
Human Coronavirus 229E Infection Inactivates Pyroptosis Executioner Gasdermin D but Ultimately Leads to Lytic Cell Death Partly Mediated by Gasdermin E
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Virus Infection
2.2. Generation of BEAS-2B GSDMD and GSDME Knock out Cells
2.3. Plaque Assay
2.4. Plasmids Used for Cell Culture Work and Transfections
2.5. Immunoblotting Analysis
2.6. Indirect Fluorescent Antibody Assay
2.7. CytoTox96® Assay
2.8. Caspases Activation Assays
2.9. Statistical Analysis
3. Results
3.1. Mpro of HCoV-229E Directly Cleaves GSDMD in an Overexpression System
3.2. HCoV-229E Mpro Cleaves GSDMD after Glutamines at Positions 29 and 193, and Resulting Fragments Lose Pyroptotic Activity
3.3. HCoV-229E Infection Causes Lytic Cell Death in BEAS-2B and Huh7 Cells
3.4. HCoV-229E Infection Induces Cleavage of Both GSDMD and GSDME
3.5. Pan-Caspase Inhibition Dampens Virus-Induced Lytic Cell Death and Sustains Release of Infectious Virus Particles Overtime
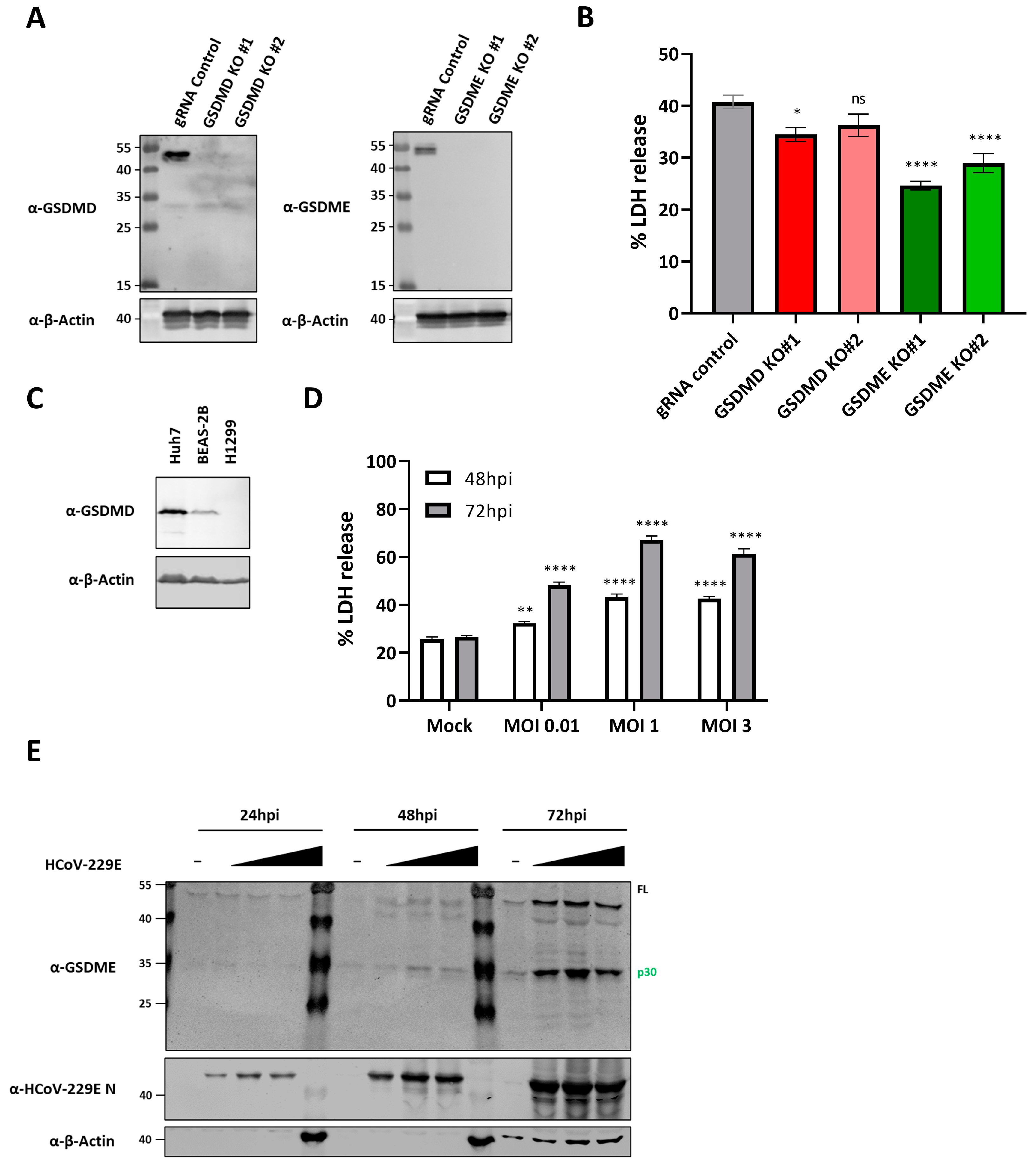
3.6. GSDME Deficiency Reduces HCoV-229E Induced Lytic Cell Death in BEAS-2B Cells
3.7. HCoV229E Infection Leads to Increased Lytic Cell Death Levels in Cells Expressing GSDMD Q29A+Q193A
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Shi, Z.L.; Guo, D.; Rottier, P.J. Coronavirus: Epidemiology, genome replication and the interactions with their hosts. Virol. Sin. 2016, 31, 1–2. [Google Scholar] [CrossRef]
- Liu, D.X.; Liang, J.Q.; Fung, T.S. Human Coronavirus-229E, -OC43, -NL63, and -HKU1 (Coronaviridae). Encycl. Virol. 2021, 2, 428–440. [Google Scholar] [CrossRef]
- Xu, R.H.; He, J.F.; Evans, M.R.; Peng, G.W.; Field, H.E.; Yu, D.W.; Lee, C.K.; Luo, H.M.; Lin, W.S.; Lin, P.; et al. Epidemiologic clues to SARS origin in China. Emerg. Infect. Dis. 2004, 10, 1030–1037. [Google Scholar] [CrossRef]
- van Boheemen, S.; de Graaf, M.; Lauber, C.; Bestebroer, T.M.; Raj, V.S.; Zaki, A.M.; Osterhaus, A.D.; Haagmans, B.L.; Gorbalenya, A.E.; Snijder, E.J.; et al. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 2012, 3, 10-1128. [Google Scholar] [CrossRef]
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284. [Google Scholar] [CrossRef]
- Hamre, D.; Procknow, J.J. A new virus isolated from the human respiratory tract. Proc. Soc. Exp. Biol. Med. 1966, 121, 190–193. [Google Scholar] [CrossRef]
- Tyrrell, D.A.; Bynoe, M.L. Cultivation of a novel type of common-cold virus in organ cultures. Br. Med. J. 1965, 1, 1467–1470. [Google Scholar] [CrossRef]
- Yeager, C.L.; Ashmun, R.A.; Williams, R.K.; Cardellichio, C.B.; Shapiro, L.H.; Look, A.T.; Holmes, K.V. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 1992, 357, 420–422. [Google Scholar] [CrossRef]
- Blau, D.M.; Holmes, K.V. Human coronavirus HCoV-229E enters susceptible cells via the endocytic pathway. Adv. Exp. Med. Biol. 2001, 494, 193–198. [Google Scholar] [CrossRef]
- Farsani, S.M.; Dijkman, R.; Jebbink, M.F.; Goossens, H.; Ieven, M.; Deijs, M.; Molenkamp, R.; van der Hoek, L. The first complete genome sequences of clinical isolates of human coronavirus 229E. Virus Genes 2012, 45, 433–439. [Google Scholar] [CrossRef]
- Thiel, V.; Herold, J.; Schelle, B.; Siddell, S.G. Infectious RNA transcribed in vitro from a cDNA copy of the human coronavirus genome cloned in vaccinia virus. J. Gen. Virol. 2001, 82, 1273–1281. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Siddell, S.G. Processing of the human coronavirus 229E replicase polyproteins by the virus-encoded 3C-like proteinase: Identification of proteolytic products and cleavage sites common to pp1a and pp1ab. J. Virol. 1999, 73, 177–185. [Google Scholar] [CrossRef]
- Lei, J.; Hilgenfeld, R. RNA-virus proteases counteracting host innate immunity. FEBS Lett. 2017, 591, 3190–3210. [Google Scholar] [CrossRef]
- Minkoff, J.M.; tenOever, B. Innate immune evasion strategies of SARS-CoV-2. Nat. Rev. Microbiol. 2023, 21, 178–194. [Google Scholar] [CrossRef]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Y.; Ratia, K.; Mesecar, A.D.; Wilkinson, K.D.; Baker, S.C. Proteolytic processing and deubiquitinating activity of papain-like proteases of human coronavirus NL63. J. Virol. 2007, 81, 6007–6018. [Google Scholar] [CrossRef]
- Mielech, A.M.; Kilianski, A.; Baez-Santos, Y.M.; Mesecar, A.D.; Baker, S.C. MERS-CoV papain-like protease has deISGylating and deubiquitinating activities. Virology 2014, 450–451, 64–70. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Bailey-Elkin, B.A.; Knaap, R.C.; Johnson, G.G.; Dalebout, T.J.; Ninaber, D.K.; van Kasteren, P.B.; Bredenbeek, P.J.; Snijder, E.J.; Kikkert, M.; Mark, B.L. Crystal structure of the Middle East respiratory syndrome coronavirus (MERS-CoV) papain-like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J. Biol. Chem. 2014, 289, 34667–34682. [Google Scholar] [CrossRef]
- Chen, S.; Tian, J.; Li, Z.; Kang, H.; Zhang, J.; Huang, J.; Yin, H.; Hu, X.; Qu, L. Feline Infectious Peritonitis Virus Nsp5 Inhibits Type I Interferon Production by Cleaving NEMO at Multiple Sites. Viruses 2020, 12, 43. [Google Scholar] [CrossRef]
- Wang, D.; Fang, L.; Shi, Y.; Zhang, H.; Gao, L.; Peng, G.; Chen, H.; Li, K.; Xiao, S. Porcine Epidemic Diarrhea Virus 3C-Like Protease Regulates Its Interferon Antagonism by Cleaving NEMO. J. Virol. 2016, 90, 2090–2101. [Google Scholar] [CrossRef]
- Wenzel, J.; Lampe, J.; Müller-Fielitz, H.; Schuster, R.; Zille, M.; Müller, K.; Krohn, M.; Körbelin, J.; Zhang, L.; Özorhan, Ü.; et al. The SARS-CoV-2 main protease M(pro) causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat. Neurosci. 2021, 24, 1522–1533. [Google Scholar] [CrossRef]
- Zhu, X.; Fang, L.; Wang, D.; Yang, Y.; Chen, J.; Ye, X.; Foda, M.F.; Xiao, S. Porcine deltacoronavirus nsp5 inhibits interferon-β production through the cleavage of NEMO. Virology 2017, 502, 33–38. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, D.; Zhou, J.; Pan, T.; Chen, J.; Yang, Y.; Lv, M.; Ye, X.; Peng, G.; Fang, L.; et al. Porcine Deltacoronavirus nsp5 Antagonizes Type I Interferon Signaling by Cleaving STAT2. J. Virol. 2017, 91, e00003-17. [Google Scholar] [CrossRef]
- Koudelka, T.; Boger, J.; Henkel, A.; Schönherr, R.; Krantz, S.; Fuchs, S.; Rodríguez, E.; Redecke, L.; Tholey, A. N-Terminomics for the Identification of In Vitro Substrates and Cleavage Site Specificity of the SARS-CoV-2 Main Protease. Proteomics 2021, 21, e2000246. [Google Scholar] [CrossRef]
- Meyer, B.; Chiaravalli, J.; Gellenoncourt, S.; Brownridge, P.; Bryne, D.P.; Daly, L.A.; Grauslys, A.; Walter, M.; Agou, F.; Chakrabarti, L.A.; et al. Characterising proteolysis during SARS-CoV-2 infection identifies viral cleavage sites and cellular targets with therapeutic potential. Nat. Commun. 2021, 12, 5553. [Google Scholar] [CrossRef]
- Pablos, I.; Machado, Y.; de Jesus, H.C.R.; Mohamud, Y.; Kappelhoff, R.; Lindskog, C.; Vlok, M.; Bell, P.A.; Butler, G.S.; Grin, P.M.; et al. Mechanistic insights into COVID-19 by global analysis of the SARS-CoV-2 3CL(pro) substrate degradome. Cell Rep. 2021, 37, 109892. [Google Scholar] [CrossRef]
- Lei, X.; Zhang, Z.; Xiao, X.; Qi, J.; He, B.; Wang, J. Enterovirus 71 Inhibits Pyroptosis through Cleavage of Gasdermin D. J. Virol. 2017, 91, e01069-17. [Google Scholar] [CrossRef]
- Zhao, G.; Li, T.; Liu, X.; Zhang, T.; Zhang, Z.; Kang, L.; Song, J.; Zhou, S.; Chen, X.; Wang, X.; et al. African swine fever virus cysteine protease pS273R inhibits pyroptosis by noncanonically cleaving gasdermin D. J. Biol. Chem. 2022, 298, 101480. [Google Scholar] [CrossRef]
- Shi, F.; Lv, Q.; Wang, T.; Xu, J.; Xu, W.; Shi, Y.; Fu, X.; Yang, T.; Yang, Y.; Zhuang, L.; et al. Coronaviruses Nsp5 Antagonizes Porcine Gasdermin D-Mediated Pyroptosis by Cleaving Pore-Forming p30 Fragment. mBio 2022, 13, e0273921. [Google Scholar] [CrossRef]
- Planès, R.; Pinilla, M.; Santoni, K.; Hessel, A.; Passemar, C.; Lay, K.; Paillette, P.; Valadão, A.C.; Robinson, K.S.; Bastard, P.; et al. Human NLRP1 is a sensor of pathogenic coronavirus 3CL proteases in lung epithelial cells. Mol. Cell 2022, 82, 2385–2400.e2389. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Aglietti, R.A.; Estevez, A.; Gupta, A.; Ramirez, M.G.; Liu, P.S.; Kayagaki, N.; Ciferri, C.; Dixit, V.M.; Dueber, E.C. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 2016, 113, 7858–7863. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Prochera, A.; Payne, L.; Smith, A.; Garlick, J.A.; Kagan, J.C. Virus-mediated inactivation of anti-apoptotic Bcl-2 family members promotes Gasdermin-E-dependent pyroptosis in barrier epithelial cells. Immunity 2021, 54, 1447–1462. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Shi, Y.; Dong, X.; Xiao, X.; Qi, J.; Ren, L.; Xiang, Z.; Zhou, Z.; Wang, J.; Lei, X. Gasdermin E is required for induction of pyroptosis and severe disease during enterovirus 71 infection. J. Biol. Chem. 2022, 298, 101850. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Cervantes-Barragan, L.; Züst, R.; Maier, R.; Sierro, S.; Janda, J.; Levy, F.; Speiser, D.; Romero, P.; Rohrlich, P.S.; Ludewig, B.; et al. Dendritic cell-specific antigen delivery by coronavirus vaccine vectors induces long-lasting protective antiviral and antitumor immunity. mBio 2010, 1, e00171-10. [Google Scholar] [CrossRef]
- Carlotti, F.; Bazuine, M.; Kekarainen, T.; Seppen, J.; Pognonec, P.; Maassen, J.A.; Hoeben, R.C. Lentiviral vectors efficiently transduce quiescent mature 3T3-L1 adipocytes. Mol. Ther. 2004, 9, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.C.; Fadl, S.; Villanueva, A.J.; Rabeh, W.M. Catalytic Dyad Residues His41 and Cys145 Impact the Catalytic Activity and Overall Conformational Fold of the Main SARS-CoV-2 Protease 3-Chymotrypsin-Like Protease. Front. Chem. 2021, 9, 692168. [Google Scholar] [CrossRef]
- Kim, Y.; Liu, H.; Kankanamalage, A.C.; Weerasekara, S.; Hua, D.H.; Groutas, W.C.; Chang, K.O.; Pedersen, N.C. Correction: Reversal of the Progression of Fatal Coronavirus Infection in Cats by a Broad-Spectrum Coronavirus Protease Inhibitor. PLoS Pathog. 2016, 12, e1005650. [Google Scholar] [CrossRef]
- Wang, Y.; Li, P.; Lavrijsen, M.; Li, Y.; Ma, Z.; Peppelenbosch, M.P.; Baig, M.S.; Pan, Q. Differing pan-coronavirus antiviral potency of boceprevir and GC376 in vitro despite discordant molecular docking predictions. Arch. Virol. 2022, 167, 1125–1130. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xia, S.; Zhang, Z.; Wu, H.; Lieberman, J. Channelling inflammation: Gasdermins in physiology and disease. Nat. Rev. Drug Discov. 2021, 20, 384–405. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, C.; Rathkey, J.K.; Yang, J.; Dubyak, G.R.; Abbott, D.W.; Xiao, T.S. Structures of the Gasdermin D C-Terminal Domains Reveal Mechanisms of Autoinhibition. Structure 2018, 26, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Mesel-Lemoine, M.; Millet, J.; Vidalain, P.O.; Law, H.; Vabret, A.; Lorin, V.; Escriou, N.; Albert, M.L.; Nal, B.; Tangy, F. A human coronavirus responsible for the common cold massively kills dendritic cells but not monocytes. J. Virol. 2012, 86, 7577–7587. [Google Scholar] [CrossRef]
- Yuan, L.; Fung, T.S.; He, J.; Chen, R.A.; Liu, D.X. Modulation of viral replication, apoptosis and antiviral response by induction and mutual regulation of EGR and AP-1 family genes during coronavirus infection. Emerg. Microbes Infect. 2022, 11, 1717–1729. [Google Scholar] [CrossRef]
- Otter, C.J.; Fausto, A.; Tan, L.H.; Khosla, A.S.; Cohen, N.A.; Weiss, S.R. Infection of primary nasal epithelial cells differentiates among lethal and seasonal human coronaviruses. Proc. Natl. Acad. Sci. USA 2023, 120, e2218083120. [Google Scholar] [CrossRef]
- Schneider, K.S.; Groß, C.J.; Dreier, R.F.; Saller, B.S.; Mishra, R.; Gorka, O.; Heilig, R.; Meunier, E.; Dick, M.S.; Ćiković, T.; et al. The Inflammasome Drives GSDMD-Independent Secondary Pyroptosis and IL-1 Release in the Absence of Caspase-1 Protease Activity. Cell Rep. 2017, 21, 3846–3859. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target. Ther. 2020, 5, 235. [Google Scholar] [CrossRef]
- Kiemer, L.; Lund, O.; Brunak, S.; Blom, N. Coronavirus 3CLpro proteinase cleavage sites: Possible relevance to SARS virus pathology. BMC Bioinform. 2004, 5, 72. [Google Scholar] [CrossRef]
- Broz, P.; Pelegrín, P.; Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020, 20, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Booty, L.M.; Bryant, C.E. Gasdermin D and Beyond—Gasdermin-mediated Pyroptosis in Bacterial Infections. J. Mol. Biol. 2022, 434, 167409. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Liu, W.C.; Chen, X.Y.; Wang, X.; Li, J.L.; Zhang, X. Gasdermin D-mediated pyroptosis: Mechanisms, diseases, and inhibitors. Front. Immunol. 2023, 14, 1178662. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Yang, C.; Diao, B.; Huang, X.; Jin, M.; Chen, L.; Yan, W.; Ning, Q.; Zheng, L.; Wu, Y.; et al. The NLRP3 Inflammasome and IL-1β Accelerate Immunologically Mediated Pathology in Experimental Viral Fulminant Hepatitis. PLoS Pathog. 2015, 11, e1005155. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, J.; Teng, Y.; Sun, H.; Tian, G.; He, L.; Li, P.; Chen, Y.; Guo, Y.; Li, J.; et al. Complement Receptor C5aR1 Inhibition Reduces Pyroptosis in hDPP4-Transgenic Mice Infected with MERS-CoV. Viruses 2019, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Nabar, N.R.; Huang, N.N.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Sefik, E.; Qu, R.; Junqueira, C.; Kaffe, E.; Mirza, H.; Zhao, J.; Brewer, J.R.; Han, A.; Steach, H.R.; Israelow, B.; et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature 2022, 606, 585–593. [Google Scholar] [CrossRef]
- Albornoz, E.A.; Amarilla, A.A.; Modhiran, N.; Parker, S.; Li, X.X.; Wijesundara, D.K.; Aguado, J.; Zamora, A.P.; McMillan, C.L.D.; Liang, B.; et al. SARS-CoV-2 drives NLRP3 inflammasome activation in human microglia through spike protein. Mol. Psychiatry 2023, 28, 2878–2893. [Google Scholar] [CrossRef]
- Tsu, B.V.; Agarwal, R.; Gokhale, N.S.; Kulsuptrakul, J.; Ryan, A.P.; Fay, E.J.; Castro, L.K.; Beierschmitt, C.; Yap, C.; Turcotte, E.A.; et al. Host-specific sensing of coronaviruses and picornaviruses by the CARD8 inflammasome. PLoS Biol. 2023, 21, e3002144. [Google Scholar] [CrossRef]
- Bauernfried, S.; Scherr, M.J.; Pichlmair, A.; Duderstadt, K.E.; Hornung, V. Human NLRP1 is a sensor for double-stranded RNA. Science 2021, 371, eabd0811. [Google Scholar] [CrossRef] [PubMed]
- Guy, C.; Baran, M.; Ribó-Molina, P.; van den Hoogen, B.G.; Bowie, A.G. Viral sensing by epithelial cells involves PKR- and caspase-3-dependent generation of gasdermin E pores. iScience 2023, 26, 107698. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Li, J.; Wang, Y.; Yu, X.; He, X.; Shi, J.; Deng, G.; Zeng, X.; Tian, G.; Li, Y.; et al. H7N9 virus infection triggers lethal cytokine storm by activating gasdermin E-mediated pyroptosis of lung alveolar epithelial cells. Natl. Sci. Rev. 2022, 9, nwab137. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, Q.; Ashraf, U.; Yang, M.; Zhu, W.; Gu, J.; Chen, Z.; Gu, C.; Si, Y.; Cao, S.; et al. Zika virus causes placental pyroptosis and associated adverse fetal outcomes by activating GSDME. Elife 2022, 11, e73792. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.N.; Kanneganti, T.D. PANoptosis in Viral Infection: The Missing Puzzle Piece in the Cell Death Field. J. Mol. Biol. 2022, 434, 167249. [Google Scholar] [CrossRef]
- Zheng, M.; Williams, E.P.; Malireddi, R.K.S.; Karki, R.; Banoth, B.; Burton, A.; Webby, R.; Channappanavar, R.; Jonsson, C.B.; Kanneganti, T.D. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J. Biol. Chem. 2020, 295, 14040–14052. [Google Scholar] [CrossRef]
- Zhu, S.; Ding, S.; Wang, P.; Wei, Z.; Pan, W.; Palm, N.W.; Yang, Y.; Yu, H.; Li, H.B.; Wang, G.; et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 2017, 546, 667–670. [Google Scholar] [CrossRef]
- Leibowitz, J.L.; Belyavskaya, E. Caspase Inhibitors Block MHV-3 Induced Apoptosis and Enhance Viral Replication and Pathogenicity. In The Nidoviruses: Coronaviruses and Arteriviruses; Lavi, E., Weiss, S.R., Hingley, S.T., Eds.; Springer: Boston, MA, USA, 2001; pp. 109–114. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martiáñez-Vendrell, X.; Bloeme-ter Horst, J.; Hutchinson, R.; Guy, C.; Bowie, A.G.; Kikkert, M. Human Coronavirus 229E Infection Inactivates Pyroptosis Executioner Gasdermin D but Ultimately Leads to Lytic Cell Death Partly Mediated by Gasdermin E. Viruses 2024, 16, 898. https://doi.org/10.3390/v16060898
Martiáñez-Vendrell X, Bloeme-ter Horst J, Hutchinson R, Guy C, Bowie AG, Kikkert M. Human Coronavirus 229E Infection Inactivates Pyroptosis Executioner Gasdermin D but Ultimately Leads to Lytic Cell Death Partly Mediated by Gasdermin E. Viruses. 2024; 16(6):898. https://doi.org/10.3390/v16060898
Chicago/Turabian StyleMartiáñez-Vendrell, Xavier, Jonna Bloeme-ter Horst, Roy Hutchinson, Coralie Guy, Andrew G. Bowie, and Marjolein Kikkert. 2024. "Human Coronavirus 229E Infection Inactivates Pyroptosis Executioner Gasdermin D but Ultimately Leads to Lytic Cell Death Partly Mediated by Gasdermin E" Viruses 16, no. 6: 898. https://doi.org/10.3390/v16060898
APA StyleMartiáñez-Vendrell, X., Bloeme-ter Horst, J., Hutchinson, R., Guy, C., Bowie, A. G., & Kikkert, M. (2024). Human Coronavirus 229E Infection Inactivates Pyroptosis Executioner Gasdermin D but Ultimately Leads to Lytic Cell Death Partly Mediated by Gasdermin E. Viruses, 16(6), 898. https://doi.org/10.3390/v16060898