
Genome-Wide Transcriptional Roles of KSHV Viral Interferon Regulatory Factors in Oral Epithelial Cells
 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
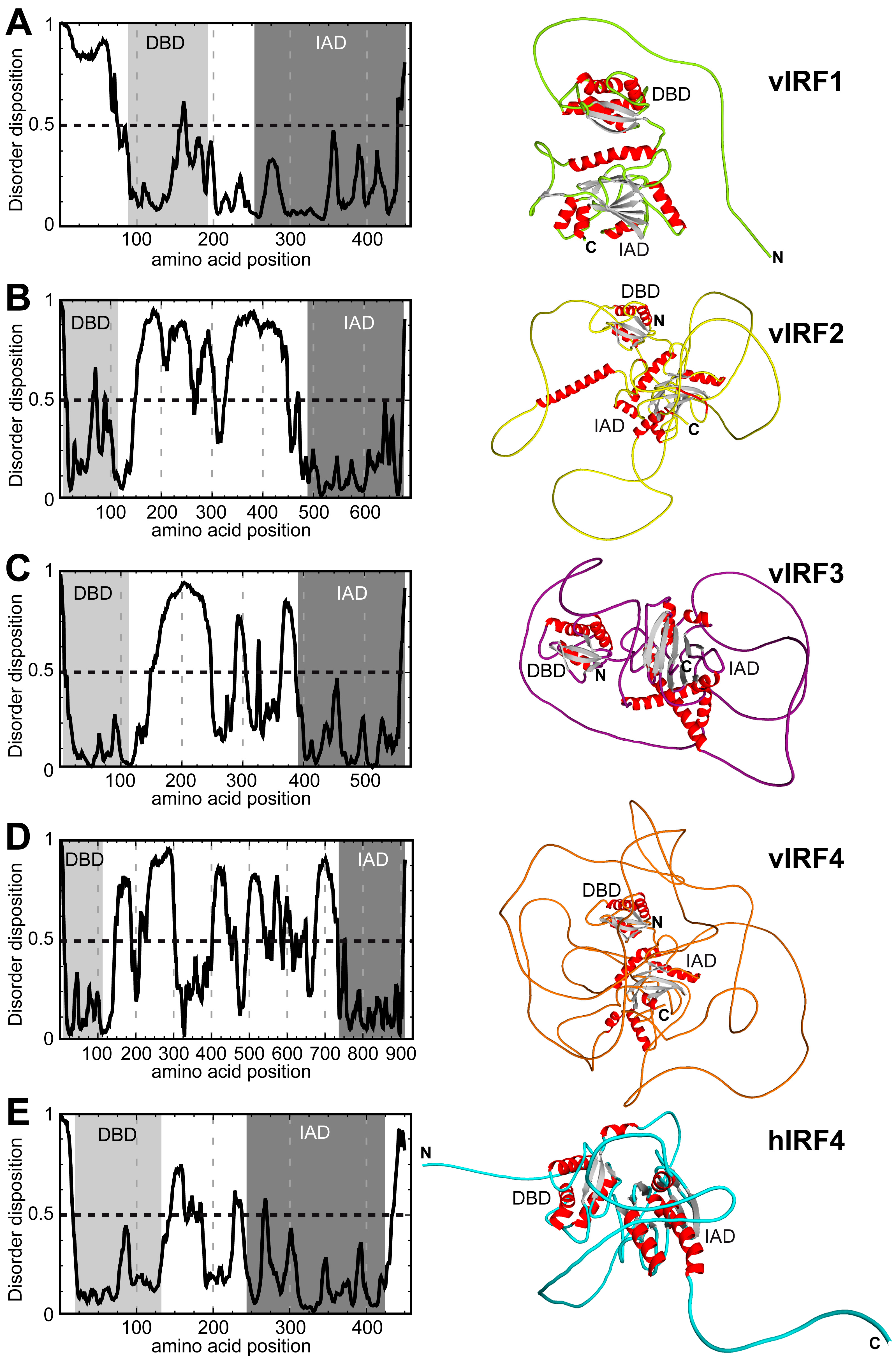
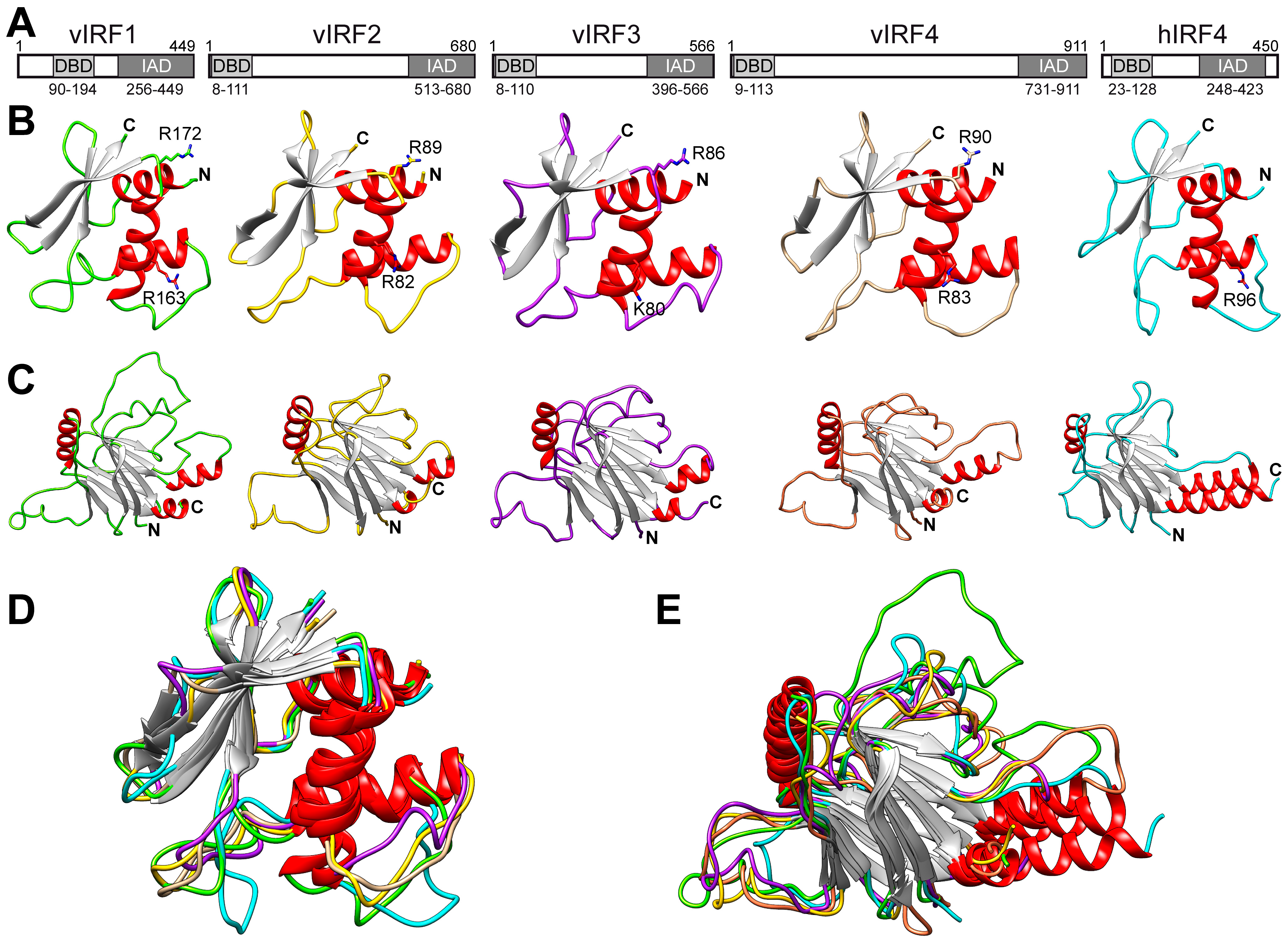
2.1. Structure Prediction and Comparison
2.2. Cell Lines
2.3. Antibodies, Plasmids and DNA Transfection
2.4. Viruses and Viral Infections
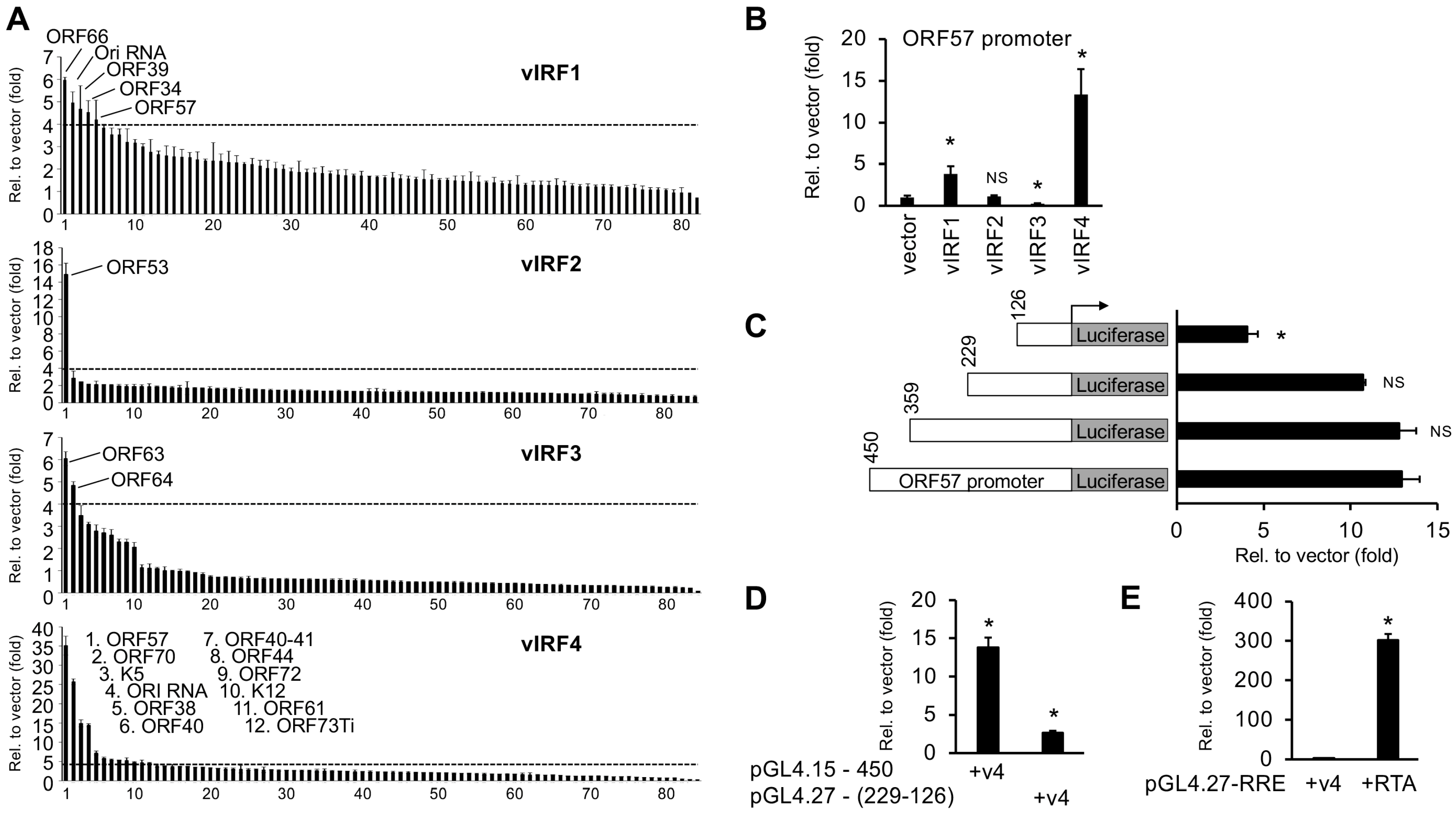
2.5. Luciferase Reporter Assay
2.6. RNA Isolation and RT-qPCR Analysis
2.7. Chromatin Immunoprecipitation Assay (ChIP)
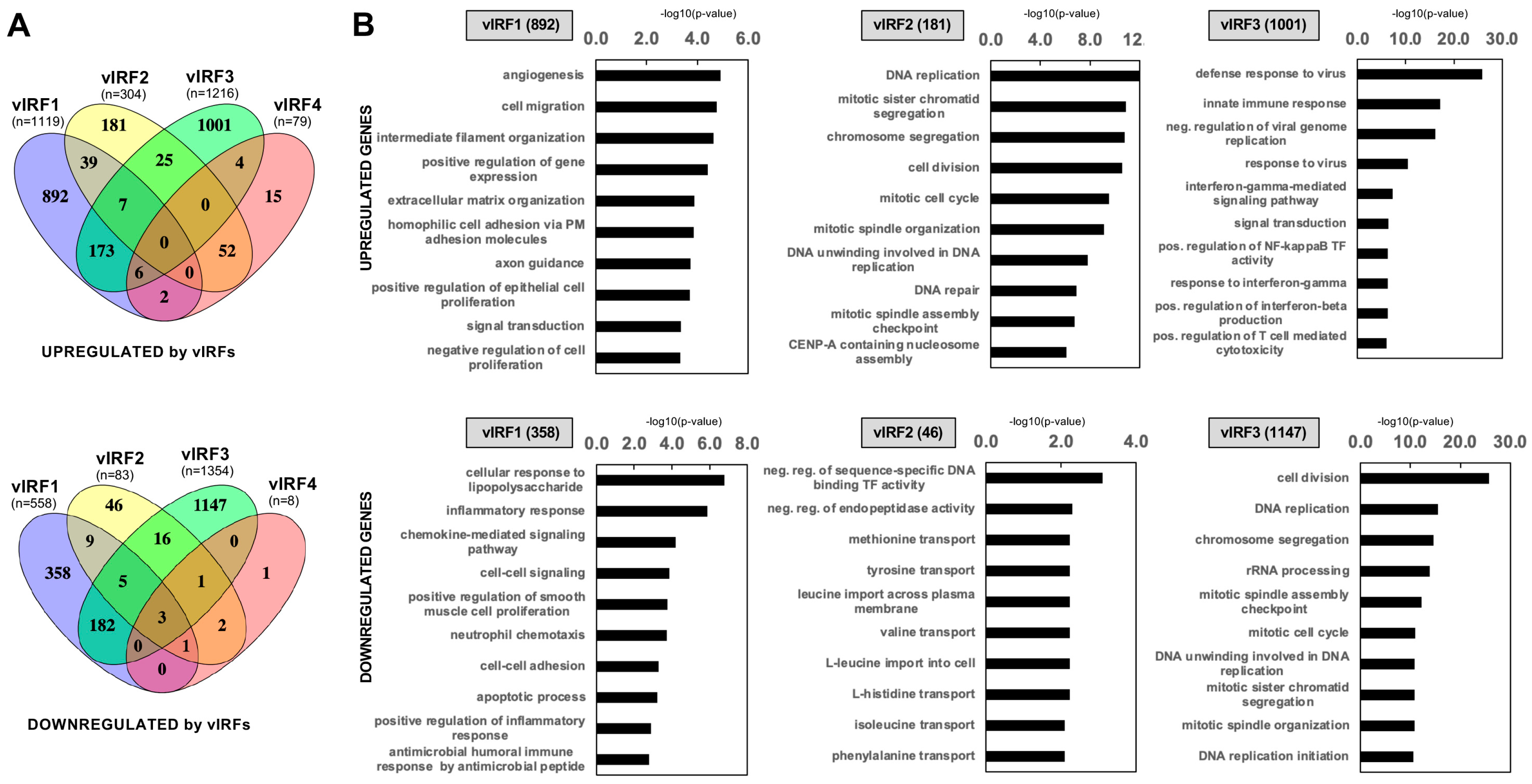
2.8. RNA-Seq Analysis
3. Results
3.1. Comparison of the Putative Tertiary Structures of vIRFs
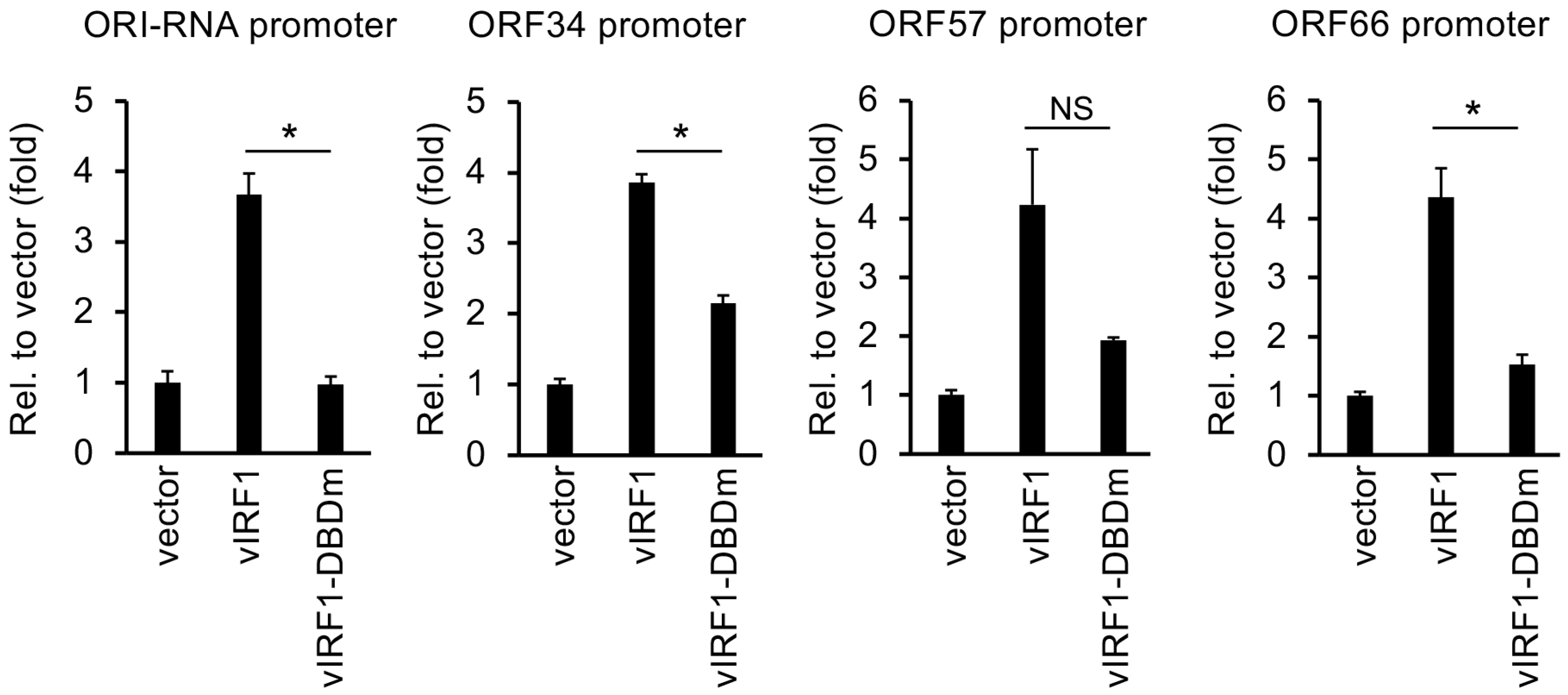
3.2. Identification of KSHV Gene Promoters Regulated by vIRFs
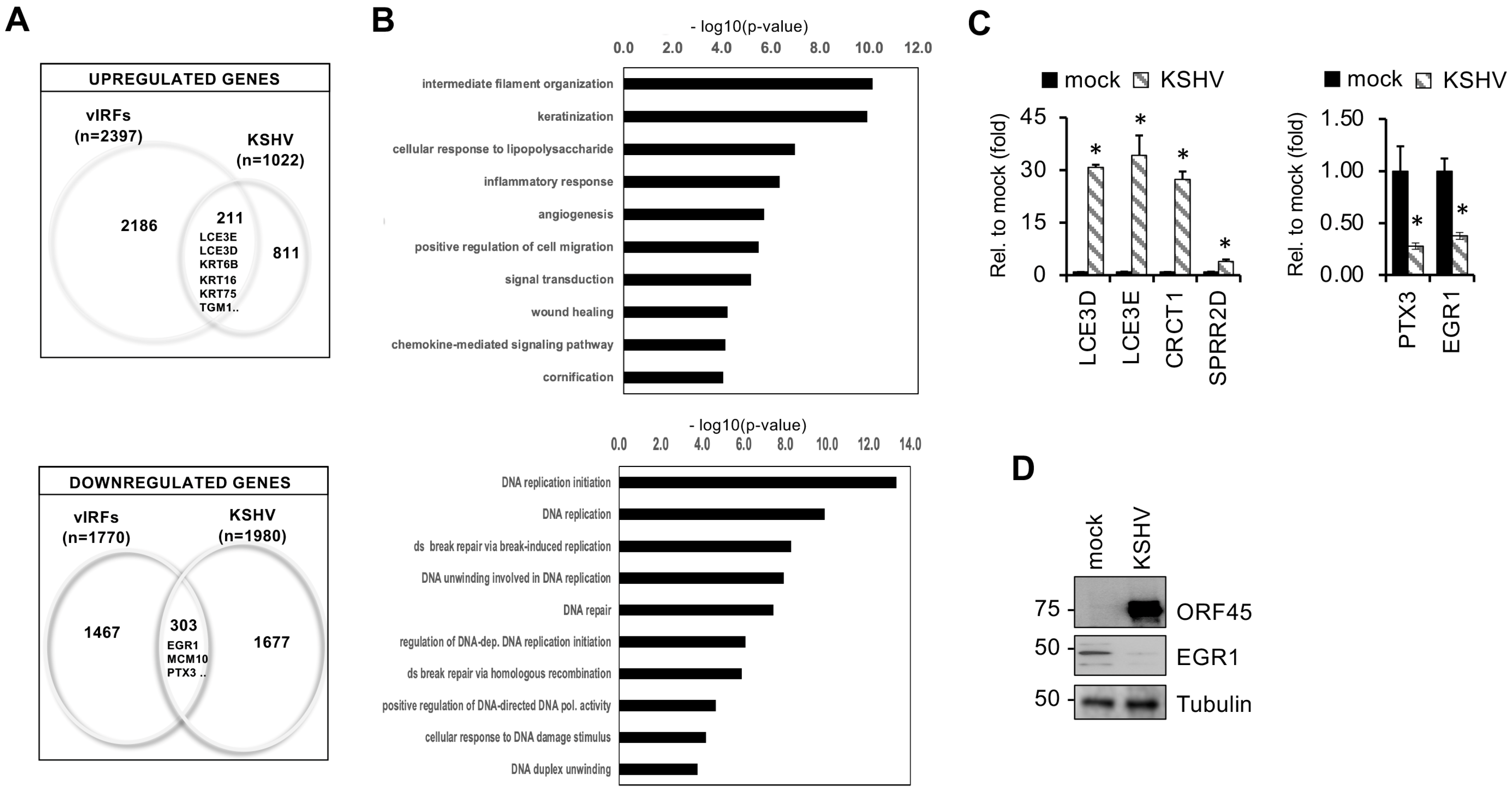
3.3. Identification of Host Genes Regulated by vIRFs in Primary Oral Epithelial Cells
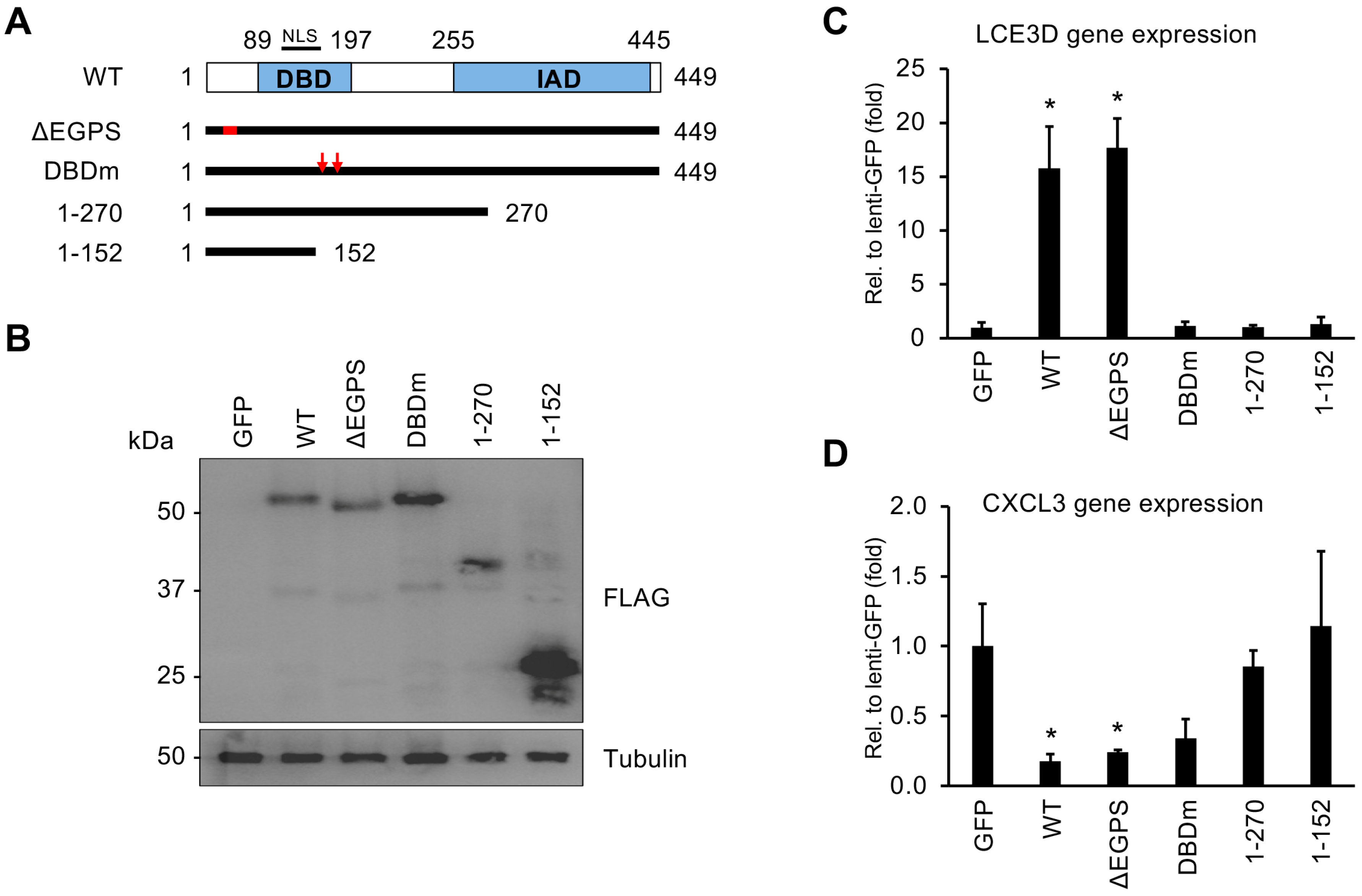
3.4. Both the DNA-Binding Domain and the C-Terminus of vIRF1 Are Required for Its Gene Regulatory Function
3.5. Mutation in the DBD of vIRF1 Compromises vIRF1-Mediated Gene Regulation Genome-Wide
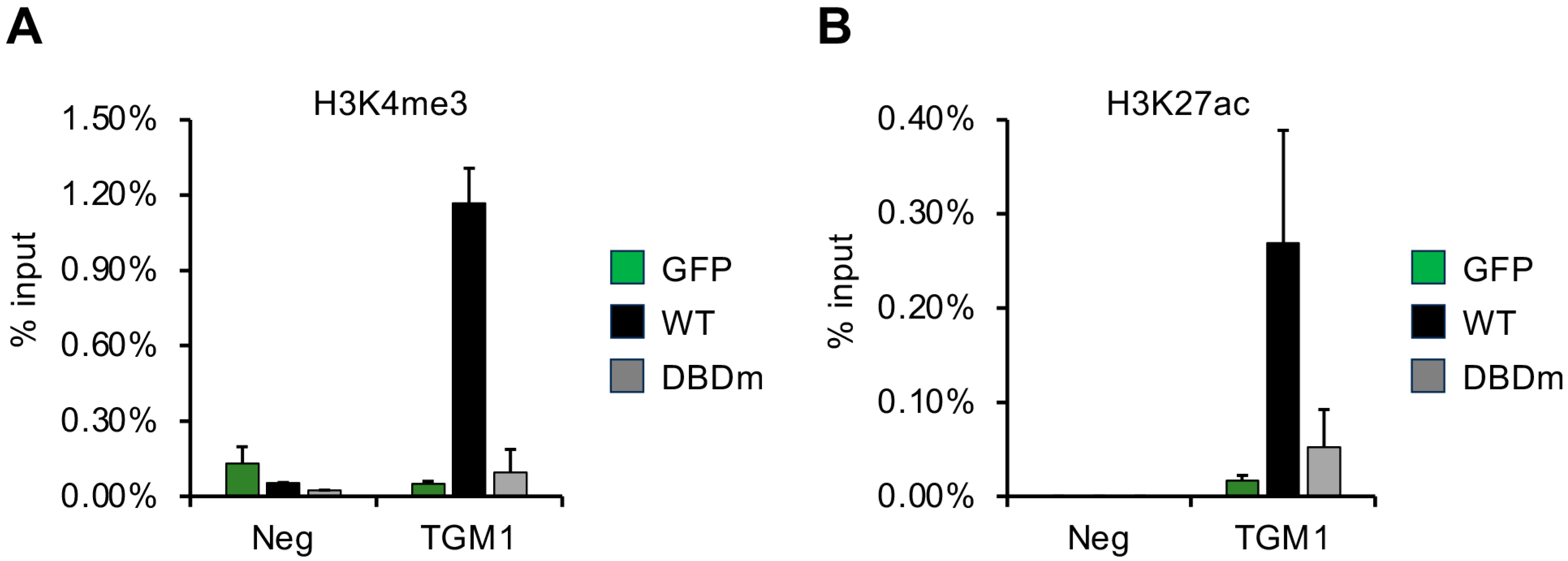
3.6. DNA-Binding Domain of vIRF1 Is Necessary to Promote Accumulation of Activating Histone Modifications on Its Target Gene
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [CrossRef] [PubMed]
- Atyeo, N.; Rodriguez, M.D.; Papp, B.; Toth, Z. Clinical Manifestations and Epigenetic Regulation of Oral Herpesvirus Infections. Viruses 2021, 13, 681. [Google Scholar] [CrossRef] [PubMed]
- Minhas, V.; Wood, C. Epidemiology and transmission of Kaposi’s sarcoma-associated herpesvirus. Viruses 2014, 6, 4178–4194. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhong, C.; Liu, D.; Yu, W.; Chen, W.; Wang, Y.; Shi, S.; Yuan, Y. Evidence for Kaposi Sarcoma Originating from Mesenchymal Stem Cell through KSHV-induced Mesenchymal-to-Endothelial Transition. Cancer Res. 2018, 78, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Rohrmus, B.; Thoma-Greber, E.M.; Bogner, J.R.; Rocken, M. Outlook in oral and cutaneous Kaposi’s sarcoma. Lancet 2000, 356, 2160. [Google Scholar] [CrossRef] [PubMed]
- Phipps, W.; Saracino, M.; Selke, S.; Huang, M.L.; Jaoko, W.; Mandaliya, K.; Wald, A.; Casper, C.; McClelland, R.S. Oral HHV-8 replication among women in Mombasa, Kenya. J. Med. Virol. 2014, 86, 1759–1765. [Google Scholar] [CrossRef] [PubMed]
- Casper, C.; Krantz, E.; Selke, S.; Kuntz, S.R.; Wang, J.; Huang, M.L.; Pauk, J.S.; Corey, L.; Wald, A. Frequent and asymptomatic oropharyngeal shedding of human herpesvirus 8 among immunocompetent men. J. Infect. Dis. 2007, 195, 30–36. [Google Scholar] [CrossRef]
- Duus, K.M.; Lentchitsky, V.; Wagenaar, T.; Grose, C.; Webster-Cyriaque, J. Wild-type Kaposi’s sarcoma-associated herpesvirus isolated from the oropharynx of immune-competent individuals has tropism for cultured oral epithelial cells. J. Virol. 2004, 78, 4074–4084. [Google Scholar] [CrossRef]
- Atyeo, N.; Chae, M.Y.; Toth, Z.; Sharma, A.; Papp, B. Kaposi’s Sarcoma-Associated Herpesvirus Immediate Early Proteins Trigger FOXQ1 Expression in Oral Epithelial Cells, Engaging in a Novel Lytic Cycle-Sustaining Positive Feedback Loop. J. Virol. 2023, 97, e0169622. [Google Scholar] [CrossRef] [PubMed]
- Broussard, G.; Damania, B. KSHV: Immune Modulation and Immunotherapy. Front. Immunol. 2019, 10, 3084. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Damania, B. The viral interferon regulatory factors of KSHV: Immunosuppressors or oncogenes? Front. Immunol. 2011, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Barnard, S.; Blackbourn, D.J.; Davison, A.J. Transcription mapping of human herpesvirus 8 genes encoding viral interferon regulatory factors. J. Gen. Virol. 2003, 84, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Jenner, R.G.; Alba, M.M.; Boshoff, C.; Kellam, P. Kaposi’s sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 2001, 75, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Rivas, C.; Thlick, A.E.; Parravicini, C.; Moore, P.S.; Chang, Y. Kaposi’s sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol. 2001, 75, 429–438. [Google Scholar] [CrossRef]
- Golas, G.; Jang, S.J.; Naik, N.G.; Alonso, J.D.; Papp, B.; Toth, Z. Comparative analysis of the viral interferon regulatory factors of KSHV for their requisite for virus production and inhibition of the type I interferon pathway. Virology 2020, 541, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Kim, M.H.; Lee, J.S.; Liang, C.; Jung, J.U. Viral interferon regulatory factors. J. Interferon Cytokine Res. 2009, 29, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.W.; Kim, D.; Jung, J.U.; Lee, H.R. KSHV-encoded viral interferon regulatory factor 4 (vIRF4) interacts with IRF7 and inhibits interferon alpha production. Biochem. Biophys. Res. Commun. 2017, 486, 700–705. [Google Scholar] [CrossRef]
- Baresova, P.; Pitha, P.M.; Lubyova, B. Distinct roles of Kaposi’s sarcoma-associated herpesvirus-encoded viral interferon regulatory factors in inflammatory response and cancer. J. Virol. 2013, 87, 9398–9410. [Google Scholar] [CrossRef]
- Myoung, J.; Lee, S.A.; Lee, H.R. Beyond Viral Interferon Regulatory Factors: Immune Evasion Strategies. J. Microbiol. Biotechnol. 2019, 29, 1873–1881. [Google Scholar] [CrossRef]
- Li, M.; Damania, B.; Alvarez, X.; Ogryzko, V.; Ozato, K.; Jung, J.U. Inhibition of p300 histone acetyltransferase by viral interferon regulatory factor. Mol. Cell. Biol. 2000, 20, 8254–8263. [Google Scholar] [CrossRef]
- Hew, K.; Dahlroth, S.L.; Venkatachalam, R.; Nasertorabi, F.; Lim, B.T.; Cornvik, T.; Nordlund, P. The crystal structure of the DNA-binding domain of vIRF-1 from the oncogenic KSHV reveals a conserved fold for DNA binding and reinforces its role as a transcription factor. Nucleic Acids Res. 2013, 41, 4295–4306. [Google Scholar] [CrossRef]
- Hu, H.; Dong, J.; Liang, D.; Gao, Z.; Bai, L.; Sun, R.; Zhang, H.; Dong, Y.; Lan, K. Genome-Wide Mapping of the Binding Sites and Structural Analysis of Kaposi’s Sarcoma-Associated Herpesvirus Viral Interferon Regulatory Factor 2 Reveal that It Is a DNA-Binding Transcription Factor. J. Virol. 2016, 90, 1158–1168. [Google Scholar] [CrossRef]
- Park, J.; Lee, M.S.; Yoo, S.M.; Jeong, K.W.; Lee, D.; Choe, J.; Seo, T. Identification of the DNA sequence interacting with Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 1. J. Virol. 2007, 81, 12680–12684. [Google Scholar] [CrossRef]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Chavoshi, S.; Egorova, O.; Lacdao, I.K.; Farhadi, S.; Sheng, Y.; Saridakis, V. Identification of Kaposi Sarcoma Herpesvirus (KSHV) vIRF1 Protein as a Novel Interaction Partner of Human Deubiquitinase USP7. J. Biol. Chem. 2016, 291, 6281–6291. [Google Scholar] [CrossRef]
- Ellison, T.J.; Izumiya, Y.; Izumiya, C.; Luciw, P.A.; Kung, H.J. A comprehensive analysis of recruitment and transactivation potential of K-Rta and K-bZIP during reactivation of Kaposi’s sarcoma-associated herpesvirus. Virology 2009, 387, 76–88. [Google Scholar] [CrossRef]
- Brulois, K.F.; Chang, H.; Lee, A.S.; Ensser, A.; Wong, L.Y.; Toth, Z.; Lee, S.H.; Lee, H.R.; Myoung, J.; Ganem, D.; et al. Construction and manipulation of a new Kaposi’s sarcoma-associated herpesvirus bacterial artificial chromosome clone. J. Virol. 2012, 86, 9708–9720. [Google Scholar] [CrossRef]
- Lee, S.C.; Naik, N.G.; Tombacz, D.; Gulyas, G.; Kakuk, B.; Boldogkoi, Z.; Hall, K.; Papp, B.; Boulant, S.; Toth, Z. Hypoxia and HIF-1alpha promote lytic de novo KSHV infection. J. Virol. 2023, 97, e0097223. [Google Scholar] [CrossRef]
- Papp, B.; Motlagh, N.; Smindak, R.J.; Jin Jang, S.; Sharma, A.; Alonso, J.D.; Toth, Z. Genome-Wide Identification of Direct RTA Targets Reveals Key Host Factors for Kaposi’s Sarcoma-Associated Herpesvirus Lytic Reactivation. J. Virol. 2019, 93, e01978-18. [Google Scholar] [CrossRef]
- Xi, X.; Persson, L.M.; O’Brien, M.W.; Mohr, I.; Wilson, A.C. Cooperation between viral interferon regulatory factor 4 and RTA to activate a subset of Kaposi’s sarcoma-associated herpesvirus lytic promoters. J. Virol. 2012, 86, 1021–1033. [Google Scholar] [CrossRef]
- Sundararaj, S.; Seneviratne, S.; Williams, S.J.; Enders, A.; Casarotto, M.G. Structural determinants of the IRF4/DNA homodimeric complex. Nucleic Acids Res. 2021, 49, 2255–2265. [Google Scholar] [CrossRef]
- Spires, L.M.; Wind, E.; Papp, B.; Toth, Z. KSHV RTA utilizes the host E3 ubiquitin ligase complex RNF20/40 to drive lytic reactivation. J. Virol. 2023, 97, e0138923. [Google Scholar] [CrossRef]
- Duan, W.; Wang, S.; Liu, S.; Wood, C. Characterization of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus-8 ORF57 promoter. Arch. Virol. 2001, 146, 403–413. [Google Scholar] [CrossRef]
- Marenholz, I.; Zirra, M.; Fischer, D.F.; Backendorf, C.; Ziegler, A.; Mischke, D. Identification of human epidermal differentiation complex (EDC)-encoded genes by subtractive hybridization of entire YACs to a gridded keratinocyte cDNA library. Genome Res. 2001, 11, 341–355. [Google Scholar] [CrossRef]
- Seo, T.; Lee, D.; Shim, Y.S.; Angell, J.E.; Chidambaram, N.V.; Kalvakolanu, D.V.; Choe, J. Viral interferon regulatory factor 1 of Kaposi’s sarcoma-associated herpesvirus interacts with a cell death regulator, GRIM19, and inhibits interferon/retinoic acid-induced cell death. J. Virol. 2002, 76, 8797–8807. [Google Scholar] [CrossRef]
- Lee, H.R.; Doganay, S.; Chung, B.; Toth, Z.; Brulois, K.; Lee, S.; Kanketayeva, Z.; Feng, P.; Ha, T.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 4 (vIRF4) targets expression of cellular IRF4 and the Myc gene to facilitate lytic replication. J. Virol. 2014, 88, 2183–2194. [Google Scholar] [CrossRef]
- Manzano, M.; Gunther, T.; Ju, H.; Nicholas, J.; Bartom, E.T.; Grundhoff, A.; Gottwein, E. Kaposi’s Sarcoma-Associated Herpesvirus Drives a Super-Enhancer-Mediated Survival Gene Expression Program in Primary Effusion Lymphoma. mBio 2020, 11, e01457-20. [Google Scholar] [CrossRef]
- Joo, C.H.; Shin, Y.C.; Gack, M.; Wu, L.; Levy, D.; Jung, J.U. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi’s sarcoma-associated herpesvirus viral IRF homolog vIRF3. J. Virol. 2007, 81, 8282–8292. [Google Scholar] [CrossRef]
- Jung, K.L.; Choi, U.Y.; Park, A.; Foo, S.S.; Kim, S.; Lee, S.A.; Jung, J.U. Single-cell analysis of Kaposi’s sarcoma-associated herpesvirus infection in three-dimensional air-liquid interface culture model. PLoS Pathog. 2022, 18, e1010775. [Google Scholar] [CrossRef]
- Seifi, A.; Weaver, E.M.; Whipple, M.E.; Ikoma, M.; Farrenberg, J.; Huang, M.L.; Vieira, J. The lytic activation of KSHV during keratinocyte differentiation is dependent upon a suprabasal position, the loss of integrin engagement, and calcium, but not the interaction of cadherins. Virology 2011, 410, 17–29. [Google Scholar] [CrossRef]
- Johnson, A.S.; Maronian, N.; Vieira, J. Activation of Kaposi’s sarcoma-associated herpesvirus lytic gene expression during epithelial differentiation. J. Virol. 2005, 79, 13769–13777. [Google Scholar] [CrossRef]
- Koch, S.; Damas, M.; Freise, A.; Hage, E.; Dhingra, A.; Ruckert, J.; Gallo, A.; Kremmer, E.; Tegge, W.; Bronstrup, M.; et al. Kaposi’s sarcoma-associated herpesvirus vIRF2 protein utilizes an IFN-dependent pathway to regulate viral early gene expression. PLoS Pathog. 2019, 15, e1007743. [Google Scholar] [CrossRef]
- Kumar, A.; Salemi, M.; Bhullar, R.; Guevara-Plunkett, S.; Lyu, Y.; Wang, K.H.; Izumiya, C.; Campbell, M.; Nakajima, K.I.; Izumiya, Y. Proximity Biotin Labeling Reveals Kaposi’s Sarcoma-Associated Herpesvirus Interferon Regulatory Factor Networks. J. Virol. 2021, 95, e02049-20. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Target | Primer Sequence in 5′ to 3′ Orientation |
|---|---|
| For RT-qPCR | |
| TGM1_F | CAGTGAAACTGCACCTCTACCT |
| TGM1_R | CTCCACTTCCTTCTTGGTCTCC |
| LCE3D_F | ATACCCTCTTCTGGCTTTCACA |
| LCE3D_R | AGGTACAGGGGTAAGGGAACAT |
| LCE3E_F | CTTCTCCTGCCTCTCTGCAC |
| LCE3E_R | TTCTTTGGGGGACACTTGGG |
| CRCT1_F | CCTGACCCCGATGTGATTTTTC |
| CRCT1_R | CAAGCCCGTGTAGACAGTAGG |
| PTX3_F | GGGACAAGCTCTTCATCATGCT |
| PTX3_R | GTCGTCCGTGGCTTGCAGCA |
| CXCL3_F | GTCCGTGGTCACTGAACTGC |
| CXCL3_R | GGGGGACCTTACATTCACACT |
| CXCL8_F | AGCTCTGTGTGAAGGTGCAGT |
| CXCL8_R | TAAATTTGGGGTGGAAAGGTT |
| EGR1_F | CAAGAGGCATACCAAGATCCA |
| EGR1_R | GGACGGGTAGGAAGAGAGAGA |
| HJURP_F | CTGCCCAAGAGCGATTCATCT |
| HJURP_R | AACGTGAAGGTCAGATGTCTG |
| MCM10_F | ACGGCGACGGTGAATCTTAT |
| MCM10_R | TCAGTTGACTGTGATGCGGG |
| SPRR2D_F | TTATCAACAGCAGCAGTGCAAG |
| SPRR2D_R | GCTGTGGACACTTTGGTGATG |
| 18S_F | TTCGAACGTCTGCCCTATCAA |
| 18S_R | GATGTGGTAGCCGTTTCTCAGG |
| For ChIP-qPCR | |
| Neg_F | CAGGATCTCCGAGAATCAGC |
| Neg_R | GAGTTGGGAGAGCTGTCAGG |
| TGM1_F | AGGGAGATCAGAGTAGGAGCAA |
| TGM1_R | AAGGTACTCTGTACCCCTGGAA |
| DBD | vIRF1 | vIRF2 | vIRF3 | vIRF4 | hIRF4 | Sequence identity (%) |
| vIRF1 | 23 | 20 | 17 | 22 | ||
| vIRF2 | 66 | 26 | 31 | 23 | ||
| vIRF3 | 73 | 87 | 25 | 15 | ||
| vIRF4 | 69 | 88 | 77 | 17 | ||
| hIRF4 | 71 | 60 | 61 | 58 | ||
| IAD | vIRF1 | vIRF2 | vIRF3 | vIRF4 | hIRF4 | |
| vIRF1 | 22 | 15 | 15 | 18 | ||
| vIRF2 | 71 | 23 | 17 | 16 | ||
| vIRF3 | 66 | 82 | 25 | 17 | ||
| vIRF4 | 71 | 71 | 62 | 14 | ||
| hIRF4 | 61 | 66 | 68 | 61 | ||
| Structural identity (%) | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, S.J.; Atyeo, N.; Mietzsch, M.; Chae, M.Y.; McKenna, R.; Toth, Z.; Papp, B. Genome-Wide Transcriptional Roles of KSHV Viral Interferon Regulatory Factors in Oral Epithelial Cells. Viruses 2024, 16, 846. https://doi.org/10.3390/v16060846
Jang SJ, Atyeo N, Mietzsch M, Chae MY, McKenna R, Toth Z, Papp B. Genome-Wide Transcriptional Roles of KSHV Viral Interferon Regulatory Factors in Oral Epithelial Cells. Viruses. 2024; 16(6):846. https://doi.org/10.3390/v16060846
Chicago/Turabian StyleJang, Seung Jin, Natalie Atyeo, Mario Mietzsch, Min Y. Chae, Robert McKenna, Zsolt Toth, and Bernadett Papp. 2024. "Genome-Wide Transcriptional Roles of KSHV Viral Interferon Regulatory Factors in Oral Epithelial Cells" Viruses 16, no. 6: 846. https://doi.org/10.3390/v16060846