R.D.; Silveira, C.S.; Aráoz, V.; Díaz, B.D.; Giannitti, F.; et al. Epidemiological and clinicopathological findings in 15 fatal outbreaks of salmonellosis in dairy calves and virulence genes in the causative Salmonella enterica Typhimurium and Dublin strains. Braz. J. Microbiol. 2023, 54, 475–490. [Google Scholar] [CrossRef]
- Castells, M.; Colina, R. Viral Enteritis in Cattle: To Well Known Viruses and Beyond. Microbiol. Res. 2021, 12, 663–682. [Google Scholar] [CrossRef]
- Woode, G.N.; Saif, L.J.; Quesada, M.; Winand, N.J.; Pohlenz, J.F.; Gourley, N.K. Comparative studies on three isolates of Breda virus of calves. Am. J. Vet. Res. 1985, 46, 1003–1010. [Google Scholar] [PubMed]
- Yoo, D.; Deregt, D. A single amino acid change within antigenic domain II of the spike protein of bovine coronavirus confers resistance to virus neutralization. Clin. Diagn. Lab. Immunol. 2001, 8, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G.M.; Rambaut, A.; Pybus, O.G.; Holmes, E.C. Rates of molecular evolution in RNA viruses: A quantitative phylogenetic analysis. J. Mol. Evol. 2002, 54, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Lu, S.; Shang, G.; Zhu, W.; Yang, J.; Liu, L.; Xu, J. Characterization and identification of a novel torovirus associated with recombinant bovine torovirus from Tibetan antelope in Qinghai-Tibet Plateau of China. Front. Microbiol. 2021, 12, 737753. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Primer Name | Sequence (5′–3′) | Position | Fragment Length |
|---|---|---|---|
| BoToV_PF-1 | AGTTCAAATGGACTACCGGGTC | 1028–1049 | 402 |
| BoToV_PR-1 | TTAGTTTCAGCAAGAGCCGGG | 1409–1429 | |
| BoToV_PF-2 | GATGTTTTGGTCAACAACCC | 3408–3429 | 255 |
| BoToV_PR-2 | CCTACCAACTGGTTTAACG | 3644–3662 | |
| BoToV_PF-3 | GGACAGAAGAGTAACAGAGC | 24,211–24,230 | 805 |
| BoToV_PR-3 | TGATGGATGAAATGTCAGGC | 24,996–25,015 | |
| BoToV_PF-4 | CCTTTACTGGTTATTGGGCC | 25,867–25,886 | 599 |
| BoToV_PR-4 | GACTCATAGGTGAATAAGGG | 26,446–26,465 | |
| BoToV_PF-5 | TACAATGCATCTACTGTTGG | 27,083–27,102 | 780 |
| BoToV_PR-5 | CCTATTAGCACGTTGTTGGG | 27,843–27,862 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castells, M.; Benítez-Galeano, M.J.; Marandino, A.; Caffarena, R.D.; Casaux, M.L.; Pérez, R.; Giannitti, F.; Colina, R. Detection and Genetic Characterization of Bovine Torovirus in Uruguay. Viruses 2024, 16, 835. https://doi.org/10.3390/v16060835
Castells M, Benítez-Galeano MJ, Marandino A, Caffarena RD, Casaux ML, Pérez R, Giannitti F, Colina R. Detection and Genetic Characterization of Bovine Torovirus in Uruguay. Viruses. 2024; 16(6):835. https://doi.org/10.3390/v16060835
Chicago/Turabian StyleCastells, Matías, María José Benítez-Galeano, Ana Marandino, Rubén Darío Caffarena, María Laura Casaux, Ruben Pérez, Federico Giannitti, and Rodney Colina. 2024. "Detection and Genetic Characterization of Bovine Torovirus in Uruguay" Viruses 16, no. 6: 835. https://doi.org/10.3390/v16060835
APA StyleCastells, M., Benítez-Galeano, M. J., Marandino, A., Caffarena, R. D., Casaux, M. L., Pérez, R., Giannitti, F., & Colina, R. (2024). Detection and Genetic Characterization of Bovine Torovirus in Uruguay. Viruses, 16(6), 835. https://doi.org/10.3390/v16060835