
cGAS-STING-TBK1 Signaling Promotes Valproic Acid-Responsive Human Cytomegalovirus Immediate-Early Transcription during Infection of Incompletely Differentiated Myeloid Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Cell Viability Assays
2.3. Western Blots
2.4. RNA Isolation and RT-qPCR
2.5. Chromatin Immunoprecipitation (ChIP) Assays
3. Results
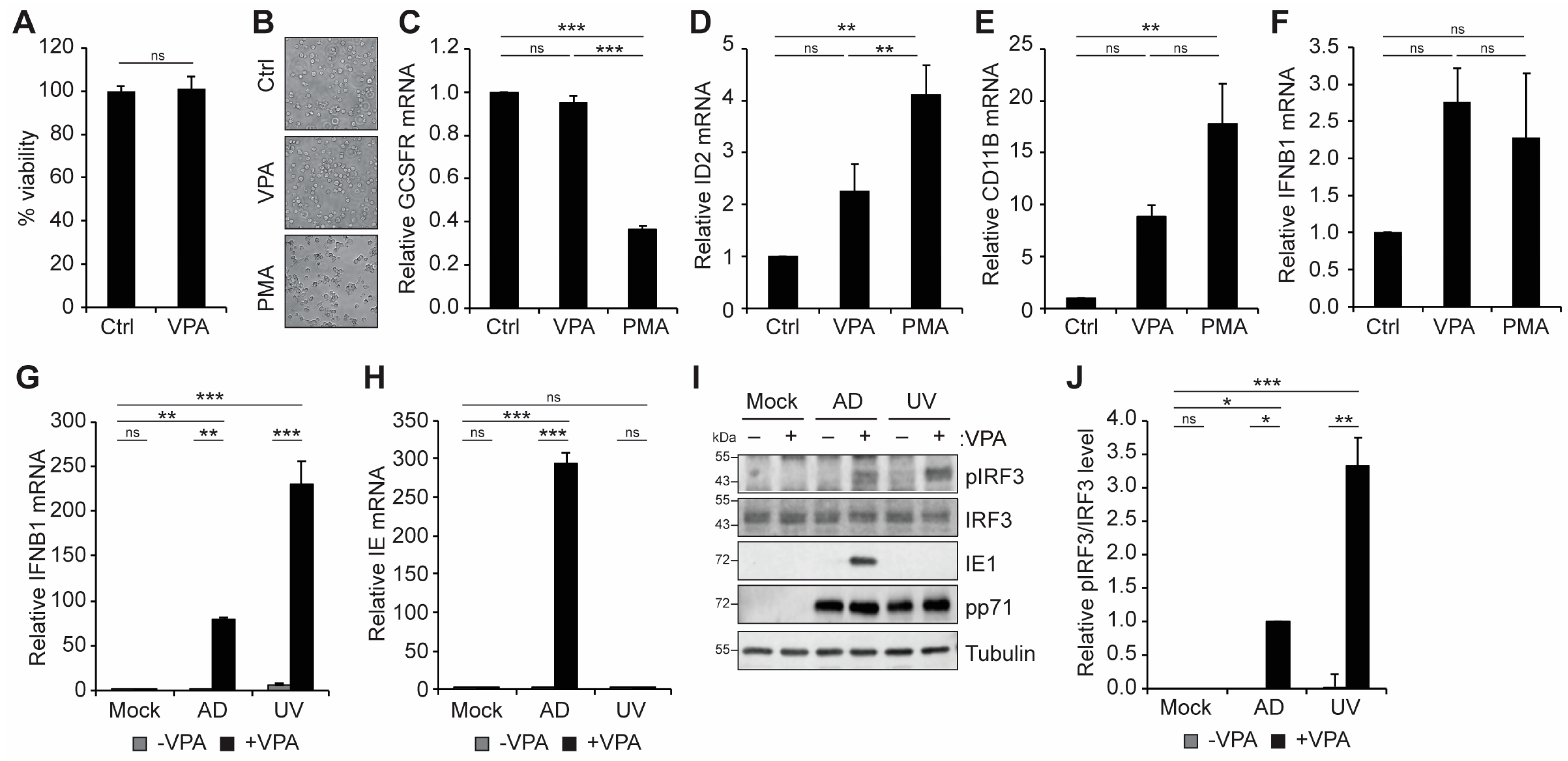
3.1. VPA Treatment Activates an IFN-I Response during HCMV Infection of Incompletely Differentiated Myeloid Cells
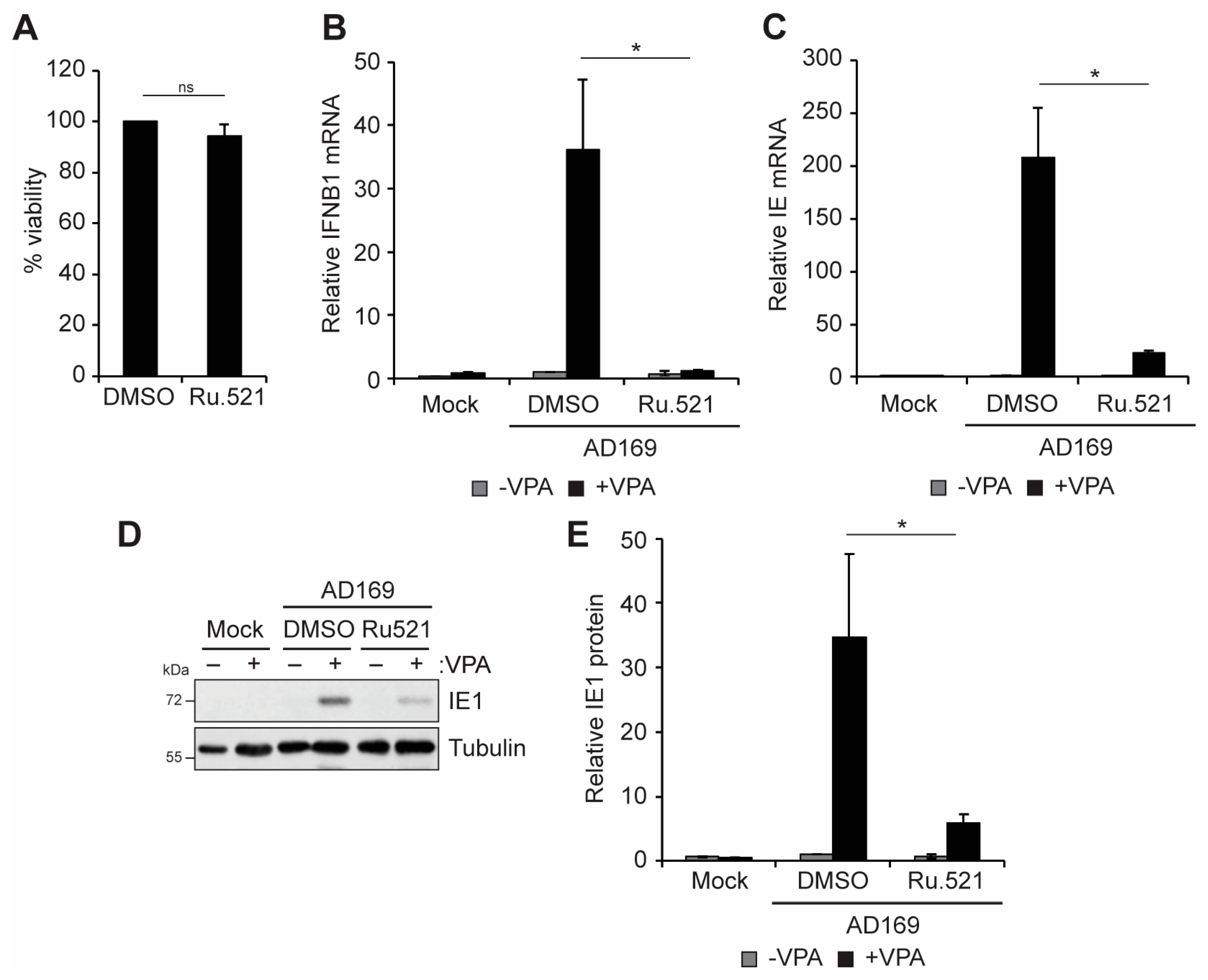
3.2. cGAS and STING Promote VPA-Responsive IFN-I and Viral IE Gene Expression during HCMV Infection of Incompletely Differentiated Myeloid Cells
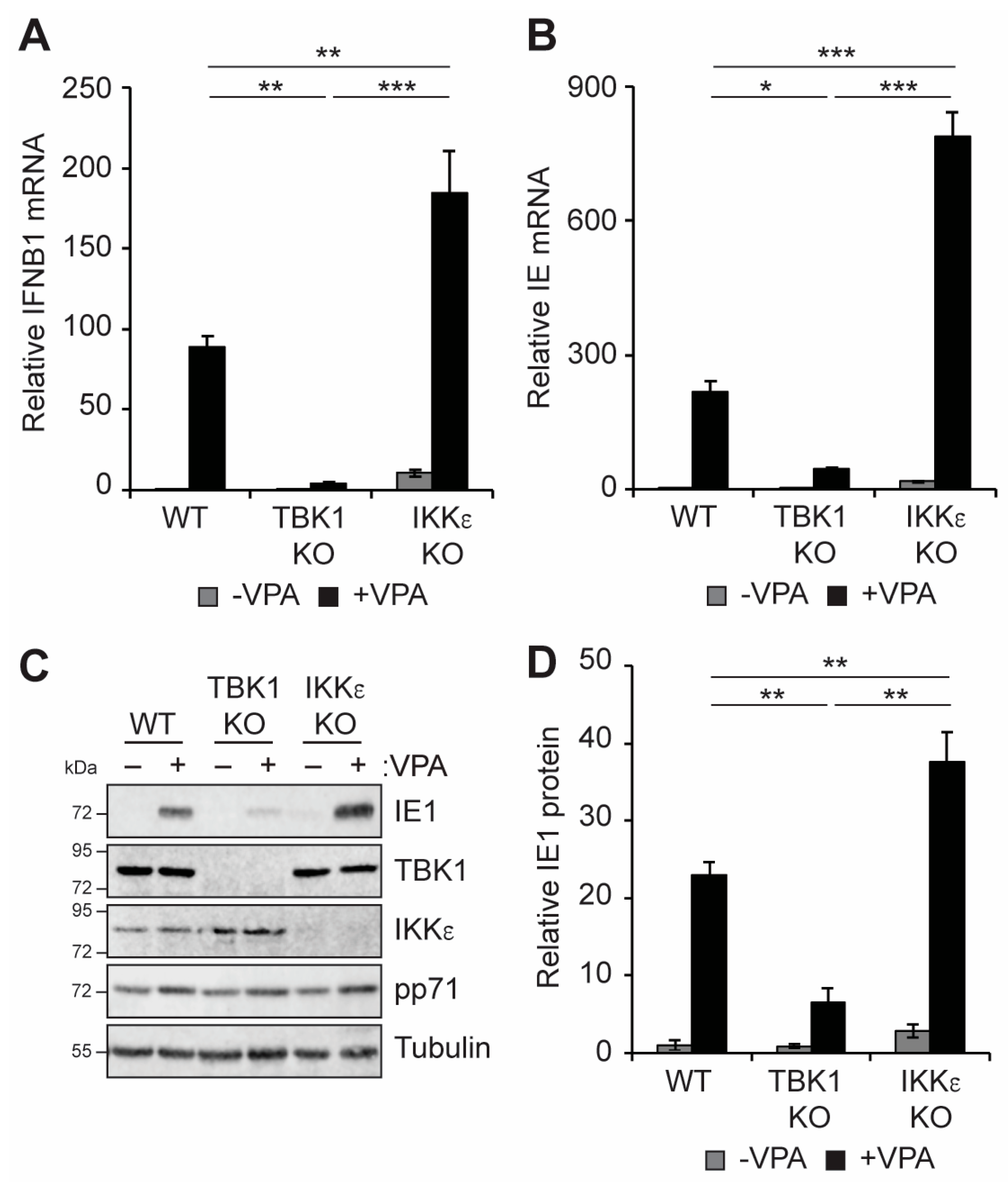
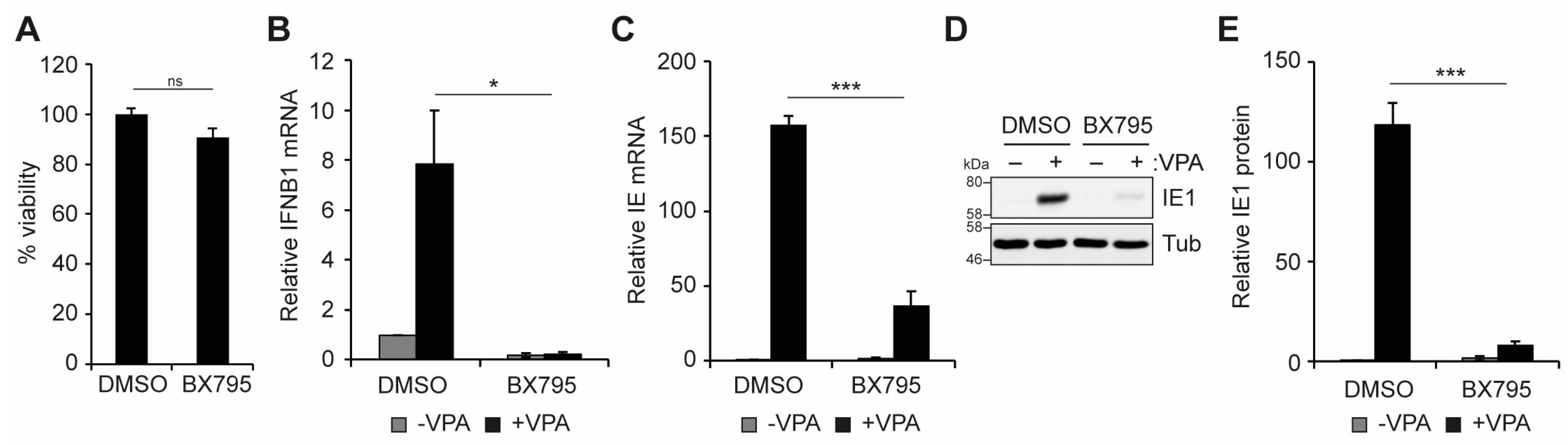
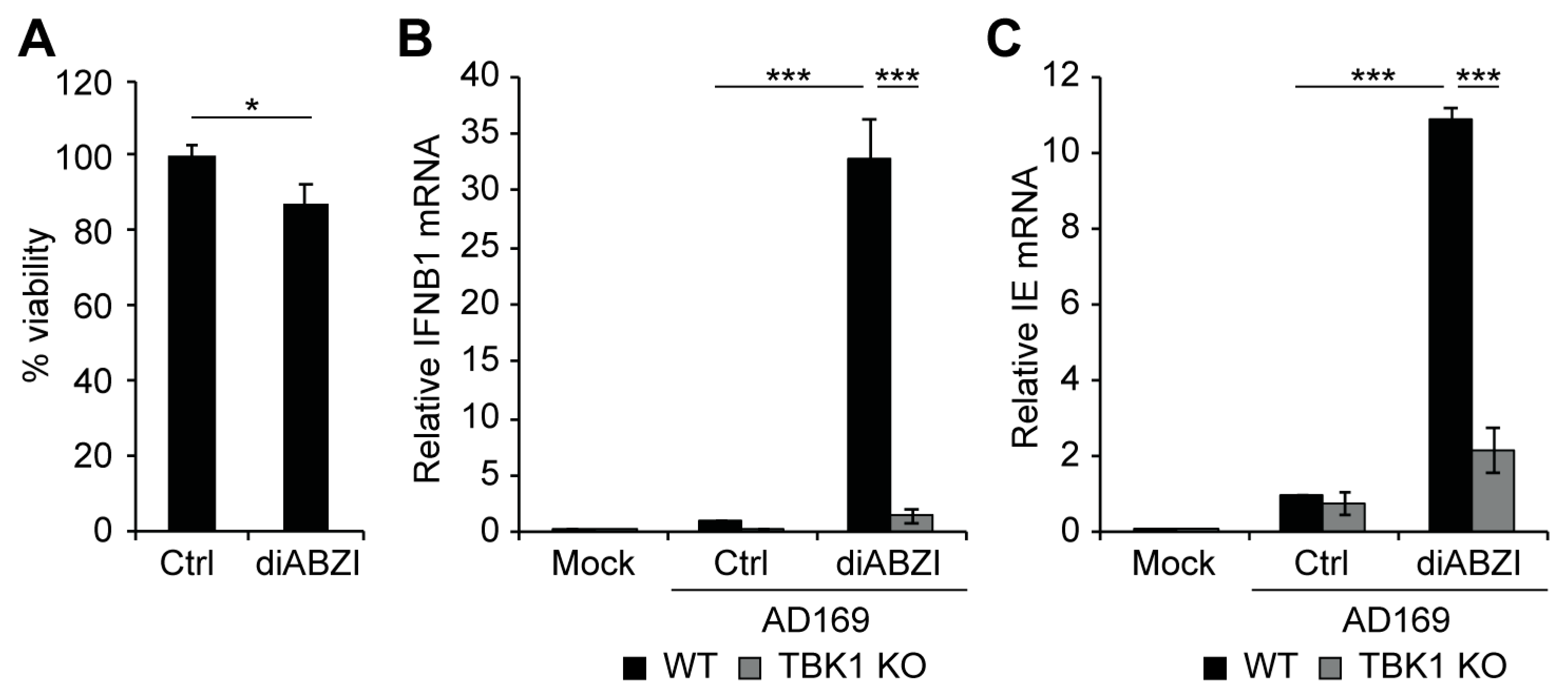
3.3. TBK1 Promotes IFN-I and VPA-Responsive Viral IE Gene Expression during HCMV Infection of Incompletely Differentiated Myeloid Cells
3.4. Activation of STING Is Sufficient to Induce Viral IE Gene Expression in Incompletely Differentiated Myeloid Cells
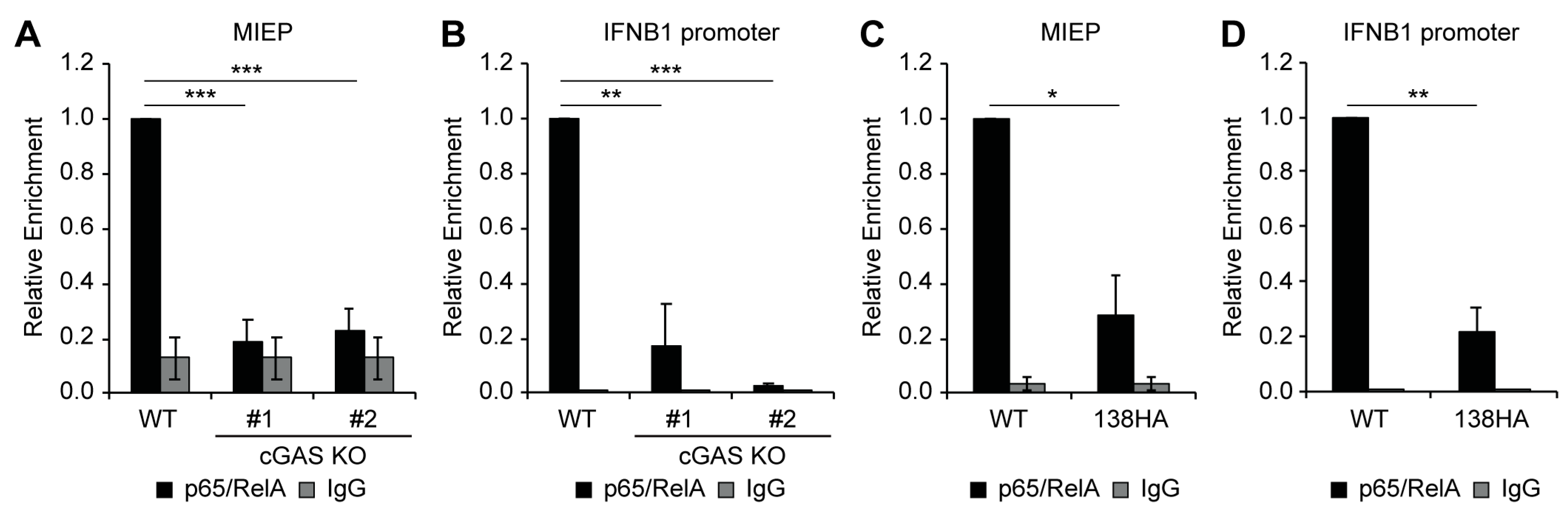
3.5. Loss of cGAS or Expression of UL138 Impairs Recruitment of NFkB to the Viral MIEP and IFNB1 Promoter
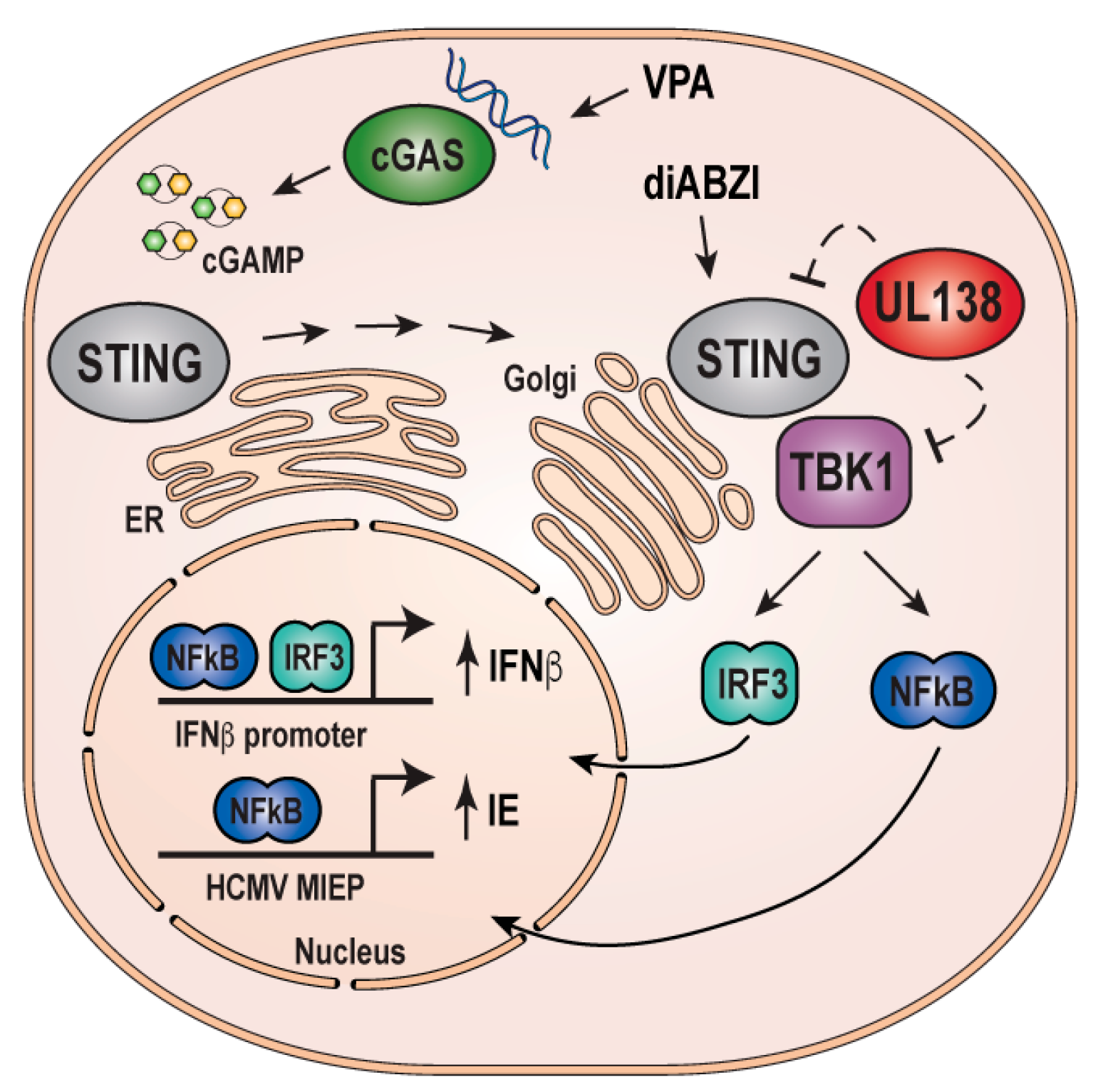
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fulkerson, H.L.; Nogalski, M.T.; Collins-McMillen, D.; Yurochko, A.D. Overview of Human Cytomegalovirus Pathogenesis. In Methods in Molecular Biology; Humana: New York, NY, USA, 2021; Volume 2244, pp. 1–18. [Google Scholar] [CrossRef]
- Griffiths, P.; Baraniak, I.; Reeves, M. The Pathogenesis of Human Cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef]
- Crawford, L.B. Hematopoietic Stem Cells and Betaherpesvirus Latency. Front. Cell. Infect. Microbiol. 2023, 13, 1189805. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, M.; Monard, S.; Sissons, P.; Sinclair, J. Detection of Endogenous Human Cytomegalovirus in CD34+ Bone Marrow Progenitors. J. Gen. Virol. 1996, 77 Pt 12, 3099–3102. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F. The Complex Biology of Human Cytomegalovirus Latency. Adv. Virus Res. 2022, 112, 31–85. [Google Scholar] [CrossRef] [PubMed]
- Wills, M.R.; Poole, E.; Lau, B.; Krishna, B.; Sinclair, J.H. The Immunology of Human Cytomegalovirus Latency: Could Latent Infection Be Cleared by Novel Immunotherapeutic Strategies? Cell. Mol. Immunol. 2015, 12, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.R.; Wills, M.R.; Sinclair, J.H. HCMV Antivirals and Strategies to Target the Latent Reservoir. Viruses 2021, 13, 817. [Google Scholar] [CrossRef] [PubMed]
- Moss, P.; Khan, N. CD8(+) T-Cell Immunity to Cytomegalovirus. Hum. Immunol. 2004, 65, 456–464. [Google Scholar] [CrossRef]
- Klenerman, P.; Oxenius, A. T Cell Responses to Cytomegalovirus. Nat. Rev. Immunol. 2016, 16, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.Y.; Jackson, S.E.; Wills, M.R. The CD4+ T Cell Response to Human Cytomegalovirus in Healthy and Immunocompromised People. Front. Cell. Infect. Microbiol. 2020, 10, 202. [Google Scholar] [CrossRef]
- Dooley, A.L.; O’Connor, C.M. Regulation of the MIE Locus During HCMV Latency and Reactivation. Pathogens 2020, 9, 869. [Google Scholar] [CrossRef]
- Meier, J.L.; Stinski, M.F. Regulation of Human Cytomegalovirus Immediate-Early Gene Expression. Intervirology 1996, 39, 331–342. [Google Scholar] [CrossRef]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly Targeted Human Cytomegalovirus-Specific CD4+ and CD8+ T Cells Dominate the Memory Compartments of Exposed Subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Stinski, M.F.; Meier, J.L. Immediate–Early Viral Gene Regulation and Function. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; ISBN 978-0-521-82714-0. [Google Scholar]
- Adamson, C.S.; Nevels, M.M. Bright and Early: Inhibiting Human Cytomegalovirus by Targeting Major Immediate-Early Gene Expression or Protein Function. Viruses 2020, 12, 110. [Google Scholar] [CrossRef]
- Crough, T.; Khanna, R. Immunobiology of Human Cytomegalovirus: From Bench to Bedside. Clin. Microbiol. Rev. 2009, 22, 76–98. [Google Scholar] [CrossRef] [PubMed]
- Matthews, S.M.; Groves, I.J.; O’Connor, C.M. Chromatin Control of Human Cytomegalovirus Infection. mBio 2023, 14, e0032623. [Google Scholar] [CrossRef]
- Kalejta, R.F.; Albright, E.R. Expanding the Known Functional Repertoire of the Human Cytomegalovirus Pp71 Protein. Front. Cell. Infect. Microbiol. 2020, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Collins-McMillen, D.; Kamil, J.; Moorman, N.; Goodrum, F. Control of Immediate Early Gene Expression for Human Cytomegalovirus Reactivation. Front. Cell. Infect. Microbiol. 2020, 10, 476. [Google Scholar] [CrossRef]
- Elder, E.; Sinclair, J. HCMV Latency: What Regulates the Regulators? Med. Microbiol. Immunol. 2019, 208, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Kalejta, R.F. Human Cytomegalovirus Enters the Primary CD34+ Hematopoietic Progenitor Cells Where It Establishes Latency by Macropinocytosis. J. Virol. 2019, 93, e00452-19. [Google Scholar] [CrossRef]
- Lee, J.-H.; Pasquarella, J.R.; Kalejta, R.F. Cell Line Models for Human Cytomegalovirus Latency Faithfully Mimic Viral Entry by Macropinocytosis and Endocytosis. J. Virol. 2019, 93, e01021-19. [Google Scholar] [CrossRef]
- Saffert, R.T.; Kalejta, R.F. Human Cytomegalovirus Gene Expression Is Silenced by Daxx-Mediated Intrinsic Immune Defense in Model Latent Infections Established in Vitro. J. Virol. 2007, 81, 9109–9120. [Google Scholar] [CrossRef] [PubMed]
- Saffert, R.T.; Penkert, R.R.; Kalejta, R.F. Cellular and Viral Control over the Initial Events of Human Cytomegalovirus Experimental Latency in CD34+ Cells. J. Virol. 2010, 84, 5594–5604. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Humby, M.S.; Miller, W.E.; O’Connor, C.M. Human Cytomegalovirus G Protein-Coupled Receptor US28 Promotes Latency by Attenuating c-Fos. Proc. Natl. Acad. Sci. USA 2019, 116, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Wass, A.B.; O’Connor, C.M. Activator Protein-1 Transactivation of the Major Immediate Early Locus Is a Determinant of Cytomegalovirus Reactivation from Latency. Proc. Natl. Acad. Sci. USA 2020, 117, 20860–20867. [Google Scholar] [CrossRef]
- Lee, S.H.; Albright, E.R.; Lee, J.-H.; Jacobs, D.; Kalejta, R.F. Cellular Defense against Latent Colonization Foiled by Human Cytomegalovirus UL138 Protein. Sci. Adv. 2015, 1, e1501164. [Google Scholar] [CrossRef]
- Elder, E.G.; Krishna, B.A.; Williamson, J.; Lim, E.Y.; Poole, E.; Sedikides, G.X.; Wills, M.; O’Connor, C.M.; Lehner, P.J.; Sinclair, J. Interferon-Responsive Genes Are Targeted during the Establishment of Human Cytomegalovirus Latency. mBio 2019, 10, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Crawford, L.B.; Caposio, P.; Kreklywich, C.; Pham, A.H.; Hancock, M.H.; Jones, T.A.; Smith, P.P.; Yurochko, A.D.; Nelson, J.A.; Streblow, D.N. Human Cytomegalovirus US28 Ligand Binding Activity Is Required for Latency in CD34+ Hematopoietic Progenitor Cells and Humanized NSG Mice. mBio 2019, 10, e01889-19. [Google Scholar] [CrossRef]
- Medica, S.; Crawford, L.B.; Denton, M.; Min, C.-K.; Jones, T.A.; Alexander, T.; Parkins, C.J.; Diggins, N.L.; Streblow, G.J.; Mayo, A.T.; et al. Proximity-Dependent Mapping of the HCMV US28 Interactome Identifies RhoGEF Signaling as a Requirement for Efficient Viral Reactivation. PLoS Pathog. 2023, 19, e1011682. [Google Scholar] [CrossRef] [PubMed]
- Elder, E.G.; Krishna, B.A.; Poole, E.; Perera, M.; Sinclair, J. Regulation of Host and Viral Promoters during Human Cytomegalovirus Latency via US28 and CTCF. J. Gen. Virol. 2021, 102, 001609. [Google Scholar] [CrossRef]
- Krishna, B.A.; Lau, B.; Jackson, S.E.; Wills, M.R.; Sinclair, J.H.; Poole, E. Transient Activation of Human Cytomegalovirus Lytic Gene Expression during Latency Allows Cytotoxic T Cell Killing of Latently Infected Cells. Sci. Rep. 2016, 6, 24674. [Google Scholar] [CrossRef]
- Groves, I.J.; Jackson, S.E.; Poole, E.L.; Nachshon, A.; Rozman, B.; Schwartz, M.; Prinjha, R.K.; Tough, D.F.; Sinclair, J.H.; Wills, M.R. Bromodomain Proteins Regulate Human Cytomegalovirus Latency and Reactivation Allowing Epigenetic Therapeutic Intervention. Proc. Natl. Acad. Sci. USA 2021, 118, e2023025118. [Google Scholar] [CrossRef]
- Murphy, J.C.; Fischle, W.; Verdin, E.; Sinclair, J.H. Control of Cytomegalovirus Lytic Gene Expression by Histone Acetylation. EMBO J. 2002, 21, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Albright, E.R.; Walter, R.M.; Saffert, R.T.; Kalejta, R.F. NFκB and Cyclic AMP Response Element Sites Mediate the Valproic Acid and UL138 Responsiveness of the Human Cytomegalovirus Major Immediate Early Enhancer and Promoter. J. Virol. 2023, 97, e0002923. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.L. Reactivation of the Human Cytomegalovirus Major Immediate-Early Regulatory Region and Viral Replication in Embryonal NTera2 Cells: Role of Trichostatin A, Retinoic Acid, and Deletion of the 21-Base-Pair Repeats and Modulator. J. Virol. 2001, 75, 1581–1593. [Google Scholar] [CrossRef]
- Nehme, Z.; Pasquereau, S.; Herbein, G. Control of Viral Infections by Epigenetic-Targeted Therapy. Clin. Epigenet. 2019, 11, 55. [Google Scholar] [CrossRef]
- Albright, E.R.; Mickelson, C.K.; Kalejta, R.F. Human Cytomegalovirus UL138 Protein Inhibits the STING Pathway and Reduces Interferon Beta mRNA Accumulation during Lytic and Latent Infections. mBio 2021, 12, e0226721. [Google Scholar] [CrossRef] [PubMed]
- Mosallanejad, K.; Kagan, J.C. Control of Innate Immunity by the cGAS-STING Pathway. Immunol. Cell Biol. 2022, 100, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Damania, B. The cGAS-STING Defense Pathway and Its Counteraction by Viruses. Cell Host Microbe 2016, 19, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.-P.; Hornung, V. Molecular Mechanisms and Cellular Functions of cGAS-STING Signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Yum, S.; Li, M.; Fang, Y.; Chen, Z.J. TBK1 Recruitment to STING Activates Both IRF3 and NF-κB That Mediate Immune Defense against Tumors and Viral Infections. Proc. Natl. Acad. Sci. USA 2021, 118, e2100225118. [Google Scholar] [CrossRef]
- Balka, K.R.; Louis, C.; Saunders, T.L.; Smith, A.M.; Calleja, D.J.; D’Silva, D.B.; Moghaddas, F.; Tailler, M.; Lawlor, K.E.; Zhan, Y.; et al. TBK1 and IKKε Act Redundantly to Mediate STING-Induced NF-κB Responses in Myeloid Cells. Cell Rep. 2020, 31, 107492. [Google Scholar] [CrossRef]
- Abe, T.; Barber, G.N. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF-κB Activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Mann, C.C.; Orzalli, M.H.; King, D.S.; Kagan, J.C.; Lee, A.S.Y.; Kranzusch, P.J. Modular Architecture of the STING C-Terminal Tail Allows Interferon and NF-κB Signaling Adaptation. Cell Rep. 2019, 27, 1165–1175. [Google Scholar] [CrossRef]
- Möser, C.V.; Stephan, H.; Altenrath, K.; Kynast, K.L.; Russe, O.Q.; Olbrich, K.; Geisslinger, G.; Niederberger, E. TANK-Binding Kinase 1 (TBK1) Modulates Inflammatory Hyperalgesia by Regulating MAP Kinases and NF-κB Dependent Genes. J. Neuroinflammation 2015, 12, 100. [Google Scholar] [CrossRef]
- Mankan, A.K.; Schmidt, T.; Chauhan, D.; Goldeck, M.; Höning, K.; Gaidt, M.; Kubarenko, A.V.; Andreeva, L.; Hopfner, K.-P.; Hornung, V. Cytosolic RNA:DNA Hybrids Activate the cGAS-STING Axis. EMBO J. 2014, 33, 2937–2946. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Shen, Y.; Shenk, T. Human Cytomegalovirus IE1 and IE2 Proteins Block Apoptosis. J. Virol. 1995, 69, 7960–7970. [Google Scholar] [CrossRef]
- Kalejta, R.F.; Bechtel, J.T.; Shenk, T. Human Cytomegalovirus Pp71 Stimulates Cell Cycle Progression by Inducing the Proteasome-Dependent Degradation of the Retinoblastoma Family of Tumor Suppressors. Mol. Cell. Biol. 2003, 23, 1885–1895. [Google Scholar] [CrossRef]
- Benjamin, D.N.; O’Donovan, T.R.; Laursen, K.B.; Orfali, N.; Cahill, M.R.; Mongan, N.P.; Gudas, L.J.; McKenna, S.L. All-Trans-Retinoic Acid Combined With Valproic Acid Can Promote Differentiation in Myeloid Leukemia Cells by an Autophagy Dependent Mechanism. Front. Oncol. 2022, 12, 848517. [Google Scholar] [CrossRef] [PubMed]
- Albright, E.R.; Kalejta, R.F. Canonical and Variant Forms of Histone H3 Are Deposited onto the Human Cytomegalovirus Genome during Lytic and Latent Infections. J. Virol. 2016, 90, 10309–10320. [Google Scholar] [CrossRef]
- Winkler, L.L.; Kalejta, R.F. The 19S Proteasome Activator Promotes Human Cytomegalovirus Immediate Early Gene Expression through Proteolytic and Nonproteolytic Mechanisms. J. Virol. 2014, 88, 11782–11790. [Google Scholar] [CrossRef]
- Gelbmann, C.B.; Kalejta, R.F. The Golgi Sorting Motifs of Human Cytomegalovirus UL138 Are Not Required for Latency Maintenance. Virus Res. 2019, 270, 197646. [Google Scholar] [CrossRef]
- Lee, S.H.; Caviness, K.; Albright, E.R.; Lee, J.-H.; Gelbmann, C.B.; Rak, M.; Goodrum, F.; Kalejta, R.F. Long and Short Isoforms of the Human Cytomegalovirus UL138 Protein Silence IE Transcription and Promote Latency. J. Virol. 2016, 90, 9483–9494. [Google Scholar] [CrossRef] [PubMed]
- Schwende, H.; Fitzke, E.; Ambs, P.; Dieter, P. Differences in the State of Differentiation of THP-1 Cells Induced by Phorbol Ester and 1,25-Dihydroxyvitamin D3. J. Leukoc. Biol. 1996, 59, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, S.; Kobayashi, Y.; Goto, Y.; Okumura, H.; Nakae, S.; Konno, T.; Tada, K. Induction of Maturation in Cultured Human Monocytic Leukemia Cells by a Phorbol Diester. Cancer Res. 1982, 42, 1530–1536. [Google Scholar] [PubMed]
- Guo, Y.-K.; Ming, S.-L.; Zeng, L.; Chang, W.-R.; Pan, J.-J.; Zhang, C.; Wan, B.; Wang, J.; Su, Y.; Yang, G.-Y.; et al. Inhibition of Histone Deacetylase 1 Suppresses Pseudorabies Virus Infection through cGAS-STING Antiviral Innate Immunity. Mol. Immunol. 2021, 136, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Paijo, J.; Döring, M.; Spanier, J.; Grabski, E.; Nooruzzaman, M.; Schmidt, T.; Witte, G.; Messerle, M.; Hornung, V.; Kaever, V.; et al. cGAS Senses Human Cytomegalovirus and Induces Type I Interferon Responses in Human Monocyte-Derived Cells. PLoS Pathog. 2016, 12, e1005546. [Google Scholar] [CrossRef]
- Vincent, J.; Adura, C.; Gao, P.; Luz, A.; Lama, L.; Asano, Y.; Okamoto, R.; Imaeda, T.; Aida, J.; Rothamel, K.; et al. Small Molecule Inhibition of cGAS Reduces Interferon Expression in Primary Macrophages from Autoimmune Mice. Nat. Commun. 2017, 8, 750. [Google Scholar] [CrossRef] [PubMed]
- Wiser, C.; Kim, B.; Vincent, J.; Ascano, M. Small Molecule Inhibition of Human cGAS Reduces Total cGAMP Output and Cytokine Expression in Cells. Sci. Rep. 2020, 10, 7604. [Google Scholar] [CrossRef]
- Clark, K.; Plater, L.; Peggie, M.; Cohen, P. Use of the Pharmacological Inhibitor BX795 to Study the Regulation and Physiological Roles of TBK1 and IkappaB Kinase Epsilon: A Distinct Upstream Kinase Mediates Ser-172 Phosphorylation and Activation. J. Biol. Chem. 2009, 284, 14136–14146. [Google Scholar] [CrossRef]
- Ramanjulu, J.M.; Pesiridis, G.S.; Yang, J.; Concha, N.; Singhaus, R.; Zhang, S.-Y.; Tran, J.-L.; Moore, P.; Lehmann, S.; Eberl, H.C.; et al. Design of Amidobenzimidazole STING Receptor Agonists with Systemic Activity. Nature 2018, 564, 439–443. [Google Scholar] [CrossRef]
- Gustems, M.; Borst, E.; Benedict, C.A.; Pérez, C.; Messerle, M.; Ghazal, P.; Angulo, A. Regulation of the Transcription and Replication Cycle of Human Cytomegalovirus Is Insensitive to Genetic Elimination of the Cognate NF-κB Binding Sites in the Enhancer. J. Virol. 2006, 80, 9899–9904. [Google Scholar] [CrossRef]
- Rozman, B.; Nachshon, A.; Levi Samia, R.; Lavi, M.; Schwartz, M.; Stern-Ginossar, N. Temporal Dynamics of HCMV Gene Expression in Lytic and Latent Infections. Cell Rep. 2022, 39, 110653. [Google Scholar] [CrossRef]
- Schwartz, M.; Shnayder, M.; Nachshon, A.; Arazi, T.; Kitsberg, Y.; Levi Samia, R.; Lavi, M.; Kuint, R.; Tsabari, R.; Stern-Ginossar, N. Molecular Characterization of Human Cytomegalovirus Infection with Single-Cell Transcriptomics. Nat. Microbiol. 2023, 8, 455–468. [Google Scholar] [CrossRef]
- Horan, K.A.; Hansen, K.; Jakobsen, M.R.; Holm, C.K.; Søby, S.; Unterholzner, L.; Thompson, M.; West, J.A.; Iversen, M.B.; Rasmussen, S.B.; et al. Proteasomal Degradation of Herpes Simplex Virus Capsids in Macrophages Releases DNA to the Cytosol for Recognition by DNA Sensors. J. Immunol. 2013, 190, 2311–2319. [Google Scholar] [CrossRef]
- Aguirre, S.; Luthra, P.; Sanchez-Aparicio, M.T.; Maestre, A.M.; Patel, J.; Lamothe, F.; Fredericks, A.C.; Tripathi, S.; Zhu, T.; Pintado-Silva, J.; et al. Dengue Virus NS2B Protein Targets cGAS for Degradation and Prevents Mitochondrial DNA Sensing during Infection. Nat. Microbiol. 2017, 2, 17037. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, Z.; Xue, Q.; Yang, F.; Li, Z.; Xue, Z.; Cao, W.; He, J.; Guo, J.; Liu, X.; et al. Innate Sensing of Picornavirus Infection Involves cGAS-STING-Mediated Antiviral Responses Triggered by Mitochondrial DNA Release. PLoS Pathog. 2023, 19, e1011132. [Google Scholar] [CrossRef]
- Moriyama, M.; Koshiba, T.; Ichinohe, T. Influenza A Virus M2 Protein Triggers Mitochondrial DNA-Mediated Antiviral Immune Responses. Nat. Commun. 2019, 10, 4624. [Google Scholar] [CrossRef]
- Jahun, A.S.; Sorgeloos, F.; Chaudhry, Y.; Arthur, S.E.; Hosmillo, M.; Georgana, I.; Izuagbe, R.; Goodfellow, I.G. Leaked Genomic and Mitochondrial DNA Contribute to the Host Response to Noroviruses in a STING-Dependent Manner. Cell Rep. 2023, 42, 112179. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Volkman, H.E.; Cambier, S.; Gray, E.E.; Stetson, D.B. Tight Nuclear Tethering of cGAS Is Essential for Preventing Autoreactivity. eLife 2019, 8, e47491. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear cGAS Suppresses DNA Repair and Promotes Tumorigenesis. Nature 2018, 563, 131–136. [Google Scholar] [CrossRef]
- Jiang, H.; Xue, X.; Panda, S.; Kawale, A.; Hooy, R.M.; Liang, F.; Sohn, J.; Sung, P.; Gekara, N.O. Chromatin-Bound cGAS Is an Inhibitor of DNA Repair and Hence Accelerates Genome Destabilization and Cell Death. EMBO J. 2019, 38, e102718. [Google Scholar] [CrossRef]
- Chen, H.; Chen, H.; Zhang, J.; Wang, Y.; Simoneau, A.; Yang, H.; Levine, A.S.; Zou, L.; Chen, Z.; Lan, L. cGAS Suppresses Genomic Instability as a Decelerator of Replication Forks. Sci. Adv. 2020, 6, eabb8941. [Google Scholar] [CrossRef]
- Wu, Y.; Song, K.; Hao, W.; Li, J.; Wang, L.; Li, S. Nuclear Soluble cGAS Senses Double-Stranded DNA Virus Infection. Commun. Biol. 2022, 5, 433. [Google Scholar] [CrossRef]
- Cui, S.; Yu, Q.; Chu, L.; Cui, Y.; Ding, M.; Wang, Q.; Wang, H.; Chen, Y.; Liu, X.; Wang, C. Nuclear cGAS Functions Non-Canonically to Enhance Antiviral Immunity via Recruiting Methyltransferase Prmt5. Cell Rep. 2020, 33, 108490. [Google Scholar] [CrossRef]
- Gentili, M.; Kowal, J.; Tkach, M.; Satoh, T.; Lahaye, X.; Conrad, C.; Boyron, M.; Lombard, B.; Durand, S.; Kroemer, G.; et al. Transmission of Innate Immune Signaling by Packaging of cGAMP in Viral Particles. Science 2015, 349, 1232–1236. [Google Scholar] [CrossRef]
- Weekes, M.P.; Tan, S.Y.L.; Poole, E.; Talbot, S.; Antrobus, R.; Smith, D.L.; Montag, C.; Gygi, S.P.; Sinclair, J.H.; Lehner, P.J. Latency-Associated Degradation of the MRP1 Drug Transporter during Latent Human Cytomegalovirus Infection. Science 2013, 340, 199–202. [Google Scholar] [CrossRef]
- Gelbmann, C.B.; Kalejta, R.F. The Membrane-Spanning Peptide and Acidic Cluster Dileucine Sorting Motif of UL138 Are Required To Downregulate MRP1 Drug Transporter Function in Human Cytomegalovirus-Infected Cells. J. Virol. 2019, 93, e00430-19. [Google Scholar] [CrossRef]
- Maltbaek, J.H.; Cambier, S.; Snyder, J.M.; Stetson, D.B. ABCC1 Transporter Exports the Immunostimulatory Cyclic Dinucleotide cGAMP. Immunity 2022, 55, 1799–1812. [Google Scholar] [CrossRef]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 Is an Innate Immune Sensor for Intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef]
- Diner, B.A.; Lum, K.K.; Cristea, I.M. The Emerging Role of Nuclear Viral DNA Sensors. J. Biol. Chem. 2015, 290, 26412–26421. [Google Scholar] [CrossRef] [PubMed]
- Dell’Oste, V.; Gatti, D.; Giorgio, A.G.; Gariglio, M.; Landolfo, S.; De Andrea, M. The Interferon-Inducible DNA-Sensor Protein IFI16: A Key Player in the Antiviral Response. New Microbiol. 2015, 38, 5–20. [Google Scholar] [PubMed]
- Orzalli, M.H.; Conwell, S.E.; Berrios, C.; DeCaprio, J.A.; Knipe, D.M. Nuclear Interferon-Inducible Protein 16 Promotes Silencing of Herpesviral and Transfected DNA. Proc. Natl. Acad. Sci. USA 2013, 110, E4492–E4501. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D. Dynamic Response of IFI16 and Promyelocytic Leukemia Nuclear Body Components to Herpes Simplex Virus 1 Infection. J. Virol. 2016, 90, 167–179. [Google Scholar] [CrossRef]
- Johnson, K.E.; Bottero, V.; Flaherty, S.; Dutta, S.; Singh, V.V.; Chandran, B. IFI16 Restricts HSV-1 Replication by Accumulating on the Hsv-1 Genome, Repressing HSV-1 Gene Expression, and Directly or Indirectly Modulating Histone Modifications. PLoS Pathog. 2014, 10, e1004503. [Google Scholar] [CrossRef] [PubMed]
- Howard, T.R.; Lum, K.K.; Kennedy, M.A.; Cristea, I.M. The Nuclear DNA Sensor IFI16 Indiscriminately Binds to and Diminishes Accessibility of the HSV-1 Genome to Suppress Infection. mSystems 2022, 7, e0019822. [Google Scholar] [CrossRef] [PubMed]
- Sodroski, C.N.; Knipe, D.M. Nuclear Interferon-Stimulated Gene Product Maintains Heterochromatin on the Herpes Simplex Viral Genome to Limit Lytic Infection. Proc. Natl. Acad. Sci. USA 2023, 120, e2310996120. [Google Scholar] [CrossRef]
- Jønsson, K.L.; Laustsen, A.; Krapp, C.; Skipper, K.A.; Thavachelvam, K.; Hotter, D.; Egedal, J.H.; Kjolby, M.; Mohammadi, P.; Prabakaran, T.; et al. IFI16 Is Required for DNA Sensing in Human Macrophages by Promoting Production and Function of cGAMP. Nat. Commun. 2017, 8, 14391. [Google Scholar] [CrossRef]
- Almine, J.F.; O’Hare, C.A.J.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS Cooperate in the Activation of STING during DNA Sensing in Human Keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef]
- Diner, B.A.; Lum, K.K.; Toettcher, J.E.; Cristea, I.M. Viral DNA Sensors IFI16 and Cyclic GMP-AMP Synthase Possess Distinct Functions in Regulating Viral Gene Expression, Immune Defenses, and Apoptotic Responses during Herpesvirus Infection. mBio 2016, 7, 10–1128. [Google Scholar] [CrossRef]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jønsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-Canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κB Signaling after Nuclear DNA Damage. Mol. Cell 2018, 71, 745–760. [Google Scholar] [CrossRef] [PubMed]
- Humby, M.S.; O’Connor, C.M. Human Cytomegalovirus US28 Is Important for Latent Infection of Hematopoietic Progenitor Cells. J. Virol. 2015, 90, 2959–2970. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.R.; Roche, K.L.; Murphy, E.A.; Sinclair, J.H. A Viral Long Non-Coding RNA Protects against Cell Death during Human Cytomegalovirus Infection of CD14+ Monocytes. Viruses 2022, 14, 246. [Google Scholar] [CrossRef]
- Perera, M.R.; Sinclair, J.H. The Human Cytomegalovirus Β2.7 Long Non-Coding RNA Prevents Induction of Reactive Oxygen Species to Maintain Viral Gene Silencing during Latency. Int. J. Mol. Sci. 2022, 23, 11017. [Google Scholar] [CrossRef]
- Andrade, B.; Jara-Gutiérrez, C.; Paz-Araos, M.; Vázquez, M.C.; Díaz, P.; Murgas, P. The Relationship between Reactive Oxygen Species and the cGAS/STING Signaling Pathway in the Inflammaging Process. Int. J. Mol. Sci. 2022, 23, 15182. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; McWhirter, S.M.; Faia, K.L.; Rowe, D.C.; Latz, E.; Golenbock, D.T.; Coyle, A.J.; Liao, S.-M.; Maniatis, T. IKKepsilon and TBK1 Are Essential Components of the IRF3 Signaling Pathway. Nat. Immunol. 2003, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Antonia, R.J.; Hagan, R.S.; Baldwin, A.S. Expanding the View of IKK: New Substrates and New Biology. Trends Cell Biol. 2021, 31, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Matsuoka, M.; Chang, T.-H.; Otsuki, N.; Noda, M.; Kimura, H.; Sakai, K.; Kato, H.; Takeda, M.; Kubota, T. Functionally Distinct Effects of the C-Terminal Regions of IKKε and TBK1 on Type I IFN Production. PLoS ONE 2014, 9, e94999. [Google Scholar] [CrossRef] [PubMed]
- Sainz, B.; LaMarca, H.L.; Garry, R.F.; Morris, C.A. Synergistic Inhibition of Human Cytomegalovirus Replication by Interferon-Alpha/Beta and Interferon-Gamma. Virol. J. 2005, 2, 14. [Google Scholar] [CrossRef]
- Paulus, C.; Krauss, S.; Nevels, M. A Human Cytomegalovirus Antagonist of Type I IFN-Dependent Signal Transducer and Activator of Transcription Signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 3840–3845. [Google Scholar] [CrossRef]
- Chowdhury, S.; Latham, K.A.; Tran, A.C.; Carroll, C.J.; Stanton, R.J.; Weekes, M.P.; Neil, S.J.D.; Swanson, C.M.; Strang, B.L. Inhibition of Human Cytomegalovirus Replication by Interferon Alpha Can Involve Multiple Anti-Viral Factors. J. Gen. Virol. 2023, 104, 001929. [Google Scholar] [CrossRef]
- Li, Y.; Shang, W.; Xiao, G.; Zhang, L.-K.; Zheng, C. A Comparative Quantitative Proteomic Analysis of HCMV-Infected Cells Highlights pUL138 as a Multifunctional Protein. Molecules 2020, 25, 2520. [Google Scholar] [CrossRef]
- Zarrella, K.; Longmire, P.; Zeltzer, S.; Collins-McMillen, D.; Hancock, M.; Buehler, J.; Reitsma, J.M.; Terhune, S.S.; Nelson, J.A.; Goodrum, F. Human Cytomegalovirus UL138 Interaction with USP1 Activates STAT1 in Infection. PLoS Pathog. 2023, 19, e1011185. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of Type-I- and Type-II-Interferon-Mediated Signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Majoros, A.; Platanitis, E.; Kernbauer-Hölzl, E.; Rosebrock, F.; Müller, M.; Decker, T. Canonical and Non-Canonical Aspects of JAK-STAT Signaling: Lessons from Interferons for Cytokine Responses. Front. Immunol. 2017, 8, 29. [Google Scholar] [CrossRef]
- Philips, R.L.; Wang, Y.; Cheon, H.; Kanno, Y.; Gadina, M.; Sartorelli, V.; Horvath, C.M.; Darnell, J.E.; Stark, G.R.; O’Shea, J.J. The JAK-STAT Pathway at 30: Much Learned, Much More to Do. Cell 2022, 185, 3857–3876. [Google Scholar] [CrossRef]
- Palermo, E.; Acchioni, C.; Di Carlo, D.; Zevini, A.; Muscolini, M.; Ferrari, M.; Castiello, L.; Virtuoso, S.; Borsetti, A.; Antonelli, G.; et al. Activation of Latent HIV-1 T Cell Reservoirs with a Combination of Innate Immune and Epigenetic Regulators. J. Virol. 2019, 93, e01194-19. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albright, E.R.; Kalejta, R.F. cGAS-STING-TBK1 Signaling Promotes Valproic Acid-Responsive Human Cytomegalovirus Immediate-Early Transcription during Infection of Incompletely Differentiated Myeloid Cells. Viruses 2024, 16, 877. https://doi.org/10.3390/v16060877
Albright ER, Kalejta RF. cGAS-STING-TBK1 Signaling Promotes Valproic Acid-Responsive Human Cytomegalovirus Immediate-Early Transcription during Infection of Incompletely Differentiated Myeloid Cells. Viruses. 2024; 16(6):877. https://doi.org/10.3390/v16060877
Chicago/Turabian StyleAlbright, Emily R., and Robert F. Kalejta. 2024. "cGAS-STING-TBK1 Signaling Promotes Valproic Acid-Responsive Human Cytomegalovirus Immediate-Early Transcription during Infection of Incompletely Differentiated Myeloid Cells" Viruses 16, no. 6: 877. https://doi.org/10.3390/v16060877
APA StyleAlbright, E. R., & Kalejta, R. F. (2024). cGAS-STING-TBK1 Signaling Promotes Valproic Acid-Responsive Human Cytomegalovirus Immediate-Early Transcription during Infection of Incompletely Differentiated Myeloid Cells. Viruses, 16(6), 877. https://doi.org/10.3390/v16060877