
Chandipura Virus Forms Cytoplasmic Inclusion Bodies through Phase Separation and Proviral Association of Cellular Protein Kinase R and Stress Granule Protein TIA-1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. CHPV Infection Induces Condensation of Viral Nucleocapsid (N), Phosphoprotein (P) and Large (L) Proteins to Form Cytoplasmic IBs
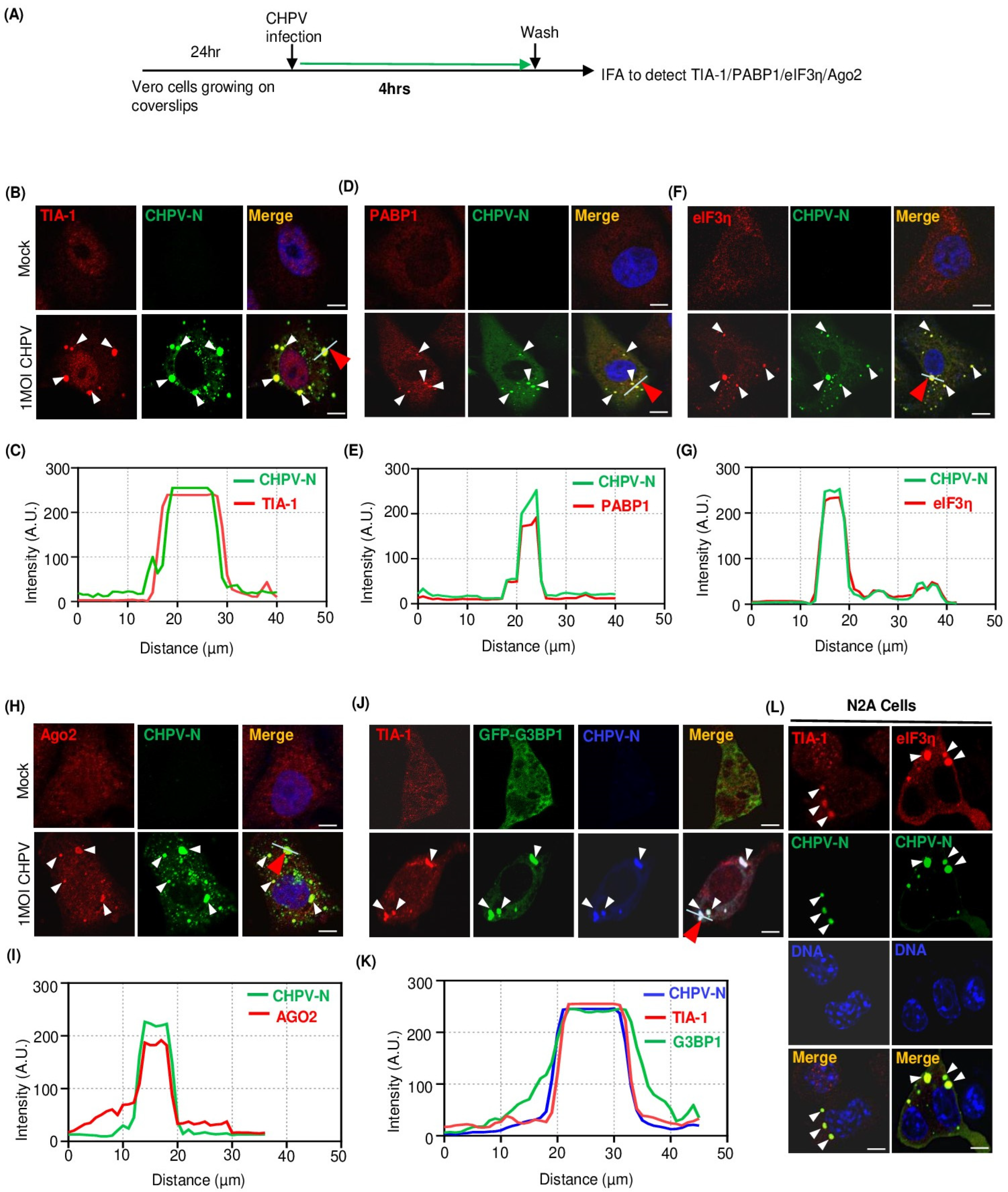
2.2. Recruitment of Multiple Stress Granule Proteins (SGPs) to CHPV-IBs
2.3. CHPV-IBs and Canonical SGs Are Distinct in Terms of Disassembly Dynamics
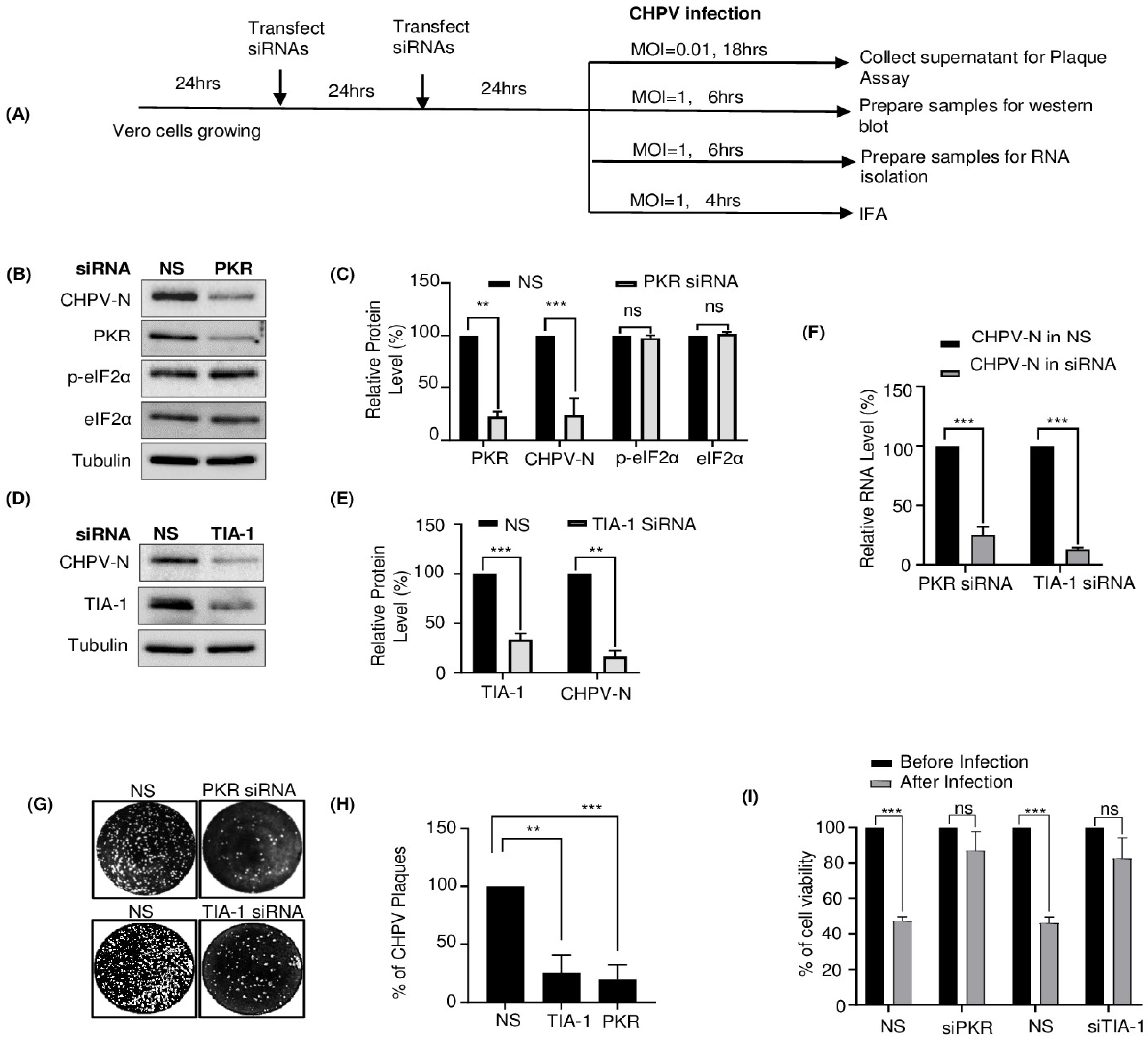
2.4. TIA-1 and PKR Play a Proviral Role in CHPV Replication
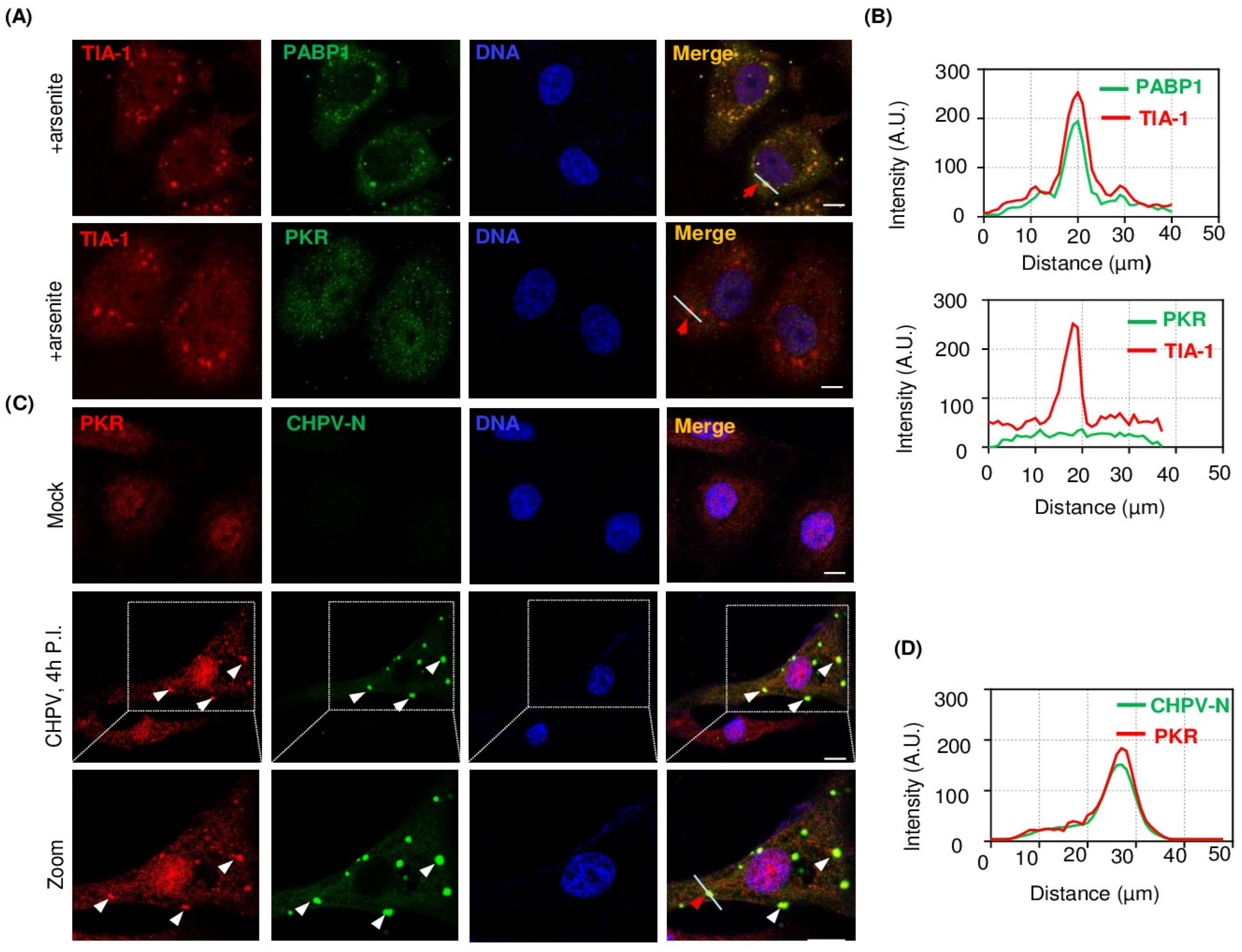
2.5. PKR Undergoes Condensation and Associates with CHPV-IBs
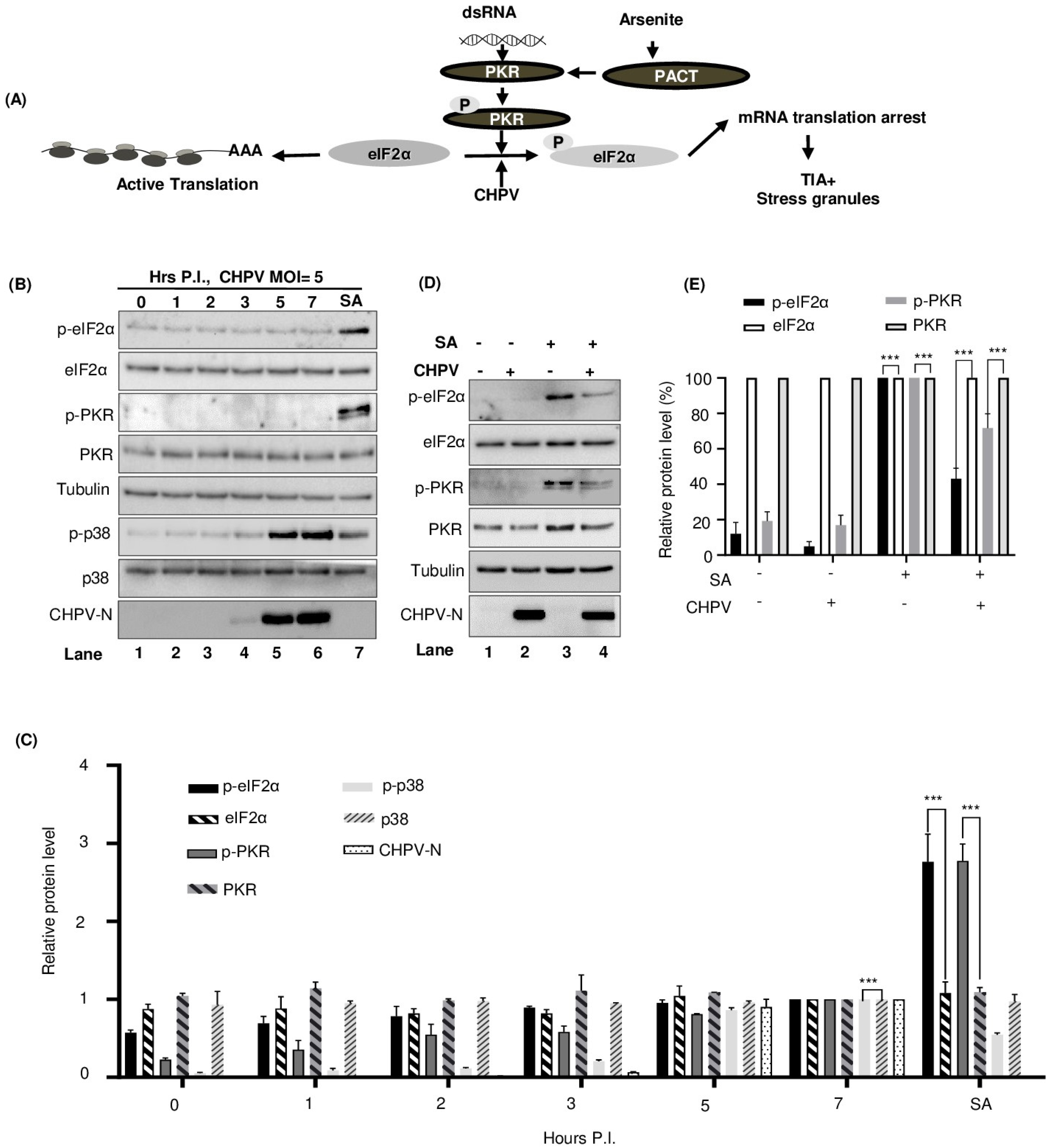
2.6. CHPV-IBs Form Independent of PKR and eIF2α Phosphorylation
3. Discussion
4. Material and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
References
- Basak, S.; Mondal, A.; Polley, S.; Mukhopadhyay, S.; Chattopadhyay, D. Reviewing Chandipura: A vesiculovirus in human epidemics. Biosci. Rep. 2007, 27, 275–298. [Google Scholar] [CrossRef]
- Rao, B.L.; Basu, A.; Wairagkar, N.S.; Gore, M.M.; Arankalle, V.A.; Thakare, J.P.; Jadi, R.S.; Rao, K.A.; Mishra, A.C. A large outbreak of acute encephalitis with high fatality rate in children in Andhra Pradesh, India, in 2003, associated with Chandipura virus. Lancet 2004, 364, 869–874. [Google Scholar] [CrossRef]
- Menghani, S.; Chikhale, R.; Raval, A.; Wadibhasme, P.; Khedekar, P. Chandipura Virus: An emerging tropical pathogen. Acta Trop. 2012, 124, 1–14. [Google Scholar] [CrossRef]
- John, T.J. Chandipura virus, encephalitis, and epidemic brain attack in India. Lancet 2004, 364, 2175. [Google Scholar] [CrossRef]
- Sharma, N.R.; Gadhave, K.; Kumar, P.; Saif, M.; Khan, M.M.; Sarkar, D.P.; Uversky, V.N.; Giri, R. Analysis of the dark proteome of Chandipura virus reveals maximum propensity for intrinsic disorder in phosphoprotein. Sci. Rep. 2021, 11, 13253. [Google Scholar] [CrossRef]
- Marriott, A.C. Complete genome sequences of Chandipura and Isfahan vesiculoviruses. Arch. Virol. 2005, 150, 671–680. [Google Scholar] [CrossRef]
- Zhang, Q.; Sharma, N.R.; Zheng, Z.-M.; Chen, M. Viral Regulation of RNA Granules in Infected Cells. Virol. Sin. 2019, 34, 175–191. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. RNA Granules. J. Cell Biol. 2006, 172, 803–808. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. RNA granules: Post-transcriptional and epigenetic modulators of gene expression. Nat. Rev. Cell Biol. 2009, 10, 430–436. [Google Scholar] [CrossRef]
- Sharma, N.R.; Zheng, Z.M. RNA Granules in Antiviral Innate Immunity: A Kaposi’s Sarcoma-Associated Herpesvirus Journey. Front. Microbiol. 2021, 12, 794431. [Google Scholar] [CrossRef]
- Kedersha, N.; Stoecklin, G.; Ayodele, M.; Yacono, P.; Lykke-Andersen, J.; Fritzler, M.J.; Scheuner, D.; Kaufman, R.J.; Golan, D.E.; Anderson, P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 2005, 169, 871–884. [Google Scholar] [CrossRef]
- Liu, J.; Rivas, F.V.; Wohlschlegel, J.; Yates, J.R., III; Parker, R.; Hannon, G.J. A role for the P-body component GW182 in microRNA function. Nat. Cell Biol. 2005, 7, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Cougot, N.; Babajko, S.; Séraphin, B. Cytoplasmic foci are sites of mRNA decay in human cells. J. Cell Biol. 2004, 165, 31–40. [Google Scholar] [CrossRef]
- Mollet, S.; Cougot, N.; Wilczynska, A.; Dautry, F.; Kress, M.; Bertrand, E.; Weil, D. Translationally repressed mRNA transiently cycles through stress granules during stress. Mol. Biol. Cell 2008, 19, 4469–4479. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. Stressful initiations. J. Cell Sci. 2002, 115, 3227–3234. [Google Scholar] [CrossRef] [PubMed]
- Panas, M.D.; Ivanov, P.; Anderson, P. Mechanistic insights into mammalian stress granule dynamics. J. Cell Biol. 2016, 215, 313–323. [Google Scholar] [CrossRef]
- Wek, R.C. Role of eIF2alpha Kinases in Translational Control and Adaptation to Cellular Stress. Cold Spring Harb. Perspect. Biol. 2018, 10, a032870. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.R.; Majerciak, V.; Kruhlak, M.J.; Zheng, Z.M. KSHV inhibits stress granule formation by viral ORF57 blocking PKR activation. PLoS Pathog. 2017, 13, e1006677. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Lloyd, R.E. Regulation of stress granules in virus systems. Trends Microbiol. 2012, 20, 175–183. [Google Scholar] [CrossRef]
- Reineke, L.C.; Lloyd, R.E. Diversion of stress granules and P-bodies during viral infection. Virology 2013, 436, 255–267. [Google Scholar] [CrossRef]
- Firth, A.E.; Brierley, I. Non-canonical translation in RNA viruses. J. Gen. Virol. 2012, 93, 1385–1409. [Google Scholar] [CrossRef]
- Khaperskyy, D.A.; Hatchette, T.F.; McCormick, C. Influenza A virus inhibits cytoplasmic stress granule formation. FASEB J. 2011, 26, 1629–1639. [Google Scholar] [CrossRef]
- Khaperskyy, D.A.; Emara, M.M.; Johnston, B.P.; Anderson, P.; Hatchette, T.F.; McCormick, C. Influenza a virus host shutoff disables antiviral stress-induced translation arrest. PLoS Pathog. 2014, 10, e1004217. [Google Scholar] [CrossRef]
- White, J.P.; Cardenas, A.M.; Marissen, W.E.; Lloyd, R.E. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2007, 2, 295–305. [Google Scholar] [CrossRef]
- Emara, M.M.; Brinton, M.A. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 9041–9046. [Google Scholar] [CrossRef]
- Valiente-Echeverría, F.; Melnychuk, L.; Vyboh, K.; Ajamian, L.; Gallouzi, I.-E.; Bernard, N.; Mouland, A.J. eEF2 and Ras-GAP SH3 domain-binding protein (G3BP1) modulate stress granule assembly during HIV-1 infection. Nat. Commun. 2014, 5, 4819. [Google Scholar] [CrossRef]
- Matsuki, H.; Takahashi, M.; Higuchi, M.; Makokha, G.N.; Oie, M.; Fujii, M. Both G3BP1 and G3BP2 contribute to stress granule formation. Genes Cells 2013, 18, 135–146. [Google Scholar] [CrossRef]
- Reineke, L.C.; Kedersha, N.; Langereis, M.A.; Kuppeveld, F.J.M.v.; Lloyd, R.E. Stress granules regulate double-stranded RNA-dependent protein kinase activation through a complex containing G3BP1 and Caprin1. MBio 2015, 6, e02486. [Google Scholar] [CrossRef]
- Panas, M.D.; Schulte, T.; Thaa, B.; Sandalova, T.; Kedersha, N.; Achour, A.; McInerney, G.M. Viral and cellular proteins containing FGDF motifs bind G3BP to block stress granule formation. PLoS Pathog. 2015, 11, e1004659. [Google Scholar] [CrossRef]
- Garaigorta, U.; Heim, M.H.; Boyd, B.; Stefan Wieland, F.V.C. Hepatitis C virus (HCV) induces formation of stress granules whose proteins regulate HCV RNA replication and virus assembly and egress. J. Cell Biol. 2012, 86, 11043–11056. [Google Scholar] [CrossRef]
- Ruggieri, A.; Dazert, E.; Metz, P.; Hofmann, S.; Bergeest, J.-P.; Mazur, J.; Bankhead, P.; Hiet, M.-S.; Kallis, S.; Alvisi, G.; et al. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe 2012, 12, 71–85. [Google Scholar] [CrossRef]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef]
- Hyman, A.A.; Weber, C.A.; Jülicher, F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef]
- Sagan, S.M.; Weber, S.C. Let’s phase it: Viruses are master architects of biomolecular condensates. Trends Biochem. Sci. 2022, 48, 229–243. [Google Scholar] [CrossRef]
- Charman, M.; Grams, N.; Kumar, N.; Halko, E.; Dybas, J.M.; Abbott, A.; Lum, K.K.; Blumenthal, D.; Tsopurashvili, E.; Weitzman, M.D. A viral biomolecular condensate coordinates assembly of progeny particles. Nature 2023, 616, 332–338. [Google Scholar] [CrossRef]
- Dolnik, O.; Gerresheim, G.K.; Biedenkopf, N. New Perspectives on the Biogenesis of Viral Inclusion Bodies in Negative-Sense RNA Virus Infections. Cells 2021, 10, 1460. [Google Scholar] [CrossRef]
- Ghosh, S.; Dutta, K.; Basu, A. Chandipura virus induces neuronal death through Fas-mediated extrinsic apoptotic pathway. J. Virol. 2013, 87, 12398–12406. [Google Scholar] [CrossRef]
- Mukesh, R.K.; Kalam, A.A.; Nag, J.; Jaikumar, V.S.; Kunnakkadan, U.; Kumar, N.A.; Suma, S.M.; Rajavelu, A.; Johnson, J.B. Chandipura virus induces cell death in cancer cell lines of human origin and promotes tumor regression in vivo. Mol. Ther. Oncolytics 2021, 23, 254–265. [Google Scholar] [CrossRef]
- Wek, R.C.; Jiang, H.-Y.; Anthony, T.G. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006, 34, 7–11. [Google Scholar] [CrossRef]
- Onomoto, K.; Yoneyama, M.; Fung, G.; Kato, H.; Fujita, T. Antiviral innate immunity and stress granule responses. Trends Immunol. 2014, 35, 420–428. [Google Scholar] [CrossRef]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.-J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef]
- Karginov, F.V.; Hannon, G.J. Remodeling of Ago2-mRNA interactions upon cellular stress reflects miRNA complementarity and correlates with altered translation rates. Genes Dev. 2013, 27, 1624–1632. [Google Scholar] [CrossRef]
- Sharma, N.R.; Wang, X.; Majerciak, V.; Ajiro, M.; Kruhlak, M.; Meyers, C.; Zheng, Z.-M. Cell Type- and Tissue Context-dependent Nuclear Distribution of Human Ago2*. J. Biol. Chem. 2015, 291, 2302–2309. [Google Scholar] [CrossRef]
- Tourrière, H.; Chebli, K.; Zekri, L.; Courselaud, B.; Blanchard, J.M.; Bertrand, E.; Tazi, J. The RasGAP-associated endoribonuclease G3BP mediates stress granule assembly. J. Cell Biol. 2023, 222, e200212128072023new. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. Stress granules: The Tao of RNA triage. Trends Biochem. Sci. 2008, 33, 141–150. [Google Scholar] [CrossRef]
- Testerink, N.; Ajat, M.; Houweling, M.; Brouwers, J.F.; Pully, V.V.; Manen, H.-J.v.; Otto, C.; Helms, J.B.; Vaandrager, A.B. Replacement of Retinyl Esters by Polyunsaturated Triacylglycerol Species in Lipid Droplets of Hepatic Stellate Cells during Activation. PLoS ONE 2012, 7, e34945. [Google Scholar] [CrossRef]
- Preuss, C.; Jelenik, T.; Bódis, K.; Müssig, K.; Burkart, V.; Szendroedi, J.; Roden, M.; Markgraf, D.F. A New Targeted Lipidomics Approach Reveals Lipid Droplets in Liver, Muscle and Heart as a Repository for Diacylglycerol and Ceramide Species in Non-Alcoholic Fatty Liver. Cells 2019, 8, 277. [Google Scholar] [CrossRef]
- de Almeida, P.E.; Toledo, D.A.M.; Rodrigues, G.S.C.; D’Avila, H. Lipid Bodies as Sites of Prostaglandin E2 Synthesis During Chagas Disease: Impact in the Parasite Escape Mechanism. Front. Microbiol. 2018, 9, 499. [Google Scholar] [CrossRef]
- Monson, E.A.; Trenerry, A.M.; Laws, J.L.; Mackenzie, J.M.; Helbig, K.J. Lipid droplets and lipid mediators in viral infection and immunity. FEMS Microbiol. Rev. 2021, 45, fuaa066. [Google Scholar] [CrossRef] [PubMed]
- Anukumar, B.; Amirthalingam, B.G.; Shelke, V.N.; Gunjikar, R.; Shewale, P. Neuro-invasion of Chandipura virus mediates pathogenesis in experimentally infected mice. Int. J. Clin. Exp. Pathol. 2013, 6, 1272–1281. [Google Scholar]
- Kedersha, N.; Cho, M.R.; Li, W.; Yacono, P.W.; Chen, S.; Gilks, N.; Golan, D.E.; Anderson, P. Dynamic shuttling of TIA-1 accompanies the recruitment of mRNA to mammalian stress granules. J. Cell Biol. 2000, 151, 1257–1268. [Google Scholar] [CrossRef]
- Buchan, J.R.; Parker, R. Eukaryotic Stress Granules: The Ins and Out of Translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef]
- Dinh, P.X.; Beura, L.K.; Das, P.B.; Panda, D.; Das, A.; Pattnaik, A.K. Induction of stress granule-like structures in vesicular stomatitis virus-infected cells. J. Virol. 2013, 87, 372–383. [Google Scholar] [CrossRef]
- Desmyter, J.; Melnick, J.L.; Rawls, W.E. Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J. Virol. 1968, 2, 955–961. [Google Scholar] [CrossRef]
- Garcia, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2α kinases: Their structures and functions. Cell Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef]
- Lloyd, R.E. Regulation of stress granules and P-bodies during RNA virus infection. Wiley Interdiscip. Rev. RNA 2013, 4, 317–331. [Google Scholar] [CrossRef]
- Patel, C.V.; Handy, I.; Goldsmith, T.; Patel, R.C. PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J. Biol. Chem. 2000, 275, 37993–37998. [Google Scholar] [CrossRef]
- Li, S.; Peters, G.A.; Ding, K.; Zhang, X.; Qin, J.; Sen, G.C. Molecular basis for PKR activation by PACT or dsRNA. Proc. Natl. Acad. Sci. USA 2006, 103, 10005–10010. [Google Scholar] [CrossRef] [PubMed]
- Romano, P.R.; Garcia-Barrio, M.T.; Zhang, X.; Wang, Q.; Taylor, D.R.; Zhang, F.; Herring, C.; Mathews, M.B.; Qin, J.; Hinnebusch, A.G. Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2alpha kinases PKR and GCN2. Mol. Cell. Biol. 1998, 18, 2282–2297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Romano, P.R.; Nagamura-Inoue, T.; Tian, B.; Dever, T.E.; Mathews, M.B.; Ozato, K.; Hinnebusch, A.G. Binding of double-stranded RNA to protein kinase PKR is required for dimerization and promotes critical autophosphorylation events in the activation loop. J. Biol. Chem. 2001, 276, 24946–24958. [Google Scholar] [CrossRef]
- Verma, A.K.; Ghosh, S.; Basu, A. Chandipura Virus Induced Neuronal Apoptosis via Calcium Signaling Mediated Oxidative Stress. Front. Microbiol. 2018, 9, 1489. [Google Scholar] [CrossRef] [PubMed]
- Anjum, F.; Mohammad, T.; Asrani, P.; Shafie, A.; Singh, S.; Yadav, D.K.; Uversky, V.N.; Hassan, M.I. Identification of intrinsically disorder regions in non-structural proteins of SARS-CoV-2: New insights into drug and vaccine resistance. Mol. Cell. Biochem. 2022, 477, 1607–1619. [Google Scholar] [CrossRef] [PubMed]
- Giri, R.; Kumar, D.; Sharma, N.; Uversky, V.N. Intrinsically Disordered Side of the Zika Virus Proteome. Front. Cell. Infect. Microbiol. 2016, 6, 144. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradović, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Kuechler, E.R.; Zhang, J.; Matalon; Dubreuil, B.; Hofmann, A.; Loewen, C.; Levy, E.D.; Gsponer, J.; Mayor, T. Proteomic analysis reveals the direct recruitment of intrinsically disordered regions to stress granules in S. cerevisiae. J. Cell Sci. 2020, 133, jcs244657. [Google Scholar] [CrossRef] [PubMed]
- Vandelli, A.; Samper, F.C.; Burgas, M.T.; Groot, N.S.d.; Tartaglia, G.G. The Interplay Between Disordered Regions in RNAs and Proteins Modulates Interactions Within Stress Granules and Processing Bodies. J. Mol. Biol. 2022, 434, 167159. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Brocca, S.; Longhi, S.; Uversky, V.N. Liaisons dangereuses: Intrinsic Disorder in Cellular Proteins Recruited to Viral Infection-Related Biocondensates. Int. J. Mol. Sci. 2023, 24, 2151. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, J.; Le Bars, R.; Lama, Z.; Scrima, N.; Lagaudriere-Gesbert, C.; Gaudin, Y.; Blondel, D. Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 2017, 8, 58. [Google Scholar] [CrossRef]
- Aulas, A.; Fay, M.M.; Lyons, S.M.; Achorn, C.A.; Kedersha, N.; Anderson, P.; Ivanov, P. Stress-specific differences in assembly and composition of stress granules and related foci. J. Cell Sci. 2017, 130, 927–937. [Google Scholar] [CrossRef]
- Li, W.; Li, Y.; Kedersha, N.; Anderson, P.; Emara, M.; Swiderek, K.M.; Moreno, G.T.; Brinton, M.A. Cell proteins TIA-1 and TIAR interact with the 3’ stem-loop of the West Nile virus complementary minus-strand RNA and facilitate virus replication. J. Virol. 2002, 76, 11989–12000. [Google Scholar] [CrossRef]
- Cesaro, T.; Michiels, T. Inhibition of PKR by Viruses. Front. Microbiol. 2021, 12, 757238. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Abel, A.M.; Nelson, E. Protein kinase R (PKR) plays a pro-viral role in porcine reproductive and respiratory syndrome virus (PRRSV) replication by modulating viral gene transcription. Arch. Virol. 2016, 161, 327–333. [Google Scholar] [CrossRef]
- Gaoa, P.; Liua, Y.; Wanga, H.; Chaia, Y.; Wenga, W.; Zhanga, Y.; Zhoua, L.; Gea, X.; Guoa, X.; Hana, J.; et al. Viral evasion of PKR restriction by reprogramming cellular stress granules. Proc. Natl. Acad. Sci. USA 2022, 119, e2201169119. [Google Scholar] [CrossRef]
- Lu, J.; O’Hara, E.B.; Trieselmann, B.A.; Romano, P.R.; Dever, T.E. The interferon-induced double-stranded RNA-activated protein kinase PKR will phosphorylate serine, threonine, or tyrosine at residue 51 in eukaryotic initiation factor 2alpha. J. Biol. Chem. 1999, 274, 32198–32203. [Google Scholar] [CrossRef]
- Rayman, J.B.; Karl, K.A.; Kandel, E.R. TIA-1 Self-Multimerization, Phase Separation, and Recruitment into Stress Granules Are Dynamically Regulated by Zn2. Cell Rep. 2018, 22, 59–71. [Google Scholar] [CrossRef]
- Sharma, N.R.; Majerciak, V.; Kruhlak, M.J.; Yu, L.; Kang, J.G.; Yang, A.; Shou, G.; Fritzler, J.M.; Zheng, Z.M. KSHV RNA-binding protein ORF57 inhibits P-body formation to promote viral multiplication by interaction with Ago2 and GW182. Nucleic Acids Res. 2019, 17, 9368–9385. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarkar, S.; Ganguly, S.; Ganguly, N.K.; Sarkar, D.P.; Sharma, N.R. Chandipura Virus Forms Cytoplasmic Inclusion Bodies through Phase Separation and Proviral Association of Cellular Protein Kinase R and Stress Granule Protein TIA-1. Viruses 2024, 16, 1027. https://doi.org/10.3390/v16071027
Sarkar S, Ganguly S, Ganguly NK, Sarkar DP, Sharma NR. Chandipura Virus Forms Cytoplasmic Inclusion Bodies through Phase Separation and Proviral Association of Cellular Protein Kinase R and Stress Granule Protein TIA-1. Viruses. 2024; 16(7):1027. https://doi.org/10.3390/v16071027
Chicago/Turabian StyleSarkar, Sharmistha, Surajit Ganguly, Nirmal K. Ganguly, Debi P. Sarkar, and Nishi Raj Sharma. 2024. "Chandipura Virus Forms Cytoplasmic Inclusion Bodies through Phase Separation and Proviral Association of Cellular Protein Kinase R and Stress Granule Protein TIA-1" Viruses 16, no. 7: 1027. https://doi.org/10.3390/v16071027
APA StyleSarkar, S., Ganguly, S., Ganguly, N. K., Sarkar, D. P., & Sharma, N. R. (2024). Chandipura Virus Forms Cytoplasmic Inclusion Bodies through Phase Separation and Proviral Association of Cellular Protein Kinase R and Stress Granule Protein TIA-1. Viruses, 16(7), 1027. https://doi.org/10.3390/v16071027