
Human Cytomegalovirus Dysregulates Cellular Dual-Specificity Tyrosine Phosphorylation-Regulated Kinases and Sonic Hedgehog Pathway Proteins in Neural Astrocyte and Placental Models
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Cell Lines and Preparation of Virus Stocks
2.2. Clinical Placentae and Placental Villous Explant Histocultures
2.3. Infection of TEV-1 Cells, and NHA Cells with CMV
2.4. Immunofluorescence
2.5. Western Blot Analysis
3. Results
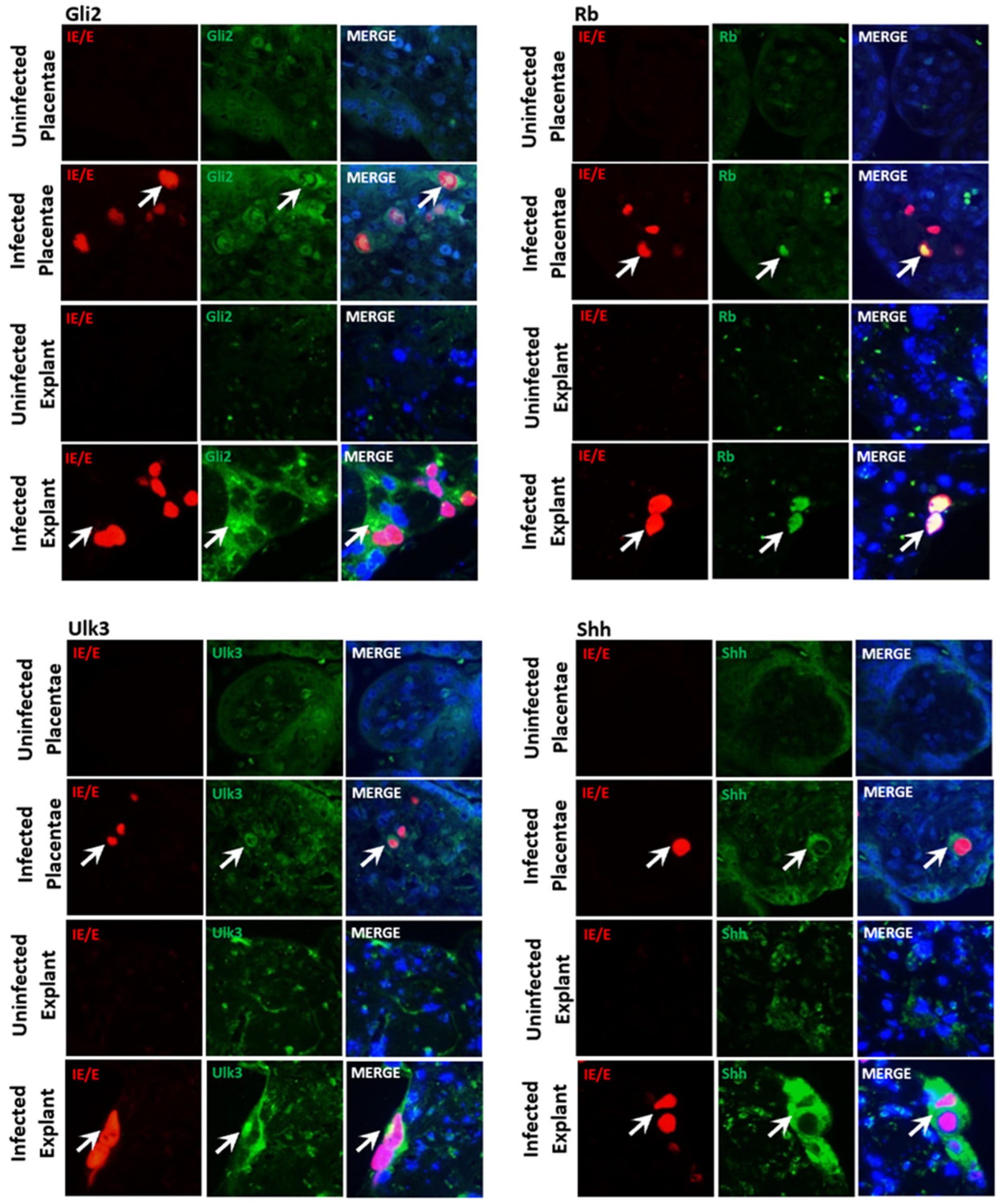
3.1. CMV Infection of Trophoblast Cells Results in Accumulation and Re-Localization of SHH Proteins
3.2. CMV-Induced Trophoblast Accumulation and Re-Localization of Rb, Ulk3, and Shh, but Not Gli2 Is Caused by Upregulation of Expression
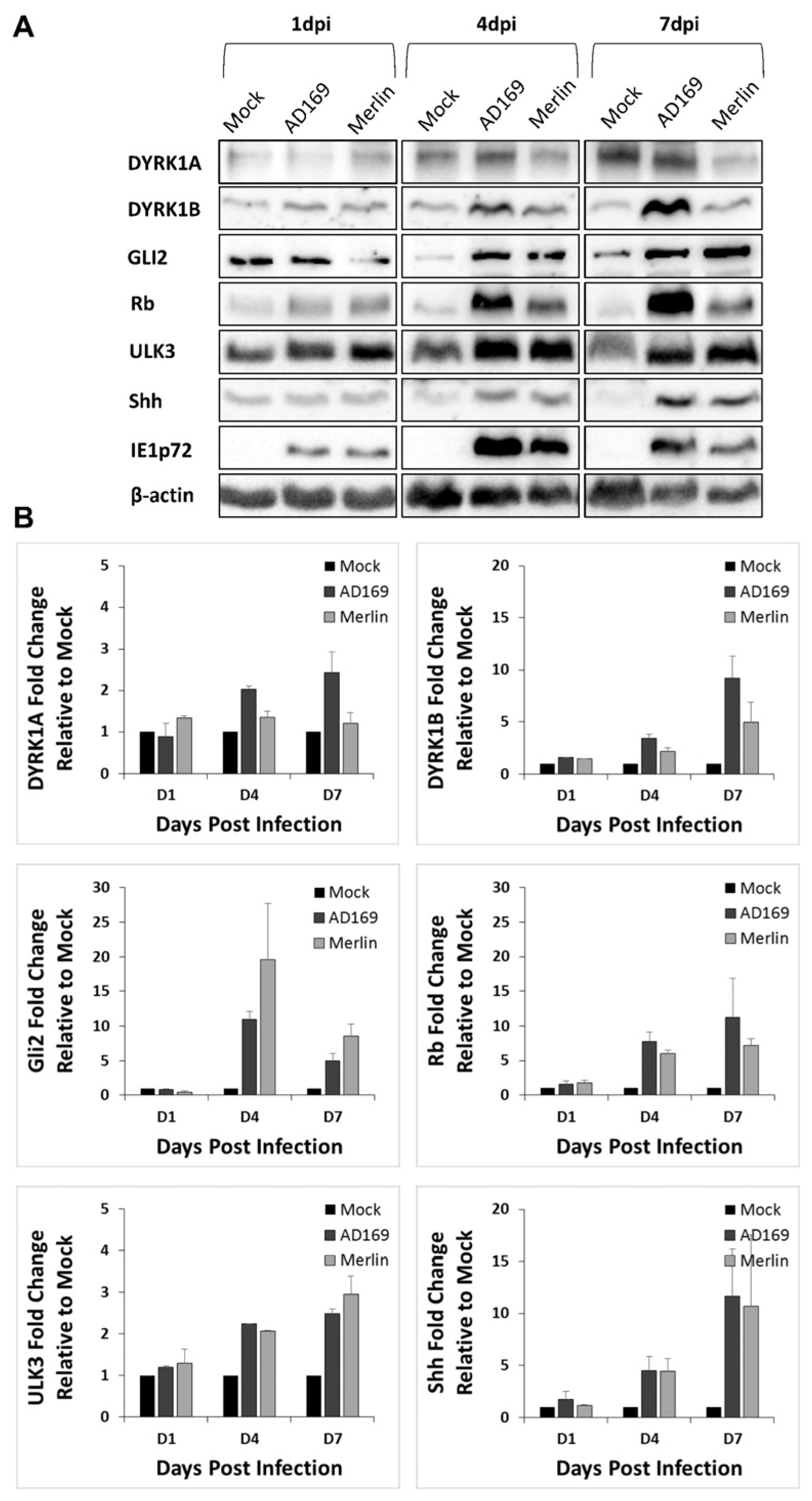
3.3. CMV Infection of Normal Human Astrocyte Cells Results in Accumulation and Re-Localization of DYRK and SHH Proteins
3.4. CMV-Induced Normal Human Astrocyte Accumulation and Re-Localization of DYRK and SHH Proteins Is Caused by Upregulated Expression
3.5. The CMV Upregulation of SHH Proteins Occurs in In Vivo and Ex Vivo Human Placental Models
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Lanzieri, T.M.; Dollard, S.C.; Bialek, S.R.; Grosse, S.D. Systematic review of the birth prevalence of congenital cytomegalovirus infection in developing countries. Int. J. Infect. Dis. 2014, 22, 44–48. [Google Scholar] [CrossRef]
- Noyola, D.E.; Demmler, G.J.; Nelson, C.T.; Griesser, C.; Williamson, W.D.; Atkins, J.T.; Rozelle, J.; Turcich, M.; Llorente, A.M.; Sellers-Vinson, S.; et al. Early predictors of neurodevelopmental outcome in symptomatic congenital cytomegalovirus infection. J. Pediatr. 2001, 138, 325–331. [Google Scholar] [CrossRef]
- Boppana, S.B.; Fowler, K.B.; Vaid, Y.; Hedlund, G.; Stagno, S.; Britt, W.J.; Pass, R.F. Neuroradiographic findings in the newborn period and long-term outcome in children with symptomatic congenital cytomegalovirus infection. Pediatrics 1997, 99, 409–414. [Google Scholar] [CrossRef]
- Cheeran, M.C.; Lokensgard, J.R.; Schleiss, M.R. Neuropathogenesis of congenital cytomegalovirus infection: Disease mechanisms and prospects for intervention. Clin. Microbiol. Rev. 2009, 22, 99–126. [Google Scholar] [CrossRef]
- Fisher, S.; Genbacev, O.; Maidji, E.; Pereira, L. Human cytomegalovirus infection of placental cytotrophoblasts in vitro and in utero: Implications for transmission and pathogenesis. J. Virol. 2000, 74, 6808–6820. [Google Scholar] [CrossRef]
- Iwasenko, J.M.; Howard, J.; Arbuckle, S.; Graf, N.; Hall, B.; Craig, M.E.; Rawlinson, W.D. Human cytomegalovirus infection is detected frequently in stillbirths and is associated with fetal thrombotic vasculopathy. J. Infect. Dis. 2011, 203, 1526–1533. [Google Scholar] [CrossRef]
- Maidji, E.; Nigro, G.; Tabata, T.; McDonagh, S.; Nozawa, N.; Shiboski, S.; Muci, S.; Anceschi, M.M.; Aziz, N.; Adler, S.P.; et al. Antibody treatment promotes compensation for human cytomegalovirus-induced pathogenesis and a hypoxia-like condition in placentas with congenital infection. Am. J. Pathol. 2010, 177, 1298–1310. [Google Scholar] [CrossRef]
- Hamilton, S.T.; Scott, G.; Naing, Z.; Iwasenko, J.; Hall, B.; Graf, N.; Arbuckle, S.; Craig, M.E.; Rawlinson, W.D. Human cytomegalovirus-induces cytokine changes in the placenta with implications for adverse pregnancy outcomes. PLoS ONE 2012, 7, e52899. [Google Scholar] [CrossRef] [PubMed]
- Marschall, M.; Feichtinger, S.; Milbradt, J. Regulatory Roles of Protein Kinases in Cytomegalovirus Replication. Adv. Virus Res. 2011, 80, 69–101. [Google Scholar] [PubMed]
- Hutterer, C.; Milbradt, J.; Hamilton, S.T.; Zaja, M.; Leban, J.; Henry, C.; Vitt, D.; Steingruber, M.; Sonntag, E.; Zeitträger, I.; et al. Inhibitors of dual-specificity tyrosine phosphorylation-regulated kinases (DYRK) exert a strong anti-herpesviral activity. Antivir. Res. 2017, 143, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Becker, W. Emerging role of DYRK family protein kinases as regulators of protein stability in cell cycle control. Cell Cycle 2012, 11, 3389–3394. [Google Scholar] [CrossRef] [PubMed]
- Litovchick, L.; Florens, L.A.; Swanson, S.K.; Washburn, M.P.; DeCaprio, J.A. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes. Dev. 2011, 25, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Abbassi, R.; Johns, T.G.; Kassiou, M.; Munoz, L. DYRK1A in neurodegeneration and cancer: Molecular basis and clinical implications. Pharmacol. Ther. 2015, 151, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Martinez, J.; Vela, E.M.; Tora-Ponsioen, M.; Ocaña, O.H.; Nieto, M.A.; Galceran, J. Attenuation of Notch signalling by the Down-syndrome-associated kinase DYRK1A. J. Cell Sci. 2009, 122, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Lauth, M. Emerging Roles of DYRK Kinases in Embryogenesis and Hedgehog Pathway Control. J. Dev. Biol. 2017, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Di Vona, C.; Bezdan, D.; Islam, A.B.; Salichs, E.; López-Bigas, N.; Ossowski, S.; de la Luna, S. Chromatin-wide profiling of DYRK1A reveals a role as a gene-specific RNA polymerase II CTD kinase. Mol. Cell 2015, 57, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Lee, H.; Argiropoulos, B.; Dorrani, N.; Mann, J.; Martinez-Agosto, J.A.; Gomez-Ospina, N.; Gallant, N.; Bernstein, J.A.; Hudgins, L.; et al. DYRK1A haploinsufficiency causes a new recognizable syndrome with microcephaly, intellectual disability, speech impairment, and distinct facies. Eur. J. Hum. Genet. 2015, 23, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Song, W.J.; Chung, K.C. Function and regulation of Dyrk1A: Towards understanding Down syndrome. Cell. Mol. Life Sci. 2009, 66, 3235–3240. [Google Scholar] [CrossRef] [PubMed]
- Møller, R.S.; Kübart, S.; Hoeltzenbein, M.; Heye, B.; Vogel, I.; Hansen, C.P.; Menzel, C.; Ullmann, R.; Tommerup, N.; Ropers, H.H.; et al. Truncation of the Down Syndrome Candidate Gene DYRK1A in Two Unrelated Patients with Microcephaly. Am. J. Hum. Genet. 2008, 82, 1165–1170. [Google Scholar] [CrossRef]
- Pozo, N.; Zahonero, C.; Fernández, P.; Liñares, J.M.; Ayuso, A.; Hagiwara, M.; Pérez, A.; Ricoy, J.R.; Hernández-Laín, A.; Sepúlveda, J.M.; et al. Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth. J. Clin. Investig. 2013, 123, 2475–2487. [Google Scholar] [CrossRef] [PubMed]
- Mercer, S.E.; Friedman, E. Mirk/Dyrk 1B. Cell Biochem. Biophys. 2006, 45, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Hu, J.; Ewton, D.Z.; Friedman, E. Mirk/dyrk1B kinase is upregulated following inhibition of mTOR. Carcinogenesis 2014, 35, 1968–1976. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.M.; Tan, K.S.; Chen, H.; Beh, T.T.; Yeo, H.C.; Ng, S.K.; Wei, S.; Lee, D.Y.; Choo, A.B.; Chan, K.K. Enhanced production of neuroprogenitors, dopaminergic neurons, and identification of target genes by overexpression of sonic hedgehog in human embryonic stem cells. Stem Cells Dev. 2012, 21, 729–741. [Google Scholar] [CrossRef]
- Agarwala, S.; Sanders, T.A.; Ragsdale, C.W. Sonic hedgehog control of size and shape in midbrain pattern formation. Science 2001, 291, 2147–2150. [Google Scholar] [CrossRef] [PubMed]
- Brooks, E.R.; Islam, M.T.; Anderson, K.V.; Zallen, J.A. Sonic hedgehog signaling directs patterned cell remodeling during cranial neural tube closure. eLife 2020, 9, 60234. [Google Scholar] [CrossRef]
- Hill, S.A.; Blaeser, A.S.; Coley, A.A.; Xie, Y.; Shepard, K.A.; Harwell, C.C.; Gao, W.J.; Garcia, A.D.R. Sonic hedgehog signaling in astrocytes mediates cell type-specific synaptic organization. eLife 2019, 8, 45545. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef]
- Pan, Y.B.; Gong, Y.; Ruan, H.F.; Pan, L.Y.; Wu, X.K.; Tang, C.; Wang, C.J.; Zhu, H.B.; Zhang, Z.M.; Tang, L.F.; et al. Sonic hedgehog through Gli2 and Gli3 is required for the proper development of placental labyrinth. Cell Death Dis. 2015, 6, e1653. [Google Scholar] [CrossRef]
- Takai, H.; Kondoh, E.; Mogami, H.; Kawasaki, K.; Chigusa, Y.; Sato, M.; Kawamura, Y.; Murakami, R.; Matsumura, N.; Konishi, I.; et al. Placental sonic hedgehog pathway regulates foetal growth via insulin-like growth factor axis in preeclampsia. J. Clin. Endocrinol. Metab. 2019, 104, 4239–4252. [Google Scholar] [CrossRef]
- Hamilton, S.T.; Hutterer, C.; Egilmezer, E.; Steingruber, M.; Milbradt, J.; Marschall, M.; Rawlinson, W.D. Human cytomegalovirus utilises cellular dual-specificity tyrosine phosphorylation-regulated kinases during placental replication. Placenta 2018, 72–73, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.T.; Marschall, M.; Rawlinson, W.D. Investigational Antiviral Therapy Models for the Prevention and Treatment of Congenital Cytomegalovirus Infection during Pregnancy. Antimicrob. Agents Chemother. 2020, 65, 01627-20. [Google Scholar] [CrossRef]
- Hutterer, C.; Hamilton, S.T.; Steingruber, M.; Zeitträger, I.; Bahsi, H.; Thuma, N.; Naing, Z.; Örfi, Z.; Örfi, L.; Socher, E.; et al. The chemical class of quinazoline compounds provides a core structure for the design of anticytomegaloviral kinase inhibitors. Antivir. Res. 2016, 134, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.T.; Scott, G.M.; Naing, Z.; Rawlinson, W.D. Human cytomegalovirus directly modulates expression of chemokine CCL2 (MCP-1) during viral replication. J. Gen. Virol. 2013, 94, 2495–2503. [Google Scholar] [CrossRef]
- Hamilton, S.T.; Milbradt, J.; Marschall, M.; Rawlinson, W.D. Human cytomegalovirus replication is strictly inhibited by siRNAs targeting UL54, UL97 or UL122/123 gene transcripts. PLoS ONE 2014, 9, e97231. [Google Scholar] [CrossRef]
- Scott, G.M.; Chow, S.S.; Craig, M.E.; Pang, C.N.; Hall, B.; Wilkins, M.R.; Rawlinson, W.D. Cytomegalovirus infection during pregnancy with maternofetal transmission induces a proinflammatory cytokine bias in placenta and amniotic fluid. J. Infect. Dis. 2012, 205, 1305–1310. [Google Scholar] [CrossRef]
- Chico, L.K.; Van Eldik, L.J.; Watterson, D.M. Targeting protein kinases in central nervous system disorders. Nat. Rev. Drug Discov. 2009, 8, 892–909. [Google Scholar] [CrossRef]
- Maloverjan, A.; Piirsoo, M.; Kasak, L.; Peil, L.; Østerlund, T.; Kogerman, P. Dual function of UNC-51-like kinase 3 (Ulk3) in the Sonic hedgehog signaling pathway. J. Biol. Chem. 2010, 285, 30079–30090. [Google Scholar] [CrossRef]
- Lu, N.; Chen, Y.; Wang, Z.; Chen, G.; Lin, Q.; Chen, Z.Y.; Li, H. Sonic hedgehog initiates cochlear hair cell regeneration through downregulation of retinoblastoma protein. Biochem. Biophys. Res. Commun. 2013, 430, 700–705. [Google Scholar] [CrossRef]
- Shao, X.; Cao, G.; Chen, D.; Liu, J.; Yu, B.; Liu, M.; Li, Y.X.; Cao, B.; Sadovsky, Y.; Wang, Y.L. Placental trophoblast syncytialization potentiates macropinocytosis via mTOR signaling to adapt to reduced amino acid supply. Proc. Natl. Acad. Sci. USA 2021, 118, e2017092118. [Google Scholar] [CrossRef]
- Red-Horse, K.; Zhou, Y.; Genbacev, O.; Prakobphol, A.; Foulk, R.; McMaster, M.; Fisher, S.J. Trophoblast differentiation during embryo implantation and formation of the maternal-fetal interface. J. Clin. Investig. 2004, 114, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Mimura, N.; Nagamatsu, T.; Morita, K.; Taguchi, A.; Toya, T.; Kumasawa, K.; Iriyama, T.; Kawana, K.; Inoue, N.; Fujii, T.; et al. Suppression of human trophoblast syncytialization by human cytomegalovirus infection. Placenta 2022, 117, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.A. Scrutinising the regulators of syncytialization and their expression in pregnancy-related conditions. Mol. Cell Endocrinol. 2016, 15, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Petitt, M.; Fong, A.; Tsuge, M.; Tabata, T.; Fang-Hoover, J.; Maidji, E.; Zydek, M.; Zhou, Y.; Inoue, N.; et al. Intrauterine growth restriction caused by underlying congenital cytomegalovirus infection. J. Infect. Dis. 2014, 209, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Dhanyamraju, P.K.; Lauth, M. DYRK1B blocks canonical and promotes non-canonical Hedgehog signaling through activation of the mTOR/AKT pathway. Oncotarget 2017, 8, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Priego, N.; Valiente, M. The Potential of Astrocytes as Immune Modulators in Brain Tumors. Front. Immunol. 2019, 10, 465833. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- McNeill, J.; Rudyk, C.; Hildebrand, M.E.; Salmaso, N. Ion Channels and Electrophysiological Properties of Astrocytes: Implications for Emergent Stimulation Technologies. Front. Cell. Neurosci. 2021, 15, 644126. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, B.; Li, L.; Qian, D.M.; Yu, H.; Xue, M.L.; Hu, M.; Song, X.X. Antiviral effects of IFIT1 in human cytomegalovirus-infected fetal astrocytes. J. Med. Virol. 2017, 89, 672–684. [Google Scholar] [CrossRef]
- Cheeran, M.C.; Gekker, G.; Hu, S.; Yager, S.L.; Peterson, P.K.; Lokensgard, J.R. CD4(+) lymphocyte-mediated suppression of cytomegalovirus expression in human astrocytes. Clin. Diagn. Lab. Immunol. 2000, 7, 710–713. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liang, Z.; Wegiel, J.; Hwang, Y.W.; Iqbal, K.; Grundke-Iqbal, I.; Ramakrishna, N.; Gong, C.X. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 2008, 22, 3224–3233. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.A.; Fu, M.; Garcia, A.D.R. Sonic hedgehog signaling in astrocytes. Cell. Mol. Life Sci. 2021, 78, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Kuan, A.T.; Wang, W.; Herbert, Z.T.; Mosto, O.; Olukoya, O.; Adam, M.; Vu, S.; Kim, M.; Tran, D.; et al. Astrocyte-neuron crosstalk through Hedgehog signaling mediates cortical synapse development. Cell Rep. 2022, 38, 110416. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Gudsnuk, K.; Kuo, S.H.; Cotrina, M.L.; Rosoklija, G.; Sosunov, A.; Sonders, M.S.; Kanter, E.; Castagna, C.; Yamamoto, A.; et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 2014, 83, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Hahn, F.; Hutterer, C.; Henry, C.; Hamilton, S.T.; Strojan, H.; Kraut, A.; Schulte, U.; Schütz, M.; Kohrt, S.; Wangen, C.; et al. Novel cytomegalovirus-inhibitory compounds of the class pyrrolopyridines show a complex pattern of target binding that suggests an unusual mechanism of antiviral activity. Antivir. Res. 2018, 159, 84–94. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Egilmezer, E.; Hamilton, S.T.; Lauw, G.; Follett, J.; Sonntag, E.; Schütz, M.; Marschall, M.; Rawlinson, W.D. Human Cytomegalovirus Dysregulates Cellular Dual-Specificity Tyrosine Phosphorylation-Regulated Kinases and Sonic Hedgehog Pathway Proteins in Neural Astrocyte and Placental Models. Viruses 2024, 16, 918. https://doi.org/10.3390/v16060918
Egilmezer E, Hamilton ST, Lauw G, Follett J, Sonntag E, Schütz M, Marschall M, Rawlinson WD. Human Cytomegalovirus Dysregulates Cellular Dual-Specificity Tyrosine Phosphorylation-Regulated Kinases and Sonic Hedgehog Pathway Proteins in Neural Astrocyte and Placental Models. Viruses. 2024; 16(6):918. https://doi.org/10.3390/v16060918
Chicago/Turabian StyleEgilmezer, Ece, Stuart T. Hamilton, Glen Lauw, Jasmine Follett, Eric Sonntag, Martin Schütz, Manfred Marschall, and William D. Rawlinson. 2024. "Human Cytomegalovirus Dysregulates Cellular Dual-Specificity Tyrosine Phosphorylation-Regulated Kinases and Sonic Hedgehog Pathway Proteins in Neural Astrocyte and Placental Models" Viruses 16, no. 6: 918. https://doi.org/10.3390/v16060918
APA StyleEgilmezer, E., Hamilton, S. T., Lauw, G., Follett, J., Sonntag, E., Schütz, M., Marschall, M., & Rawlinson, W. D. (2024). Human Cytomegalovirus Dysregulates Cellular Dual-Specificity Tyrosine Phosphorylation-Regulated Kinases and Sonic Hedgehog Pathway Proteins in Neural Astrocyte and Placental Models. Viruses, 16(6), 918. https://doi.org/10.3390/v16060918