
Discovery of Two Novel Viruses of the Willow-Carrot Aphid, Cavariella aegopodii

Abstract
:1. Introduction
2. Materials and Methods
2.1. Insect Sampling and Species Definition
2.2. RNA Extraction and Sequencing
2.3. The cDNA and Small RNA Library Were Produced through the Illumina TruSeq
2.4. Virus Genome Verification and Small Interfering RNA Detection
2.5. Determination of Viral Genome Termini and Transcript Abundance
2.6. Virus Structure Analysis
2.7. Identification of the Virus Taxonomic Status
3. Results
3.1. Discovery of New RNA Viruses Fragments in C. aegopodii
3.2. The Genome Structure and RNA-Seq Read Coverage of CAVLV1 and CAIV1
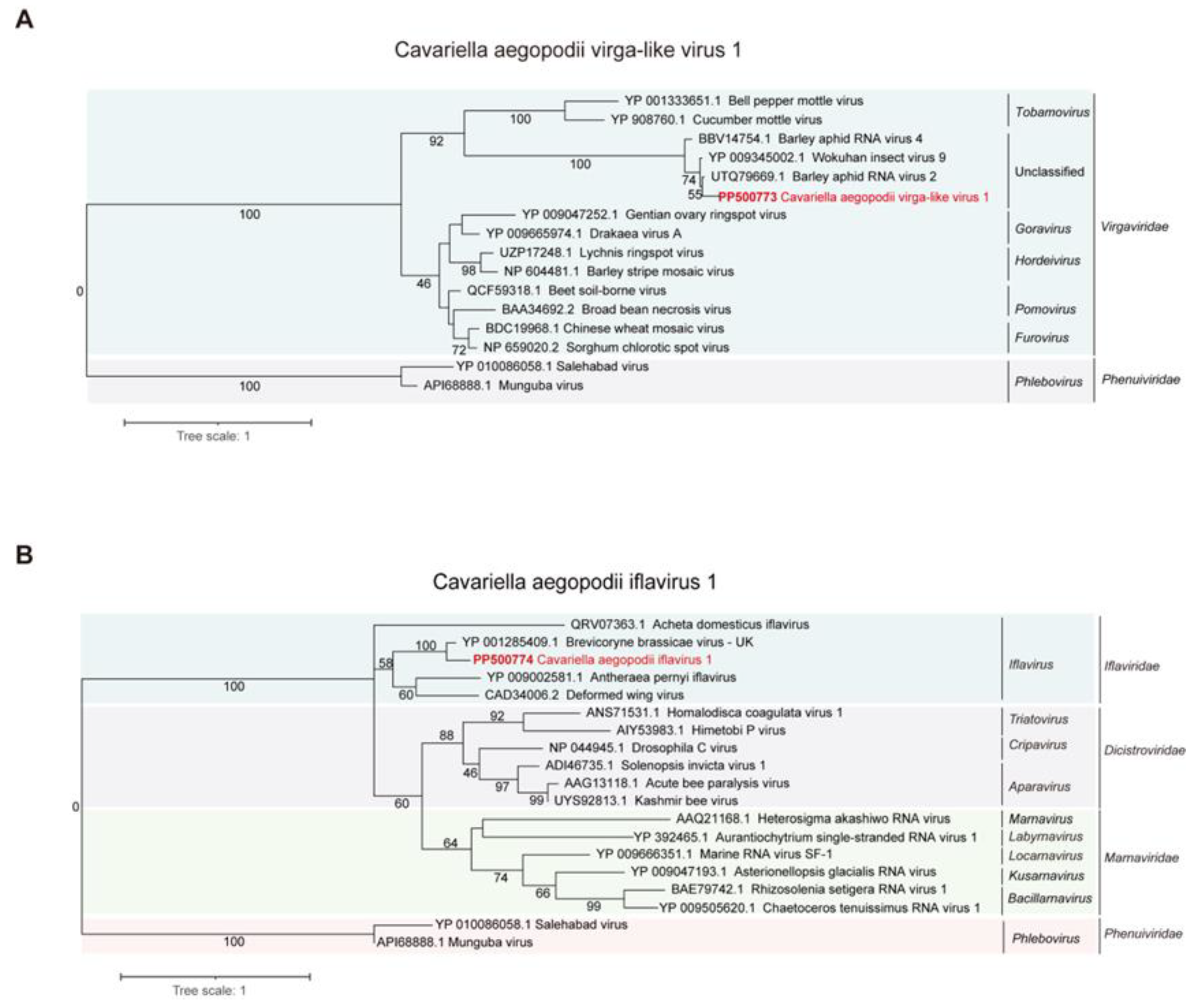
3.3. Phylogenetic Analysis of CAVLV1 and CAIV1
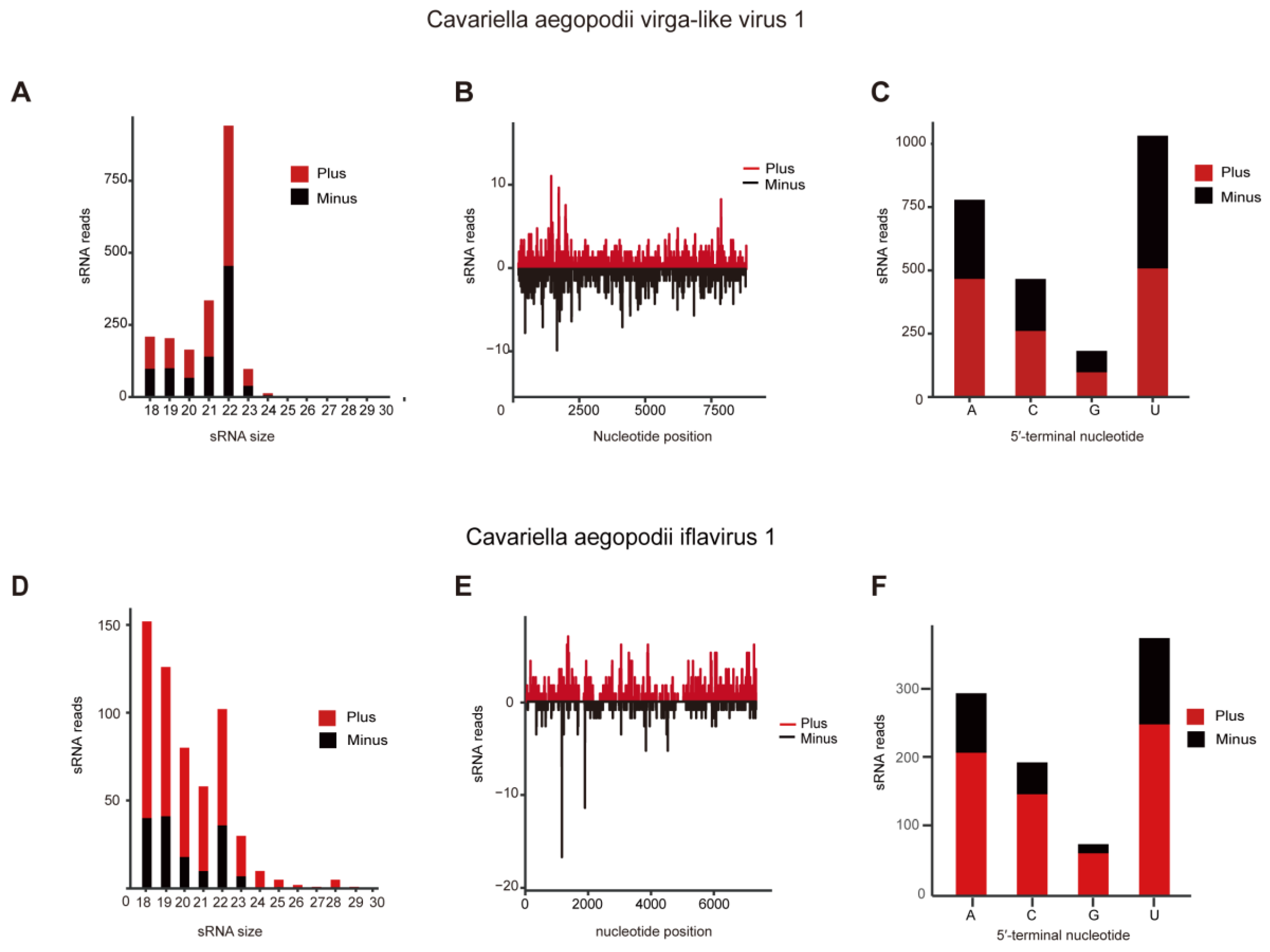
3.4. SiRNA Analyses of CAVLV1 and CAIV1
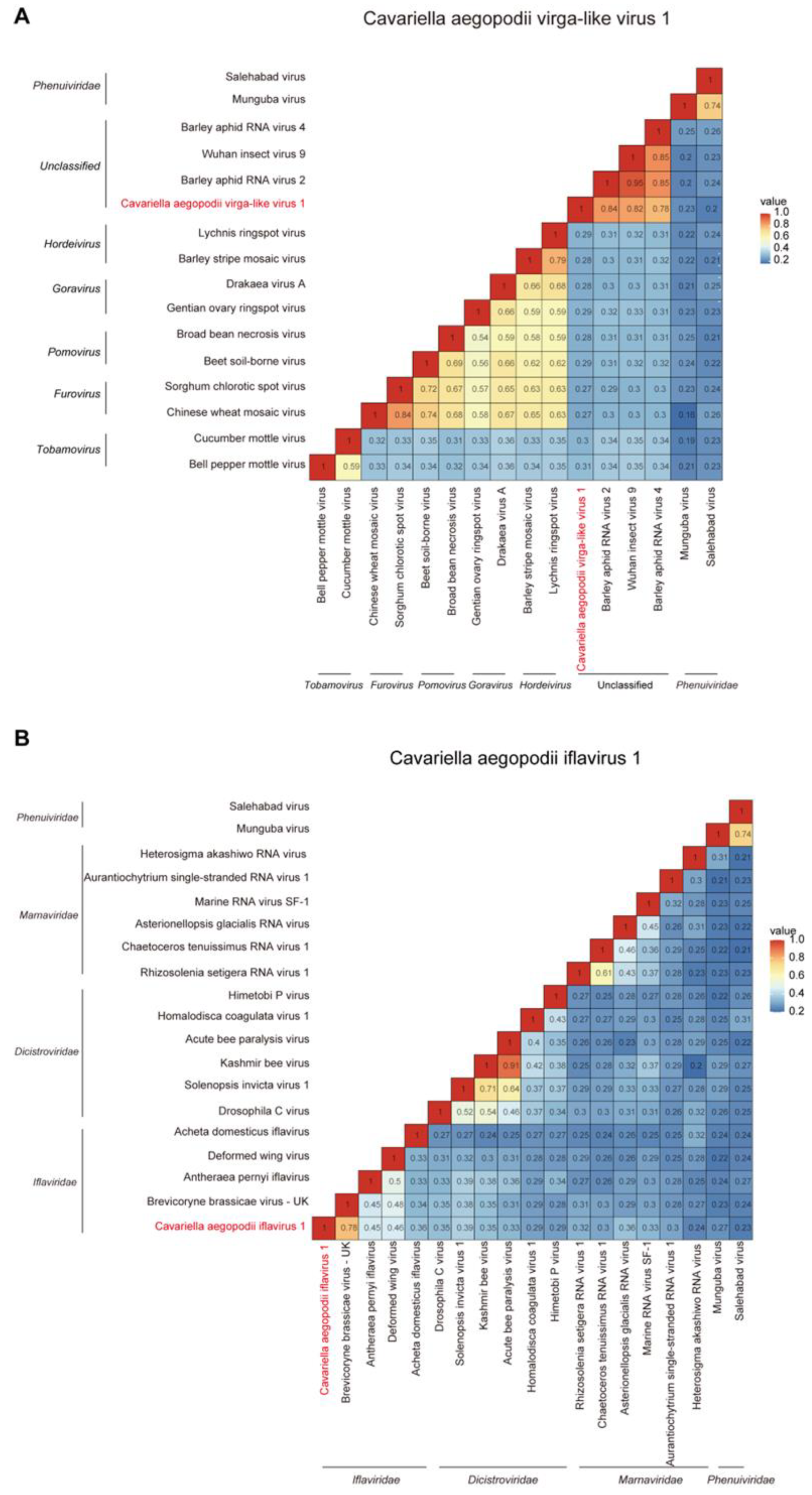
3.5. The Evolutionary Distances of between CAVLV1 or CAIV1 and Other Similar Viruses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, C.-X.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Kang, Y.-J.; Chen, L.-J.; Qin, X.-C.; Xu, J.; Holmes, E.C.; Zhang, Y.-Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, V.L.; Long, M.T. Insect-specific viruses: An overview and their relationship to arboviruses of concern to humans and animals. Virology 2021, 557, 34–43. [Google Scholar] [CrossRef]
- Nouri, S.; Matsumura, E.E.; Kuo, Y.-W.; Falk, B.W. Insect-specific viruses: From discovery to potential translational applications. Curr. Opin. Virol. 2018, 33, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Pang, R.; Cheng, T.; Xue, L.; Zeng, H.; Lei, T.; Chen, M.; Wu, S.; Ding, Y.; Zhang, J. Abundant and diverse RNA viruses in insects revealed by RNA-Seq analysis: Ecological and evolutionary implications. Msystems 2020, 5, e00039-20. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J. Studies on the aphid, Cavariella aegopodii Scop: I. On willow and carrot. Ann. Appl. Biol. 1965, 56, 429–438. [Google Scholar] [CrossRef]
- Ghosh, D.; Medda, P.; Chakrabarti, S. Holocycly, seasonal activity, morphometry and natural enemies of willow aphid, Cavariella aegopodii (Scopoli) (Homoptera: Aphididae) in the Indian region. Proc. Anim. Sci. 1986, 95, 181–186. [Google Scholar] [CrossRef]
- Dunn, J. The susceptibility of verieties of carrot to attack by the aphid, Cavariella aegopodii (Scop.). Ann. Appl. Biol. 1970, 66, 301–312. [Google Scholar] [CrossRef]
- Elnagar, S.; Murant, A.F. Relations of carrot red leaf and carrot mottle viruses with their aphid vector, Cavariella aegopodii. Ann. Appl. Biol. 1978, 89, 237–244. [Google Scholar] [CrossRef]
- Valles, S.; Chen, Y.; Firth, A.; Guérin, D.A.; Hashimoto, Y.; Herrero, S.; De Miranda, J.; Ryabov, E.; ICTV Report Consortium. ICTV virus taxonomy profile: Iflaviridae. J. Gen. Virol. 2017, 98, 527–528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ye, Z.-X.; Feng, X.-X.; Xu, Z.-T.; Chen, J.-P.; Zhang, C.-X.; Li, J.-M. Prevalence of Reversed Genome Organizations for Viruses in the Family Iflaviridae, Order Picornavirales. Microbiol. Spectr. 2023, 11, e0473822. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Antoniw, J.F.; Kreuze, J. Virgaviridae: A new family of rod-shaped plant viruses. Arch. Virol. 2009, 154, 1967–1972. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Adkins, S.; Bragard, C.; Gilmer, D.; Li, D.; MacFarlane, S.A.; Wong, S.-M.; Melcher, U.; Ratti, C.; Ryu, K.H. ICTV virus taxonomy profile: Virgaviridae. J. Gen. Virol. 2017, 98, 1999–2000. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Chiba, S.; Maruyama, K.; Andika, I.B.; Suzuki, N. A novel insect-infecting virga/nege-like virus group and its pervasive endogenization into insect genomes. Virus Res. 2019, 262, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- He, Y.-J.; Ye, Z.-X.; Zhang, C.-X.; Li, J.-M.; Chen, J.-P.; Lu, G. An RNA virome analysis of the pink-winged grasshopper Atractomorpha sinensis. Insects 2022, 14, 9. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Ye, Z.-X.; He, Y.-J.; Zhang, Y.; Wang, X.; Huang, H.-J.; Zhuo, J.-C.; Sun, Z.-T.; Yan, F.; Chen, J.-P. Discovery of two novel negeviruses in a dungfly collected from the Arctic. Viruses 2020, 12, 692. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]
- Ryabov, E.V. A novel virus isolated from the aphid Brevicoryne brassicae with similarity to Hymenoptera picorna-like viruses. J. Gen. Virol. 2007, 88, 2590–2595. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Ji, N.; Bai, L.; Ma, J.; Li, Z. Aphid Viruses: A Brief View of a Long History. Front. Insect Sci. 2022, 2, 846716. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Guo, J.; Ye, Z.; Zhao, Z.; Chen, J.; Yang, J. Molecular characterization of a novel virga-like virus associated with wheat. Arch. Virol. 2022, 167, 1909–1913. [Google Scholar] [CrossRef] [PubMed]
- Geng, P.; Li, W.; Lin, L.; de Miranda, J.R.; Emrich, S.; An, L.; Terenius, O. Genetic characterization of a novel iflavirus associated with vomiting disease in the chinese oak silkmoth Antheraea pernyi. PLoS ONE 2014, 9, e92107. [Google Scholar] [CrossRef] [PubMed]
- Fujita, R.; Kato, F.; Kobayashi, D.; Murota, K.; Takasaki, T.; Tajima, S.; Lim, C.-K.; Saijo, M.; Isawa, H.; Sawabe, K. Persistent viruses in mosquito cultured cell line suppress multiplication of flaviviruses. Heliyon 2018, 4, e00736. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.I.; Villinger, J.; Muthoni, J.N.; Dobel-Ober, L.; Hughes, G.L. Exploiting insect-specific viruses as a novel strategy to control vector-borne disease. Curr. Opin. Insect Sci. 2020, 39, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Meeus, I.; Cappelle, K.; Piot, N.; Smagghe, G. The immune response of the small interfering RNA pathway in the defense against bee viruses. Curr. Opin. Insect Sci. 2014, 6, 22–27. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Names | Accession | Length (nt) | Classification | Abundance | E-Value | RdRP Protein Identities | Top BlastP Hit Virus | Virus Reference |
|---|---|---|---|---|---|---|---|---|
| Cavariella aegopodii virga-like virus 1 (CAVLV1) | PP500773 | 8853 | virga-like virus | 2459 | 0.0 | 79.29% | Hubei Wuhan insect virus 9 | [2] |
| Cavariella aegopodii iflavirus 1 (CAIV1) | PP500774 | 7550 | Iflavirus | 997 | 0.0 | 74.26% | Brevicoryne brassicae virus–UK | [26] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiao, G.; Ye, Z.; Feng, K.; Zhang, C.; Chen, J.; Li, J.; He, Y. Discovery of Two Novel Viruses of the Willow-Carrot Aphid, Cavariella aegopodii. Viruses 2024, 16, 919. https://doi.org/10.3390/v16060919
Jiao G, Ye Z, Feng K, Zhang C, Chen J, Li J, He Y. Discovery of Two Novel Viruses of the Willow-Carrot Aphid, Cavariella aegopodii. Viruses. 2024; 16(6):919. https://doi.org/10.3390/v16060919
Chicago/Turabian StyleJiao, Gaoyang, Zhuangxin Ye, Kehui Feng, Chuanxi Zhang, Jianping Chen, Junmin Li, and Yujuan He. 2024. "Discovery of Two Novel Viruses of the Willow-Carrot Aphid, Cavariella aegopodii" Viruses 16, no. 6: 919. https://doi.org/10.3390/v16060919