
Identifying Protein Interactions with Viral DNA Genomes during Virus Infection

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of Approaches to Study Protein–DNA Interactions in Cells
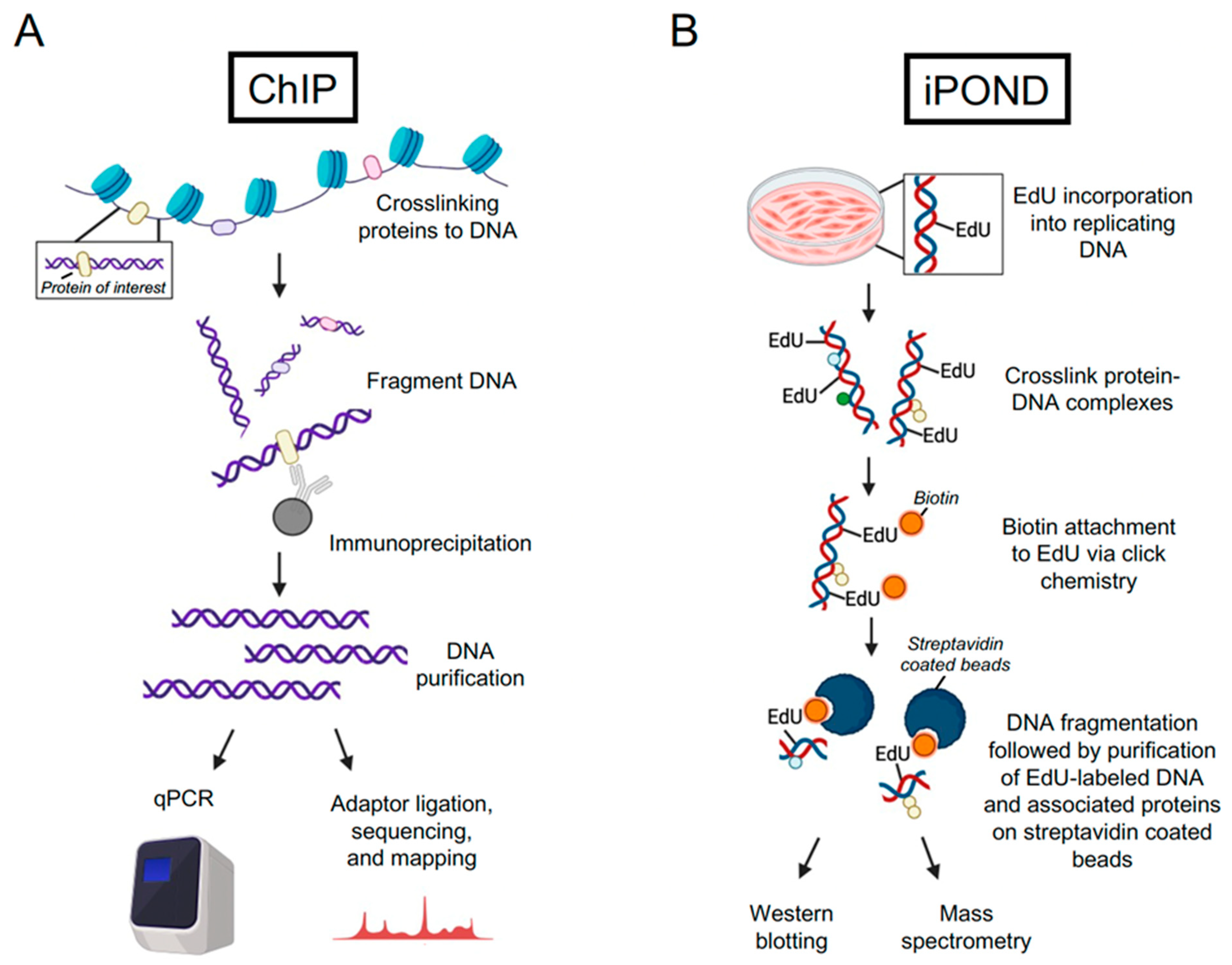
2.1. Chromatin Immunoprecipitation (ChIP) and Cleavage under Targets and Release Using Nuclease (CUT&RUN)
2.2. Unbiased Techniques to Identify Proteins Associated with Nascent DNA
2.3. Validation of Protein–DNA Interactions
3. iPOND as a Tool to Identify Host Factors Associated with Viral Genomes
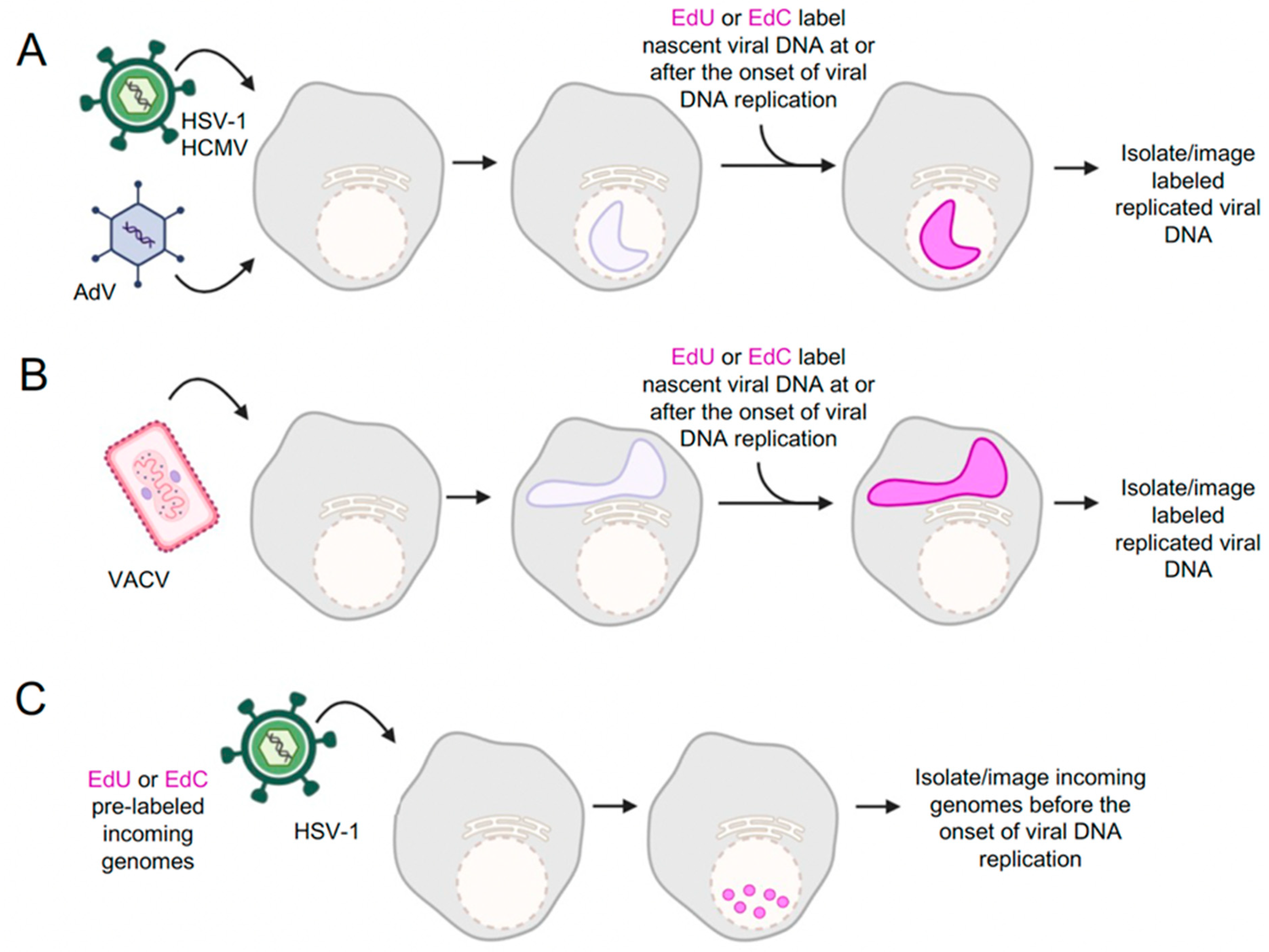
4. Cellular Proteins That Interact with Viral DNA Genomes
4.1. Transcription Factors
4.2. Replication Factors
4.3. DNA Damage Response (DDR) and Repair Proteins
4.4. Antiviral Restriction Factors
5. Accumulation of Host Proteins to Specific Sub-Compartments during Infection
5.1. Accumulation of Activated ATM within Viral Replication Compartments (VRCs)
5.2. The Role of ATR Signaling during Viral Infection
5.3. Association of PML NBs with Incoming Genomes
5.4. Selective Recruitment of Key Regulators of DDR
5.5. Dynamic Interactions of RNA Polymerase II with Replicating Viruses
6. Three-dimensional Genome Techniques to Evaluate Interactions during Viral Infection
7. Future Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Flint, J.; Hatziioannou, T.; Racaniello, V.R.; Rall, G.F.; Skalka, A.M. Principles of Virology, 5th ed.; Wiley & American Society for Microbiology: Hoboken, NJ, USA, 2020. [Google Scholar]
- Knipe, D.M.; Howley, P.M. Fields Virology, 6th ed.; Wolters Kluwer/Lippincott Williams & Wilkins Health: Philadelphia, PA, USA, 2013; Volume 2. [Google Scholar]
- Fay, N.; Pante, N. Nuclear entry of DNA viruses. Front. Microbiol. 2015, 6, 467. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F.; Fassati, A. Nuclear import of viral DNA genomes. Traffic 2003, 4, 136–143. [Google Scholar] [CrossRef]
- Hellman, L.M.; Fried, M.G. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2007, 2, 1849–1861. [Google Scholar] [CrossRef]
- Fuxman Bass, J.I.; Reece-Hoyes, J.S.; Walhout, A.J. Gene-Centered Yeast One-Hybrid Assays. Cold Spring Harb. Protoc. 2016, 2016, top077669. [Google Scholar] [CrossRef] [PubMed]
- Ouwerkerk, P.B.; Meijer, A.H. Yeast one-hybrid screening for DNA-protein interactions. Curr. Protoc. Mol. Biol. 2001, 55, 12.12.1–12.12.12. [Google Scholar] [CrossRef] [PubMed]
- Rigaut, G.; Shevchenko, A.; Rutz, B.; Wilm, M.; Mann, M.; Séraphin, B. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 1999, 17, 1030–1032. [Google Scholar] [CrossRef]
- Galas, D.J.; Schmitz, A. DNAse footprinting: A simple method for the detection of protein-DNA binding specificity. Nucleic Acids Res. 1978, 5, 3157–3170. [Google Scholar] [CrossRef] [PubMed]
- Lum, K.K.; Cristea, I.M. Proteomic approaches to uncovering virus-host protein interactions during the progression of viral infection. Expert Rev. Proteom. 2016, 13, 325–340. [Google Scholar] [CrossRef]
- Cristea, I.M.; Carroll, J.-W.N.; Rout, M.P.; Rice, C.M.; Chait, B.T.; MacDonald, M.R. Tracking and elucidating alphavirus-host protein interactions. J. Biol. Chem. 2006, 281, 30269–30278. [Google Scholar] [CrossRef]
- Taylor, T.J.; Knipe, D.M. Proteomics of herpes simplex virus replication compartments: Association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J. Virol. 2004, 78, 5856–5866. [Google Scholar] [CrossRef]
- Gilmour, D.S.; Lis, J.T. In vivo interactions of RNA polymerase II with genes of Drosophila melanogaster. Mol. Cell Biol. 1985, 5, 2009–2018. [Google Scholar] [PubMed]
- Kim, T.H.; Dekker, J. ChIP-seq. Cold Spring Harb. Protoc. 2018, 2018, prot082610. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Dekker, J. ChIP-Quantitative Polymerase Chain Reaction (ChIP-qPCR). Cold Spring Harb. Protoc. 2018, 2018, prot082628. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 2017, 6, e21856. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Durussel, T.; Laemmli, U.K. ChIC and ChEC: Genomic mapping of chromatin proteins. Mol. Cell 2004, 16, 147–157. [Google Scholar] [PubMed]
- Hainer, S.J.; Bošković, A.; McCannell, K.N.; Rando, O.J.; Fazzio, T.G. Profiling of Pluripotency Factors in Single Cells and Early Embryos. Cell 2019, 177, 1319–1329.e11. [Google Scholar] [CrossRef] [PubMed]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019, 10, 1930. [Google Scholar] [PubMed]
- Sirbu, B.M.; Couch, F.B.; Feigerle, J.T.; Bhaskara, S.; Hiebert, S.W.; Cortez, D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011, 25, 1320–1327. [Google Scholar] [CrossRef] [PubMed]
- Kliszczak, A.E.; Rainey, M.D.; Harhen, B.; Boisvert, F.M.; Santocanale, C. DNA mediated chromatin pull-down for the study of chromatin replication. Sci. Rep. 2011, 1, 95. [Google Scholar] [CrossRef]
- Alabert, C.; Bukowski-Wills, J.-C.; Lee, S.-B.; Kustatscher, G.; Nakamura, K.; Alves, F.D.L.; Menard, P.; Mejlvang, J.; Rappsilber, J.; Groth, A. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat. Cell Biol. 2014, 16, 281–293. [Google Scholar] [CrossRef]
- Sirbu, B.M.; Couch, F.B.; Cortez, D. Monitoring the spatiotemporal dynamics of proteins at replication forks and in assembled chromatin using isolation of proteins on nascent DNA. Nat. Protoc. 2012, 7, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Contreras, A.J.; Ruppen, I.; Nieto-Soler, M.; Murga, M.; Rodriguez-Acebes, S.; Remeseiro, S.; Rodrigo-Perez, S.; Rojas, A.M.; Mendez, J.; Muñoz, J.; et al. A proteomic characterization of factors enriched at nascent DNA molecules. Cell Rep. 2013, 3, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, B.M.; McDonald, W.H.; Dungrawala, H.; Badu-Nkansah, A.; Kavanaugh, G.M.; Chen, Y.; Tabb, D.L.; Cortez, D. Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem. 2013, 288, 31458–31467. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.H.; El Hassan, M.A.; Bremner, R. A rapid and efficient method to purify proteins at replication forks under native conditions. Biotechniques 2013, 55, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Wiest, N.E.; Tomkinson, A.E. Optimization of Native and Formaldehyde iPOND Techniques for Use in Suspension Cells. Methods Enzymol. 2017, 591, 1–32. [Google Scholar] [PubMed]
- Ramachandran, S.; Henikoff, S. MINCE-Seq: Mapping In Vivo Nascent Chromatin with EdU and Sequencing. Methods Mol. Biol. 2018, 1832, 159–168. [Google Scholar] [PubMed]
- Salic, A.; Mitchison, T.J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 2415–2420. [Google Scholar] [CrossRef] [PubMed]
- Dembowski, J.A.; DeLuca, N.A. Selective recruitment of nuclear factors to productively replicating herpes simplex virus genomes. PLoS Pathog. 2015, 11, e1004939. [Google Scholar] [CrossRef] [PubMed]
- Reyes, E.D.; Kulej, K.; Pancholi, N.J.; Akhtar, L.N.; Avgousti, D.C.; Kim, E.T.; Bricker, D.K.; Spruce, L.A.; Koniski, S.A.; Seeholzer, S.H.; et al. Identifying Host Factors Associated with DNA Replicated during Virus Infection. Mol. Cell. Proteom. 2017, 16, 2079–2097. [Google Scholar] [CrossRef]
- Dembowski, J.A.; Dremel, S.E.; DeLuca, N.A. Replication-Coupled Recruitment of Viral and Cellular Factors to Herpes Simplex Virus Type 1 Replication Forks for the Maintenance and Expression of Viral Genomes. PLoS Pathog. 2017, 13, e1006166. [Google Scholar] [CrossRef]
- Senkevich, T.G.; Katsafanas, G.C.; Weisberg, A.; Olano, L.R.; Moss, B. Identification of Vaccinia Virus Replisome and Transcriptome Proteins by Isolation of Proteins on Nascent DNA Coupled with Mass Spectrometry. J. Virol. 2017, 91, 01015-17. [Google Scholar] [CrossRef] [PubMed]
- Dembowski, J.A.; DeLuca, N.A. Temporal Viral Genome-Protein Interactions Define Distinct Stages of Productive Herpesviral Infection. mBio 2018, 9, 01182-18. [Google Scholar] [CrossRef] [PubMed]
- Dembowski, J.A.; Deluca, N.A. Purification of Viral DNA for the Identification of Associated Viral and Cellular Proteins. J. Vis. Exp. 2017, 126, e56374. [Google Scholar]
- Fox, H.L.; Dembowski, J.A.; DeLuca, N.A. A Herpesviral Immediate Early Protein Promotes Transcription Elongation of Viral Transcripts. mBio 2017, 8, 00745-17. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.T.; Dybas, J.M.; Kulej, K.; Reyes, E.D.; Price, A.M.; Akhtar, L.N.; Orr, A.; Garcia, B.A.; Boutell, C.; Weitzman, M.D. Comparative proteomics identifies Schlafen 5 (SLFN5) as a herpes simplex virus restriction factor that suppresses viral transcription. Nat. Microbiol. 2021, 6, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.C.; Dybas, J.M.; Hughes, J.; Weitzman, M.D.; Boutell, C. The HSV-1 ubiquitin ligase ICP0: Modifying the cellular proteome to promote infection. Virus Res. 2020, 285, 198015. [Google Scholar] [CrossRef] [PubMed]
- Boutell, C.; Sadis, S.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 2002, 76, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Perez, R.D.; Frey, T.R.; Burton, E.M.; Mannemuddhu, S.; Haley, J.D.; McIntosh, M.T.; Bhaduri-McIntosh, S. Novel replisome-associated proteins at cellular replication forks in EBV-transformed B lymphocytes. PLoS Pathog. 2019, 15, e1008228. [Google Scholar] [CrossRef] [PubMed]
- Manska, S.; Rossetto, C.C. Identification of cellular proteins associated with human cytomegalovirus (HCMV) DNA replication suggests novel cellular and viral interactions. Virology 2022, 566, 26–41. [Google Scholar] [CrossRef]
- Dabral, P.; Uppal, T.; Rossetto, C.C.; Verma, S.C. Minichromosome Maintenance Proteins Cooperate with LANA during the G1/S Phase of the Cell Cycle to Support Viral DNA Replication. J. Virol. 2019, 93, 02256-18. [Google Scholar] [CrossRef]
- Dybas, J.M.; Lum, K.K.; Kulej, K.; Reyes, E.D.; Lauman, R.; Charman, M.; Purman, C.E.; Steinbock, R.T.; Grams, N.; Price, A.M.; et al. Adenovirus Remodeling of the Host Proteome and Host Factors Associated with Viral Genomes. mSystems 2021, 6, e0046821. [Google Scholar] [CrossRef] [PubMed]
- Batterson, W.; Roizman, B. Characterization of the herpes simplex virion-associated factor responsible for the induction of alpha genes. J. Virol. 1983, 46, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 1974, 14, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis: Sequential transition of polypeptide synthesis requires functional viral polypeptides. Proc. Natl. Acad. Sci. USA 1975, 72, 1276–1280. [Google Scholar] [CrossRef] [PubMed]
- Heath, J.R.; Dembowski, J.A. Fashionably late: Temporal regulation of HSV-1 late gene transcription. PLoS Pathog. 2022, 18, e1010536. [Google Scholar] [CrossRef] [PubMed]
- Dremel, S.E.; DeLuca, N.A. Herpes simplex viral nucleoprotein creates a competitive transcriptional environment facilitating robust viral transcription and host shut off. Elife 2019, 8, e51109. [Google Scholar] [CrossRef] [PubMed]
- Sampath, P.; Deluca, N.A. Binding of ICP4, TATA-binding protein, and RNA polymerase II to herpes simplex virus type 1 immediate-early, early, and late promoters in virus-infected cells. J. Virol. 2008, 82, 2339–2349. [Google Scholar] [CrossRef] [PubMed]
- Lester, J.T.; DeLuca, N.A. Herpes simplex virus 1 ICP4 forms complexes with TFIID and mediator in virus-infected cells. J. Virol. 2011, 85, 5733–5744. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Bates, P.; Rivera-Gonzalez, R.; Gu, B.; A DeLuca, N. ICP4, the major transcriptional regulatory protein of herpes simplex virus type 1, forms a tripartite complex with TATA-binding protein and TFIIB. J. Virol. 1993, 67, 4676–4687. [Google Scholar] [CrossRef]
- Ding, X.; Neumann, D.M.; Zhu, L. Host factors associated with either VP16 or VP16-induced complex differentially affect HSV-1 lytic infection. Rev. Med. Virol. 2022, 32, e2394. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, Q.; Wang, M.; Chen, S.; Jia, R.; Yang, Q.; Zhu, D.; Liu, M.; Zhao, X.; Zhang, S. Multifaceted Roles of ICP22/ORF63 Proteins in the Life Cycle of Human Herpesviruses. Front. Microbiol. 2021, 12, 668461. [Google Scholar] [CrossRef] [PubMed]
- Rice, S.A.; Davido, D.J. HSV-1 ICP22: Hijacking host nuclear functions to enhance viral infection. Future Microbiol. 2013, 8, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Charman, M.; Herrmann, C.; Weitzman, M.D. Viral and cellular interactions during adenovirus DNA replication. FEBS Lett. 2019, 593, 3531–3550. [Google Scholar] [CrossRef] [PubMed]
- Boyer, T.G.; Martin, M.E.D.; Lees, E.; Ricciardi, R.P.; Berk, A.J. Mammalian Srb/Mediator complex is targeted by adenovirus E1A protein. Nature 1999, 399, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Ornelles, D.A. Inactivating intracellular antiviral responses during adenovirus infection. Oncogene 2005, 24, 7686–7696. [Google Scholar] [CrossRef] [PubMed]
- Broyles, S.S. Vaccinia virus transcription. J. Gen. Virol. 2003, 84, 2293–2303. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.F.; Oswald, B.W.; Dellis, S. Vaccinia virus late transcription is activated in vitro by cellular heterogeneous nuclear ribonucleoproteins. J. Biol. Chem. 2001, 276, 40680–40686. [Google Scholar] [CrossRef] [PubMed]
- Katsafanas, G.C.; Moss, B. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2007, 2, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Packard, J.E.; Dembowski, J.A. HSV-1 DNA Replication-Coordinated Regulation by Viral and Cellular Factors. Viruses 2021, 13, 2015. [Google Scholar] [CrossRef] [PubMed]
- Weller, S.K.; Coen, D.M. Herpes simplex viruses: Mechanisms of DNA replication. Cold Spring Harb. Perspect. Biol. 2012, 4, a013011. [Google Scholar] [CrossRef]
- Greseth, M.D.; Traktman, P. The Life Cycle of the Vaccinia Virus Genome. Annu. Rev. Virol. 2022, 9, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Strzalka, W.; Ziemienowicz, A. Proliferating cell nuclear antigen (PCNA): A key factor in DNA replication and cell cycle regulation. Ann. Bot. 2011, 107, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Sanders, I.; Boyer, M.; Fraser, N.W. Early nucleosome deposition on, and replication of, HSV DNA requires cell factor PCNA. J. Neurovirol. 2015, 21, 358–369. [Google Scholar] [CrossRef]
- Packard, J.E.; Williams, M.R.; Fromuth, D.P.; Dembowski, J.A. Proliferating cell nuclear antigen inhibitors block distinct stages of herpes simplex virus infection. PLoS Pathog. 2023, 19, e1011539. [Google Scholar] [CrossRef] [PubMed]
- Postigo, A.; Ramsden, A.E.; Howell, M.; Way, M. Cytoplasmic ATR Activation Promotes Vaccinia Virus Genome Replication. Cell Rep. 2017, 19, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Nussenzweig, A.; Takeda, S.; Austin, C. Human topoisomerases and their roles in genome stability and organization. Nat. Rev. Mol. Cell Biol. 2022, 23, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Li, J.; Irwin, C.R.; Jenkins, H.; DeLange, L.; Evans, D.H. Vaccinia virus DNA ligase recruits cellular topoisomerase II to sites of viral replication and assembly. J. Virol. 2008, 82, 5922–5932. [Google Scholar] [CrossRef] [PubMed]
- Schaack, J.; Schedl, P.; Shenk, T. Topoisomerase I and II cleavage of adenovirus DNA in vivo: Both topoisomerase activities appear to be required for adenovirus DNA replication. J. Virol. 1990, 64, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Turnell, A.S.; Grand, R.J. DNA viruses and the cellular DNA-damage response. J. Gen. Virol. 2012, 93, 2076–2097. [Google Scholar] [CrossRef]
- Weitzman, M.D.; Lilley, C.E.; Chaurushiya, M.S. Genomes in conflict: Maintaining genome integrity during virus infection. Annu. Rev. Microbiol. 2010, 64, 61–81. [Google Scholar] [CrossRef] [PubMed]
- Luftig, M.A. Viruses and the DNA Damage Response: Activation and Antagonism. Annu. Rev. Virol. 2014, 1, 605–625. [Google Scholar] [CrossRef]
- Weitzman, M.D.; Fradet-Turcotte, A. Virus DNA Replication and the Host DNA Damage Response. Annu. Rev. Virol. 2018, 5, 141–164. [Google Scholar] [CrossRef] [PubMed]
- Mertens, M.E.; Knipe, D.M. Herpes Simplex Virus 1 Manipulates Host Cell Antiviral and Proviral DNA Damage Responses. mBio 2021, 12, 03552-20. [Google Scholar] [CrossRef] [PubMed]
- Dybas, J.M.; Herrmann, C.; Weitzman, M.D. Ubiquitination at the interface of tumor viruses and DNA damage responses. Curr. Opin. Virol. 2018, 32, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Lilley, C.E.; Carson, C.T.; Muotri, A.R.; Gage, F.H.; Weitzman, M.D. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 2005, 102, 5844–5849. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Carson, C.T.; Weitzman, M.D. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 2002, 418, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.L.; Kennedy, M.A.; Hutton, J.E.; Liu, D.; Song, B.; Phelan, B.; Cristea, I.M. Systematic profiling of protein complex dynamics reveals DNA-PK phosphorylation of IFI16 en route to herpesvirus immunity. Sci. Adv. 2021, 7, eabg6680. [Google Scholar] [CrossRef] [PubMed]
- Mohni, K.N.; Mastrocola, A.S.; Bai, P.; Weller, S.K.; Heinen, C.D. DNA mismatch repair proteins are required for efficient herpes simplex virus 1 replication. J. Virol. 2011, 85, 12241–12253. [Google Scholar] [CrossRef]
- Chaudhuri, M.; Song, L.; Parris, D.S. The herpes simplex virus type 1 DNA polymerase processivity factor increases fidelity without altering pre-steady-state rate constants for polymerization or excision. J. Biol. Chem. 2003, 278, 8996–9004. [Google Scholar] [CrossRef]
- Karttunen, H.; Savas, J.N.; McKinney, C.; Chen, Y.-H.; Yates, J.R.; Hukkanen, V.; Huang, T.T.; Mohr, I. Co-opting the Fanconi anemia genomic stability pathway enables herpesvirus DNA synthesis and productive growth. Mol. Cell 2014, 55, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Hanzlikova, H.; Kalasova, I.; Demin, A.A.; Pennicott, L.E.; Cihlarova, Z.; Caldecott, K.W. The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol. Cell 2018, 71, 319–331.e3. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zheng, C. When PARPs Meet Antiviral Innate Immunity. Trends Microbiol. 2021, 29, 776–778. [Google Scholar] [CrossRef]
- Grady, S.L.; Hwang, J.; Vastag, L.; Rabinowitz, J.D.; Shenk, T. Herpes simplex virus 1 infection activates poly(ADP-ribose) polymerase and triggers the degradation of poly(ADP-ribose) glycohydrolase. J. Virol. 2012, 86, 8259–8268. [Google Scholar] [CrossRef]
- Chung, W.C.; Song, M.J. Virus-Host Interplay between Poly (ADP-Ribose) Polymerase 1 and Oncogenic Gammaherpesviruses. Front. Microbiol. 2021, 12, 811671. [Google Scholar] [CrossRef]
- Everett, R.D. DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 2001, 20, 7266–7273. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; de The, H. PML nuclear bodies: From architecture to function. Curr. Opin. Cell Biol. 2018, 52, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Sourvinos, G.; Leiper, C.; Clements, J.B.; Orr, A. Formation of nuclear foci of the herpes simplex virus type 1 regulatory protein ICP4 at early times of infection: Localization, dynamics, recruitment of ICP27, and evidence for the de novo induction of ND10-like complexes. J. Virol. 2004, 78, 1903–1917. [Google Scholar] [CrossRef]
- Ishov, A.M.; Maul, G.G. The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J. Cell Biol. 1996, 134, 815–826. [Google Scholar] [CrossRef]
- Hofmann, S.; Stubbe, M.; Mai, J.; Schreiner, S. Double-edged role of PML nuclear bodies during human adenovirus infection. Virus Res. 2021, 295, 198280. [Google Scholar] [CrossRef] [PubMed]
- Maul, G.G.; Ishov, A.M.; Everett, R.D. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 1996, 217, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Everett, R.D.; Weitzman, M.D. The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog. 2011, 7, e1002084. [Google Scholar] [CrossRef] [PubMed]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Landry, S.; Suh, J.; Panier, S.; Everett, R.D.; Stewart, G.S.; Durocher, D.; Weitzman, M.D. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 2010, 29, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Chaurushiya, M.S.; Lilley, C.E.; Aslanian, A.; Meisenhelder, J.; Scott, D.C.; Landry, S.; Ticau, S.; Boutell, C.; Yates, J.R.; Schulman, B.A.; et al. Viral E3 ubiquitin ligase-mediated degradation of a cellular E3: Viral mimicry of a cellular phosphorylation mark targets the RNF8 FHA domain. Mol. Cell 2012, 46, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Charman, M.; Weitzman, M.D. Replication Compartments of DNA Viruses in the Nucleus: Location, Location, Location. Viruses 2020, 12, 151. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, N.; Bai, P.; Buchek, G.; Korza, G.; Weller, S.K. Physical interaction between the herpes simplex virus type 1 exonuclease, UL12, and the DNA double-strand break-sensing MRN complex. J. Virol. 2010, 84, 12504–12514. [Google Scholar] [CrossRef] [PubMed]
- Petrini, J.H.; Stracker, T.H. The cellular response to DNA double-strand breaks: Defining the sensors and mediators. Trends Cell Biol. 2003, 13, 458–462. [Google Scholar] [CrossRef]
- Pancholi, N.J.; Weitzman, M.D. Serotype-specific restriction of wild-type adenoviruses by the cellular Mre11-Rad50-Nbs1 complex. Virology 2018, 518, 221–231. [Google Scholar] [CrossRef]
- Alekseev, O.; Donegan, W.E.; Donovan, K.R.; Limonnik, V.; Azizkhan-Clifford, J. HSV-1 Hijacks the Host DNA Damage Response in Corneal Epithelial Cells through ICP4-Mediated Activation of ATM. Investig. Ophthalmol. Vis. Sci. 2020, 61, 39. [Google Scholar] [CrossRef]
- Heiser, K.; Nicholas, C.; Garcea, R.L. Activation of DNA damage repair pathways by murine polyomavirus. Virology 2016, 497, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.L.; Verhalen, B.; Jiang, M. Polyomavirus interaction with the DNA damage response. Virol. Sin. 2015, 30, 122–129. [Google Scholar] [CrossRef]
- Erickson, K.D.; Garcea, R.L. Viral replication centers and the DNA damage response in JC virus-infected cells. Virology 2019, 528, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.E.; Weller, S.K. Herpes simplex virus type I disrupts the ATR-dependent DNA-damage response during lytic infection. J. Cell Sci. 2006, 119, 2695–2703. [Google Scholar] [CrossRef]
- Livingston, C.M.; Ifrim, M.F.; Cowan, A.E.; Weller, S.K. Virus-Induced Chaperone-Enriched (VICE) domains function as nuclear protein quality control centers during HSV-1 infection. PLoS Pathog. 2009, 5, e1000619. [Google Scholar] [CrossRef]
- Mohni, K.N.; Livingston, C.M.; Cortez, D.; Weller, S.K. ATR and ATRIP are recruited to herpes simplex virus type 1 replication compartments even though ATR signaling is disabled. J. Virol. 2010, 84, 12152–12164. [Google Scholar] [CrossRef] [PubMed]
- Mohni, K.N.; Dee, A.R.; Smith, S.; Schumacher, A.J.; Weller, S.K. Efficient herpes simplex virus 1 replication requires cellular ATR pathway proteins. J. Virol. 2013, 87, 531–542. [Google Scholar] [CrossRef]
- Edwards, T.G.; Bloom, D.C.; Fisher, C. The ATM and Rad3-Related (ATR) Protein Kinase Pathway Is Activated by Herpes Simplex Virus 1 and Required for Efficient Viral Replication. J. Virol. 2018, 92, 01884-17. [Google Scholar] [CrossRef]
- Burkham, J.; Coen, D.M.; Weller, S.K. ND10 protein PML is recruited to herpes simplex virus type 1 prereplicative sites and replication compartments in the presence of viral DNA polymerase. J. Virol. 1998, 72, 10100–10107. [Google Scholar] [CrossRef]
- Burkham, J.; Coen, D.M.; Hwang, C.B.C.; Weller, S.K. Interactions of herpes simplex virus type 1 with ND10 and recruitment of PML to replication compartments. J. Virol. 2001, 75, 2353–2367. [Google Scholar] [CrossRef]
- Wilkinson, D.E.; Weller, S.K. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J. Virol. 2004, 78, 4783–4796. [Google Scholar] [CrossRef] [PubMed]
- Mok, M.T.; Cheng, A.S.; Henderson, B.R. The ubiquitin ligases RNF8 and RNF168 display rapid but distinct dynamics at DNA repair foci in living cells. Int. J. Biochem. Cell Biol. 2014, 57, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Croke, M.; Neumann, M.A.; Grotsky, D.A.; Kreienkamp, R.; Yaddanapudi, S.C.; Gonzalo, S. Differences in 53BP1 and BRCA1 regulation between cycling and non-cycling cells. Cell Cycle 2013, 12, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.C.; Li, X.; Vladmirova, O.; Li, Z.-R.; Chen, G.-J.; Xiao, Y.; Li, L.-H.; Lu, D.-F.; Han, H.-B.; Zhou, J.-M. Selective recruitment of host factors by HSV-1 replication centers. Dongwuxue Yanjiu 2015, 36, 142–151. [Google Scholar] [PubMed]
- Tanwar, V.S.; Jose, C.C.; Cuddapah, S. Role of CTCF in DNA damage response. Mutat. Res. Rev. Mutat. Res. 2019, 780, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Ohkuma, Y. Phosphorylation of the C-terminal domain of RNA polymerase II plays central roles in the integrated events of eucaryotic gene expression. J. Biochem. 2007, 141, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.; Mirny, L. The 3D Genome as Moderator of Chromosomal Communication. Cell 2016, 164, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Gorkin, D.U.; Ren, B. Chromatin Domains: The Unit of Chromosome Organization. Mol. Cell 2016, 62, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Liang, W.; Wang, S.; Wang, H.; Li, X.; Meng, Q.; Zhao, Y.; Zheng, C. When 3D genome technology meets viral infection, including SARS-CoV-2. J. Med. Virol. 2022, 94, 5627–5639. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Cournac, A.; Palumbo, G.A.; Marbouty, M.; Mortaza, S.; Thierry, A.; Cairo, S.; Lavigne, M.; Koszul, R.; Neuveut, C. Tridimensional infiltration of DNA viruses into the host genome shows preferential contact with active chromatin. Nat. Commun. 2018, 9, 4268. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Catez, F.; Masumoto, H.; Lomonte, P. Centromere architecture breakdown induced by the viral E3 ubiquitin ligase ICP0 protein of herpes simplex virus type 1. PLoS ONE 2012, 7, e44227. [Google Scholar] [CrossRef] [PubMed]
- Catez, F.; Picard, C.; Held, K.; Gross, S.; Rousseau, A.; Theil, D.; Sawtell, N.; Labetoulle, M.; Lomonte, P. HSV-1 genome subnuclear positioning and associations with host-cell PML-NBs and centromeres regulate LAT locus transcription during latency in neurons. PLoS Pathog. 2012, 8, e1002852. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Kim, E.T.; Vladimirova, O.; Dheekollu, J.; Wang, Z.; Newhart, A.; Liu, D.; Myers, J.L.; Hensley, S.E.; Moffat, J.; et al. HSV-1 remodels host telomeres to facilitate viral replication. Cell Rep. 2014, 9, 2263–2278. [Google Scholar] [CrossRef] [PubMed]
- Lippert, T.P.; Marzec, P.; Idilli, A.I.; Sarek, G.; Vancevska, A.; Bower, M.; Farrell, P.J.; Ojala, P.M.; Feldhahn, N.; Boulton, S.J. Oncogenic herpesvirus KSHV triggers hallmarks of alternative lengthening of telomeres. Nat. Commun. 2021, 12, 512. [Google Scholar] [CrossRef] [PubMed]
- Schang, L.M.; Hu, M.Y.; Cortes, E.F.; Sun, K. Chromatin-mediated epigenetic regulation of HSV-1 transcription as a potential target in antiviral therapy. Antivir. Res. 2021, 192, 105103. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Cullen, B.R. Epigenetic and epitranscriptomic regulation of viral replication. Nat. Rev. Microbiol. 2020, 18, 559–570. [Google Scholar] [CrossRef] [PubMed]
- Bloom, D.C.; Giordani, N.V.; Kwiatkowski, D.L. Epigenetic regulation of latent HSV-1 gene expression. Biochim. Biophys. Acta 2010, 1799, 246–256. [Google Scholar] [CrossRef]
- Lu, X.; Triezenberg, S.J. Chromatin assembly on herpes simplex virus genomes during lytic infection. Biochim. Biophys. Acta 2010, 1799, 217–222. [Google Scholar] [CrossRef]
- Placek, B.J.; Berger, S.L. Chromatin dynamics during herpes simplex virus-1 lytic infection. Biochim. Biophys. Acta 2010, 1799, 223–227. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Packard, J.E.; Kumar, N.; Weitzman, M.D.; Dembowski, J.A. Identifying Protein Interactions with Viral DNA Genomes during Virus Infection. Viruses 2024, 16, 845. https://doi.org/10.3390/v16060845
Packard JE, Kumar N, Weitzman MD, Dembowski JA. Identifying Protein Interactions with Viral DNA Genomes during Virus Infection. Viruses. 2024; 16(6):845. https://doi.org/10.3390/v16060845
Chicago/Turabian StylePackard, Jessica E., Namrata Kumar, Matthew D. Weitzman, and Jill A. Dembowski. 2024. "Identifying Protein Interactions with Viral DNA Genomes during Virus Infection" Viruses 16, no. 6: 845. https://doi.org/10.3390/v16060845