
Elucidation of the Epitranscriptomic RNA Modification Landscape of Chikungunya Virus
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Viruses and Infection Conditions
2.3. Poly(A)+ RNA-Selection and Isolation of gRNA and sgRNA
2.4. Quantitation of RNA Modifications by LC-MS/MS Analysis
2.5. Generation of In Vitro Transcribed (IVT) CHIKV RNA
2.6. IFN Pre-Treatment
2.7. siRNA Knockdown and ADAR1 Overexpression
2.8. Western Blotting
2.9. Two-Step Quantitative Reverse Transcription PCR (qRT-PCR)
2.10. Cell Viability Assay
2.11. Library Preparation for RNA-Seq
2.12. ADAR1 Editing Analyses
2.13. Statistical Analysis and Chart Production
3. Results
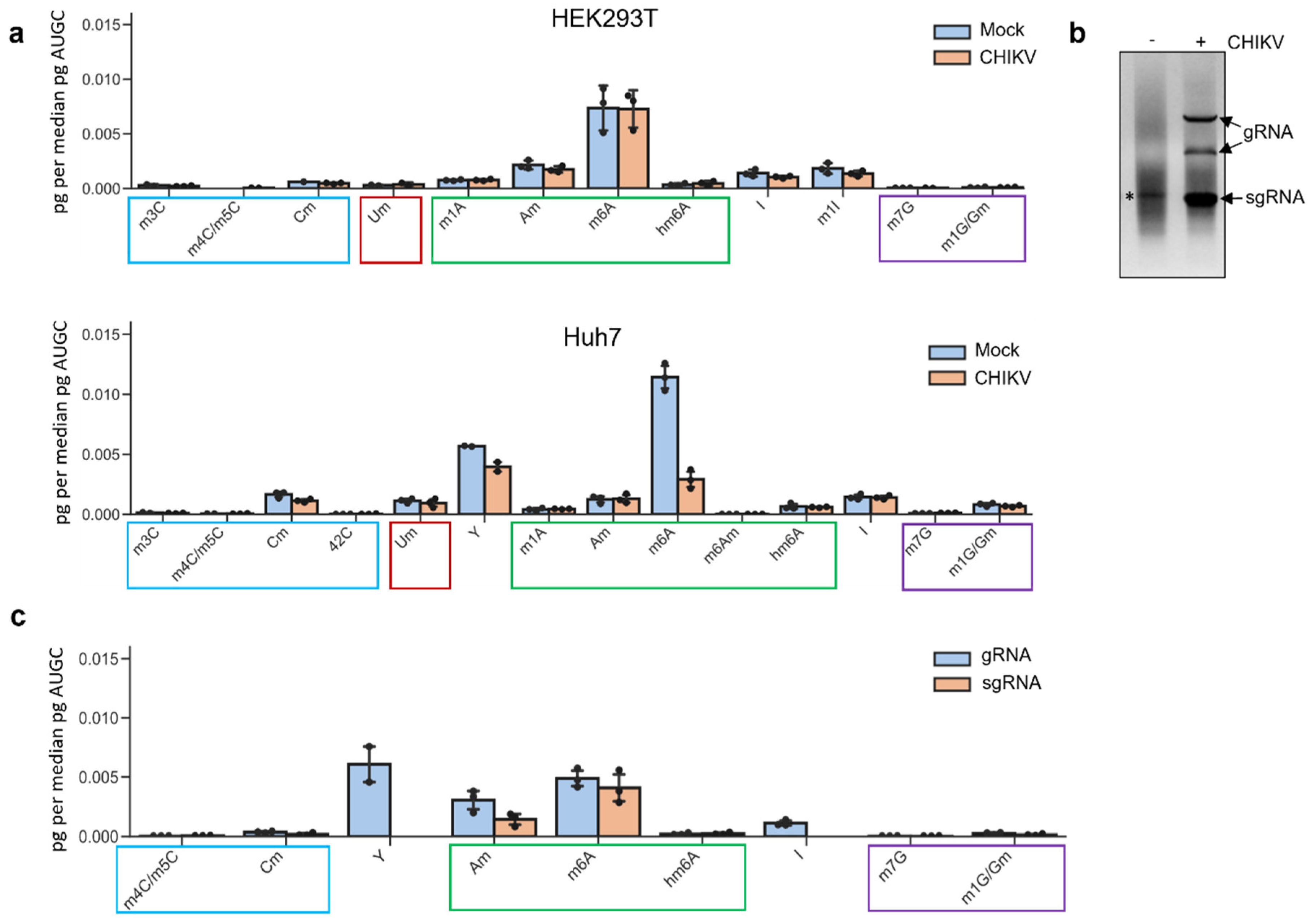
3.1. Mass Spectrometry Analyses Reveal Inosine Enrichment in the CHIKV Genomic RNA but Not in the Subgenomic One
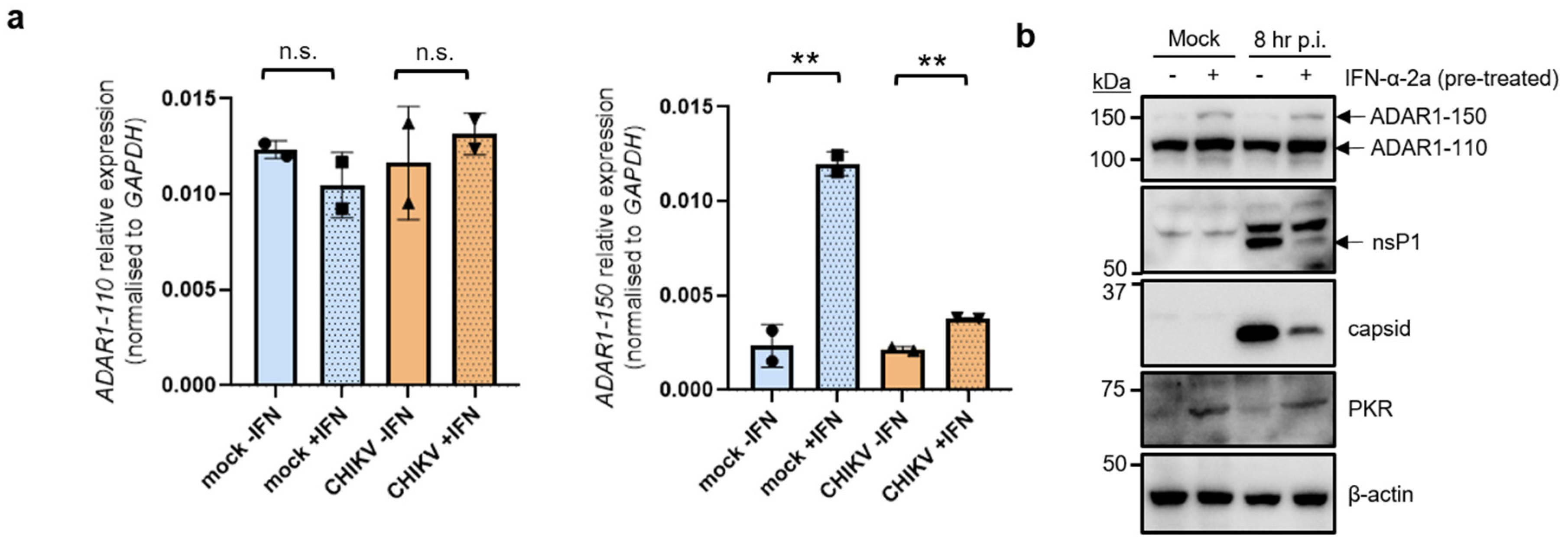
3.2. CHIKV Infection Does Not Alter the Induction of ADAR1 Isoforms
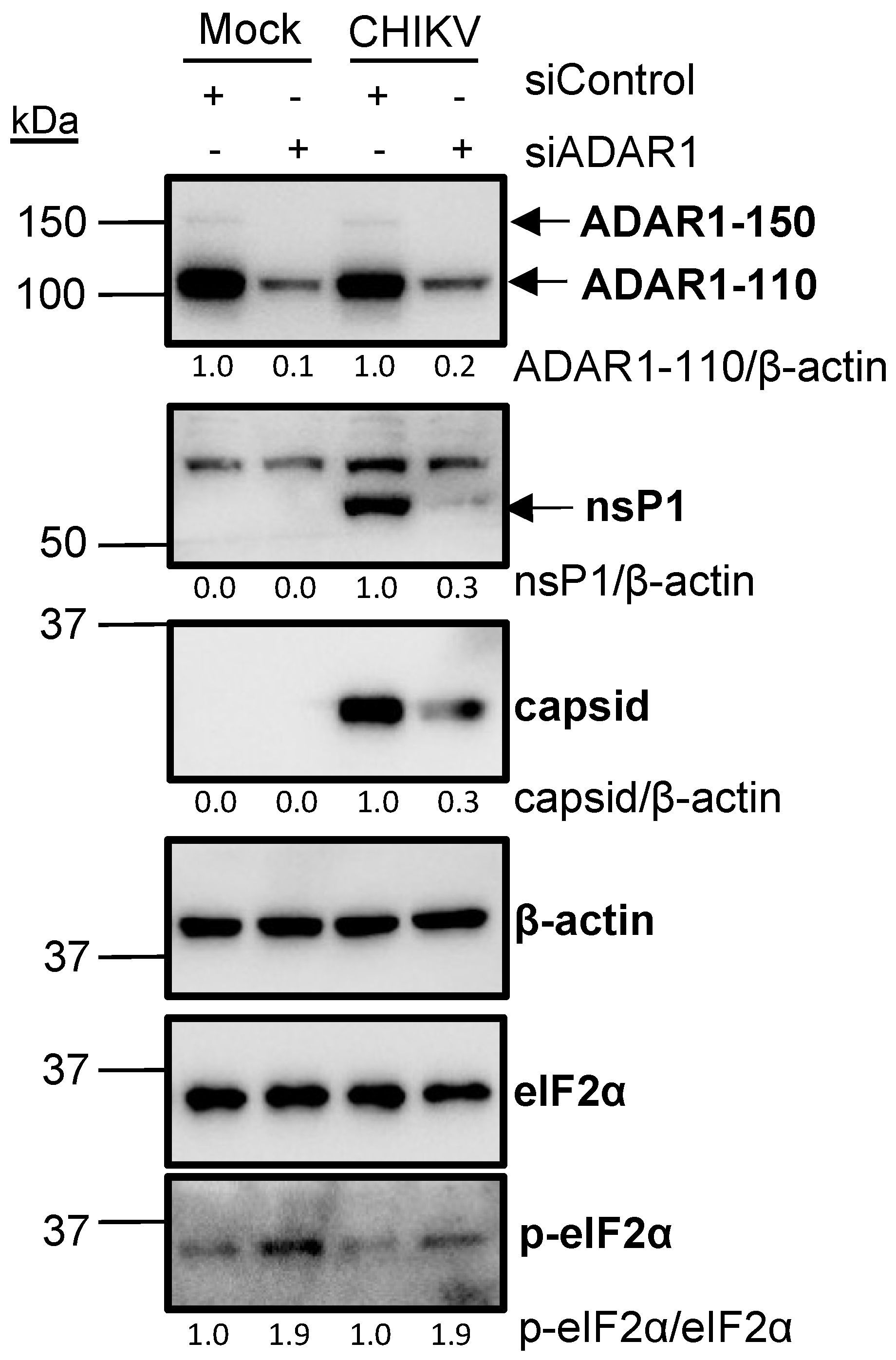
3.3. ADAR1 Knockdown Inhibits CHIKV RNA and Protein Levels, and Virion Production
3.4. ADAR1 Knockdown Leads to Phosphorylation of Eukaryotic Translation Initiation Factor 2α (eIF2α)
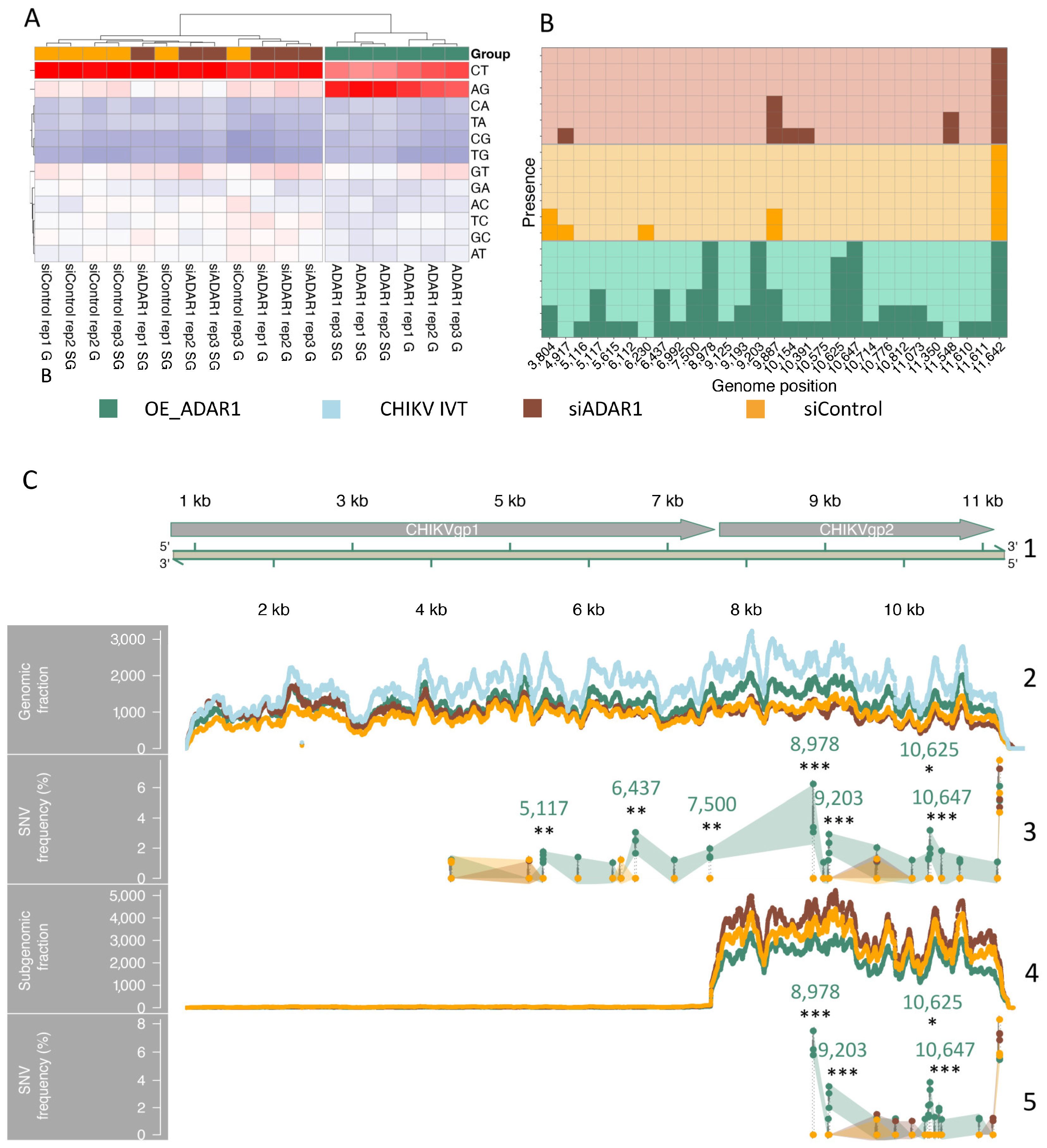
3.5. RNA-Seq Analyses Detect Adenosine to Inosine (A-to-I) Editing in CHIKV RNAs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA Modification Landscape in Human Disease. RNA 2017, 23, 1754–1769. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Harada, B.T.; Behm, M.; He, C. RNA Modifications Modulate Gene Expression during Development. Science 2018, 361, 1346–1349. [Google Scholar] [CrossRef] [PubMed]
- Motorin, Y.; Helm, M. RNA Nucleotide Methylation. WIREs RNA 2011, 2, 611–631. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the Human and Mouse m6A RNA Methylomes Revealed by m6A-Seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wei, J.; He, C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, Writing and Erasing mRNA Methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, L.; Li, X. Detection Technologies for RNA Modifications. Exp. Mol. Med. 2022, 54, 1601–1616. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, W.; Netzband, R.; Bonenfant, G.; Biegel, J.M.; Miller, C.; Fuchs, G.; Henderson, E.; Arra, M.; Canki, M.; Fabris, D.; et al. Positive-Sense RNA Viruses Reveal the Complexity and Dynamics of the Cellular and Viral Epitranscriptomes during Infection. Nucleic Acids Res. 2018, 46, 5776–5791. [Google Scholar] [CrossRef] [PubMed]
- Baquero-Perez, B.; Geers, D.; Díez, J. From A to m(6)A: The Emerging Viral Epitranscriptome. Viruses 2021, 13, 1049. [Google Scholar] [CrossRef] [PubMed]
- Safra, M.; Sas-Chen, A.; Nir, R.; Winkler, R.; Nachshon, A.; Bar-Yaacov, D.; Erlacher, M.; Rossmanith, W.; Stern-Ginossar, N.; Schwartz, S. The m1A Landscape on Cytosolic and Mitochondrial mRNA at Single-Base Resolution. Nature 2017, 551, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Baquero-Pérez, B.; Yonchev, I.D.; Delgado-Tejedor, A.; Medina, R.; Puig-Torrents, M.; Sudbery, I.; Begik, O.; Wilson, S.A.; Novoa, E.M.; Díez, J. N6-Methyladenosine Modification Is Not a General Trait of Viral RNA Genomes. Nat. Commun. 2024, 15, 1964. [Google Scholar] [CrossRef] [PubMed]
- Grozhik, A.V.; Olarerin-George, A.O.; Sindelar, M.; Li, X.; Gross, S.S.; Jaffrey, S.R. Antibody Cross-Reactivity Accounts for Widespread Appearance of m1A in 5′UTRs. Nat. Commun. 2019, 10, 5126. [Google Scholar] [CrossRef] [PubMed]
- Sas-Chen, A.; Thomas, J.M.; Matzov, D.; Taoka, M.; Nance, K.D.; Nir, R.; Bryson, K.M.; Shachar, R.; Liman, G.L.S.; Burkhart, B.W.; et al. Dynamic RNA Acetylation Revealed by Quantitative Cross-Evolutionary Mapping. Nature 2020, 583, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Šimonová, A.; Svojanovská, B.; Trylčová, J.; Hubálek, M.; Moravčík, O.; Zavřel, M.; Pávová, M.; Hodek, J.; Weber, J.; Cvačka, J.; et al. LC/MS Analysis and Deep Sequencing Reveal the Accurate RNA Composition in the HIV-1 Virion. Sci. Rep. 2019, 9, 8697. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, N.S.; McIntyre, A.B.R.; McFadden, M.J.; Roder, A.E.; Kennedy, E.M.; Gandara, J.A.; Hopcraft, S.E.; Quicke, K.M.; Vazquez, C.; Willer, J.; et al. N6-Methyladenosine in Flaviviridae Viral RNA Genomes Regulates Infection. Cell Host Microbe 2016, 20, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Jungfleisch, J.; Böttcher, R.; Talló-Parra, M.; Pérez-Vilaró, G.; Merits, A.; Novoa, E.M.; Díez, J. CHIKV Infection Reprograms Codon Optimality to Favor Viral RNA Translation by Altering the tRNA Epitranscriptome. Nat. Commun. 2022, 13, 4725. [Google Scholar] [CrossRef] [PubMed]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’Donovan, C. MetaboLights: A Resource Evolving in Response to the Needs of Its Scientific Community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinform. Oxf. Engl. 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinform. Oxf. Engl. 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A Sequence-Quality Aware, Ultra-Sensitive Variant Caller for Uncovering Cell-Population Heterogeneity from High-Throughput Sequencing Datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef] [PubMed]
- Picardi, E.; Pesole, G. REDItools: High-Throughput RNA Editing Detection Made Easy. Bioinformatics 2013, 29, 1813–1814. [Google Scholar] [CrossRef] [PubMed]
- van der Auwera, G.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra; O’Reilly Media, Incorporated: Sebastopol, CA, USA, 2020; ISBN 978-1-4919-7519-0. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Porath, H.T.; Carmi, S.; Levanon, E.Y. A Genome-Wide Map of Hyper-Edited RNA Reveals Numerous New Sites. Nat. Commun. 2014, 5, 4726. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, R.A.M.; Chen, Z.J. Ggplot2: Elegant Graphics for Data Analysis (2nd ed.). Meas. Interdiscip. Res. Perspect. 2019, 17, 160–167. [Google Scholar] [CrossRef]
- Hahne, F.; Ivanek, R. Visualizing Genomic Data Using Gviz and Bioconductor. Methods Mol. Biol. 2016, 1418, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Shishodia, S.; Zhang, D.; El-Sagheer, A.H.; Brown, T.; Claridge, T.D.W.; Schofield, C.J.; Hopkinson, R.J. NMR Analyses on N-Hydroxymethylated Nucleobases—Implications for Formaldehyde Toxicity and Nucleic Acid Demethylases. Org. Biomol. Chem. 2018, 16, 4021–4032. [Google Scholar] [CrossRef] [PubMed]
- Alseth, I.; Dalhus, B.; Bjørås, M. Inosine in DNA and RNA. Curr. Opin. Genet. Dev. 2014, 26, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Lamers, M.M.; van den Hoogen, B.G.; Haagmans, B.L. ADAR1: “Editor-in-Chief” of Cytoplasmic Innate Immunity. Front. Immunol. 2019, 10, 1763. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.E. Adenosine Deaminase Acting on RNA (ADAR1), a Suppressor of Double-Stranded RNA–Triggered Innate Immune Responses. J. Biol. Chem. 2019, 294, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, W.; Nishikura, K. Adenosine-to-Inosine RNA Editing and Human Disease. Genome Med. 2013, 5, 105. [Google Scholar] [CrossRef] [PubMed]
- Piontkivska, H.; Wales-McGrath, B.; Miyamoto, M.; Wayne, M.L. ADAR Editing in Viruses: An Evolutionary Force to Reckon With. Genome Biol. Evol. 2021, 13, evab240. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Yu, Y.; Wu, X.; Acevedo, A.; Luo, J.-D.; Wang, J.; Schneider, W.M.; Hurwitz, B.; Rosenberg, B.R.; Chung, H.; et al. Decoupling Expression and Editing Preferences of ADAR1 P150 and P110 Isoforms. Proc. Natl. Acad. Sci. USA 2021, 118, e2021757118. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.B.; Sumner, R.P.; Rodriguez-Plata, M.T.; Rasaiyaah, J.; Milne, R.S.; Thrasher, A.J.; Qasim, W.; Towers, G.J. Lentiviral Vector Production Titer Is Not Limited in HEK293T by Induced Intracellular Innate Immunity. Mol. Ther. Methods Clin. Dev. 2020, 17, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Baird, T.D.; Wek, R.C. Eukaryotic Initiation Factor 2 Phosphorylation and Translational Control in Metabolism. Adv. Nutr. 2012, 3, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Yang, C.; Zhao, F.; Huang, Y.; Lin, Y.; Huang, C.; Ma, X.; Du, J.; Wang, Y.; Long, G.; et al. Double-Stranded RNA Deaminase ADAR1 Promotes the Zika Virus Replication by Inhibiting the Activation of Protein Kinase PKR. J. Biol. Chem. 2019, 294, 18168–18180. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Tejedor, A.; Medina, R.; Begik, O.; Cozzuto, L.; Ponomarenko, J.; Novoa, E.M. Native RNA Nanopore Sequencing Reveals Antibiotic-Induced Loss of rRNA Modifications in the A- and P-Sites. bioRxiv 2023. [Google Scholar] [CrossRef]
- Leger, A.; Amaral, P.P.; Pandolfini, L.; Capitanchik, C.; Capraro, F.; Miano, V.; Migliori, V.; Toolan-Kerr, P.; Sideri, T.; Enright, A.J.; et al. RNA Modifications Detection by Comparative Nanopore Direct RNA Sequencing. Nat. Commun. 2021, 12, 7198. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, C.K.; George, C.X.; Samuel, C.E. Adenosine Deaminases Acting on RNA (ADARs) and Viral Infections. Annu. Rev. Virol. 2021, 8, 239–264. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.X.; Chao, M.; Hsieh, S.Y.; Sureau, C.; Nishikura, K.; Taylor, J. A Specific Base Transition Occurs on Replicating Hepatitis Delta Virus RNA. J. Virol. 1990, 64, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.L.; Chen, P.J.; Tu, S.J.; Wang, C.J.; Chen, D.S. The Large Form of Hepatitis Delta Antigen Is Crucial for Assembly of Hepatitis Delta Virus. Proc. Natl. Acad. Sci. USA 1991, 88, 8490–8494. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.K.; Lazinski, D.W. Replicating Hepatitis Delta Virus RNA Is Edited in the Nucleus by the Small Form of ADAR1. Proc. Natl. Acad. Sci. USA 2002, 99, 15118–15123. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.B.; Samuel, C.E. Expression and Regulation by Interferon of a Double-Stranded-RNA-Specific Adenosine Deaminase from Human Cells: Evidence for Two Forms of the Deaminase. Mol. Cell Biol. 1995, 15, 5376–5388. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, R.; Schmid, A.; Eschle, D.; Baczko, K.; ter Meulen, V.; Billeter, M.A. Biased Hypermutation and Other Genetic Changes in Defective Measles Viruses in Human Brain Infections. Cell 1988, 55, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, S.; Galeano, F.; Locatelli, F.; Gallo, A. ADARs and the Balance Game between Virus Infection and Innate Immune Cell Response. Curr. Issues Mol. Biol. 2015, 17, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Carmichael, G.G. Nuclear Antisense RNA Induces Extensive Adenosine Modifications and Nuclear Retention of Target Transcripts. Proc. Natl. Acad. Sci. USA 1997, 94, 3542–3547. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, C.R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie, S.F. HNRNPA2B1 Is a Mediator of m(6)A-Dependent Nuclear RNA Processing Events. Cell 2015, 162, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.; Snyder, J.M.; Connelly, C.; Henao-Mejia, J.; Sidrauski, C.; Stetson, D.B. Protein Kinase R and the Integrated Stress Response Drive Immunopathology Caused by Mutations in the RNA Deaminase ADAR1. Immunity 2021, 54, 1948–1960. [Google Scholar] [CrossRef]
- de Reuver, R.; Maelfait, J. Novel Insights into Double-Stranded RNA-Mediated Immunopathology. Nat. Rev. Immunol. 2024, 24, 235–249. [Google Scholar] [CrossRef]
- Wang, Y.; Samuel, C.E. Adenosine Deaminase ADAR1 Increases Gene Expression at the Translational Level by Decreasing Protein Kinase PKR-Dependent eIF-2alpha Phosphorylation. J. Mol. Biol. 2009, 393, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A Diverse Range of Gene Products Are Effectors of the Type I Interferon Antiviral Response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Clavarino, G.; Cláudio, N.; Couderc, T.; Dalet, A.; Judith, D.; Camosseto, V.; Schmidt, E.K.; Wenger, T.; Lecuit, M.; Gatti, E.; et al. Induction of GADD34 Is Necessary for dsRNA-Dependent Interferon-β Production and Participates in the Control of Chikungunya Virus Infection. PLoS Pathog. 2012, 8, e1002708. [Google Scholar] [CrossRef] [PubMed]
- Begik, O.; Mattick, J.S.; Novoa, E.M. Exploring the Epitranscriptome by Native RNA Sequencing. RNA 2022, 28, 1430–1439. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baquero-Pérez, B.; Bortoletto, E.; Rosani, U.; Delgado-Tejedor, A.; Medina, R.; Novoa, E.M.; Venier, P.; Díez, J. Elucidation of the Epitranscriptomic RNA Modification Landscape of Chikungunya Virus. Viruses 2024, 16, 945. https://doi.org/10.3390/v16060945
Baquero-Pérez B, Bortoletto E, Rosani U, Delgado-Tejedor A, Medina R, Novoa EM, Venier P, Díez J. Elucidation of the Epitranscriptomic RNA Modification Landscape of Chikungunya Virus. Viruses. 2024; 16(6):945. https://doi.org/10.3390/v16060945
Chicago/Turabian StyleBaquero-Pérez, Belinda, Enrico Bortoletto, Umberto Rosani, Anna Delgado-Tejedor, Rebeca Medina, Eva Maria Novoa, Paola Venier, and Juana Díez. 2024. "Elucidation of the Epitranscriptomic RNA Modification Landscape of Chikungunya Virus" Viruses 16, no. 6: 945. https://doi.org/10.3390/v16060945