
Molecular Mechanisms Involved in the B Cell Growth and Clonogenic Activity of HIV-1 Matrix Protein p17 Variants

 ,
,  ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Modeling of refp17 and vp17s
2.2. Classic Molecular Dynamics
2.3. Accelerated Molecular Dynamics
2.4. Molecular Dynamics Data Analysis
2.5. Cell Cultures
2.6. Recombinant Proteins and Monoclonal Antibodies to p17
2.7. B Cell Colony Formation Assay
2.8. Statistical Analysis
3. Results
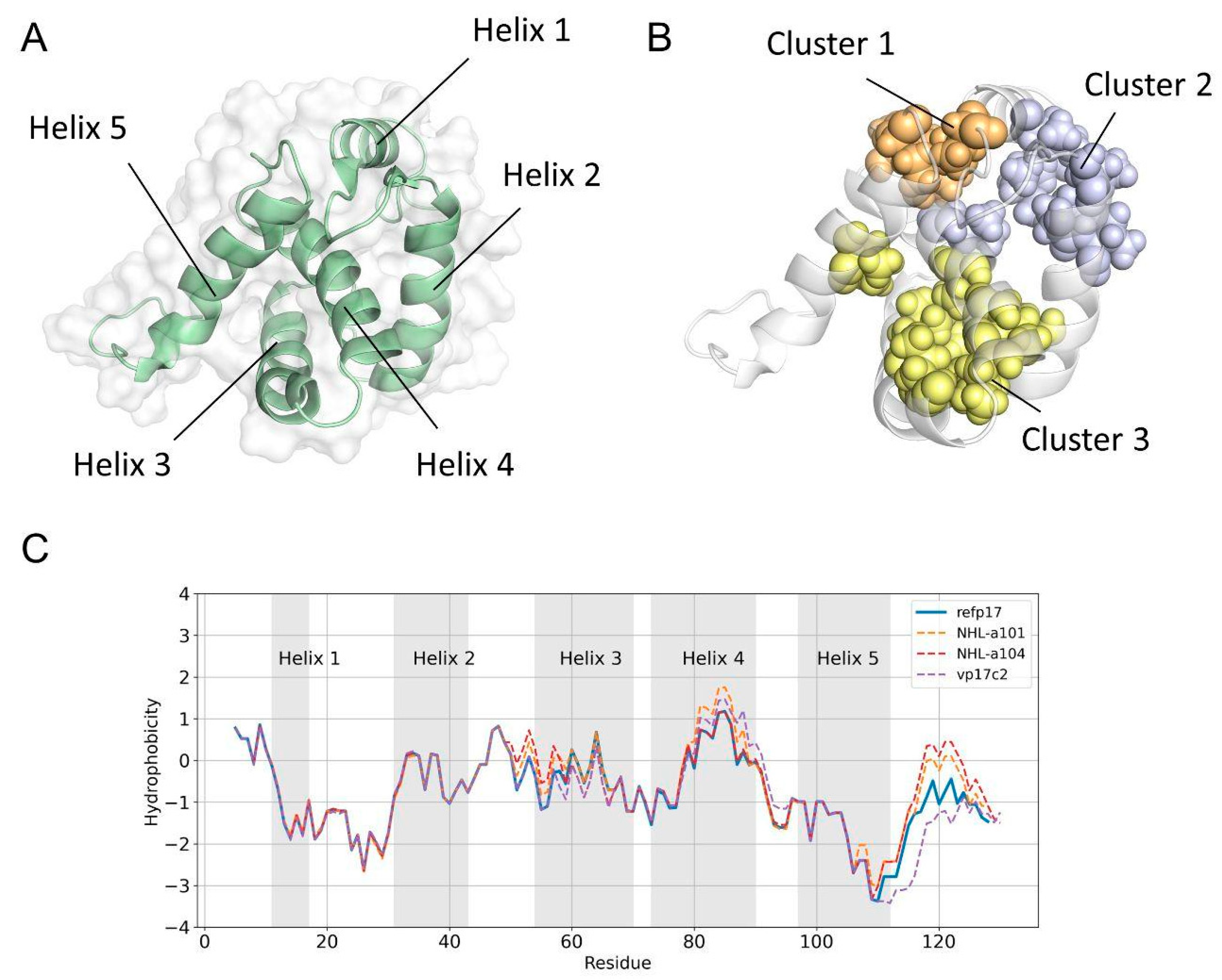
3.1. A Double Amino Acid Insertion Induces Changes in the Hydrophobic Profile of vp17s
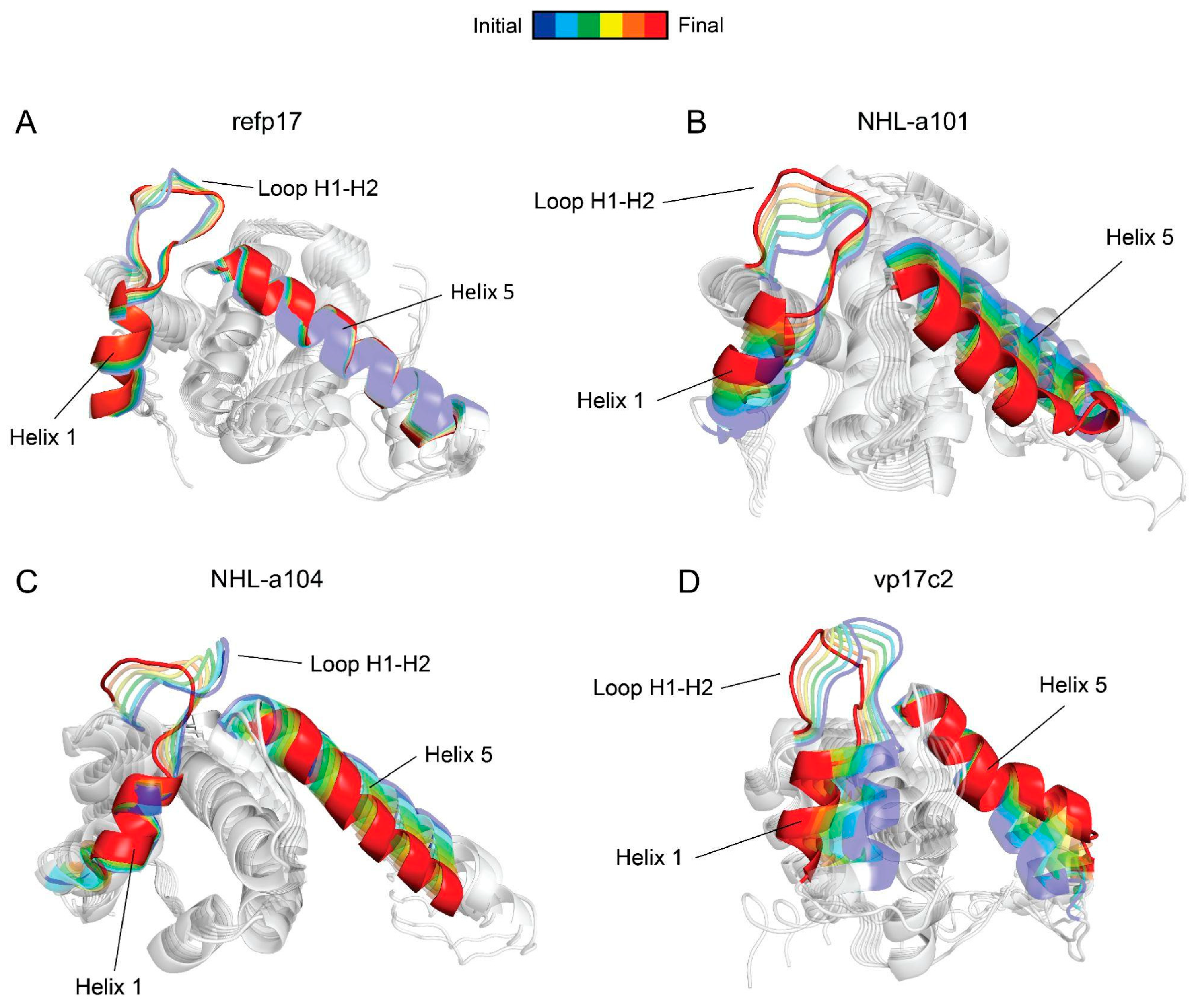
3.2. Refp17 and vp17s Show Critical Behavioral Differences
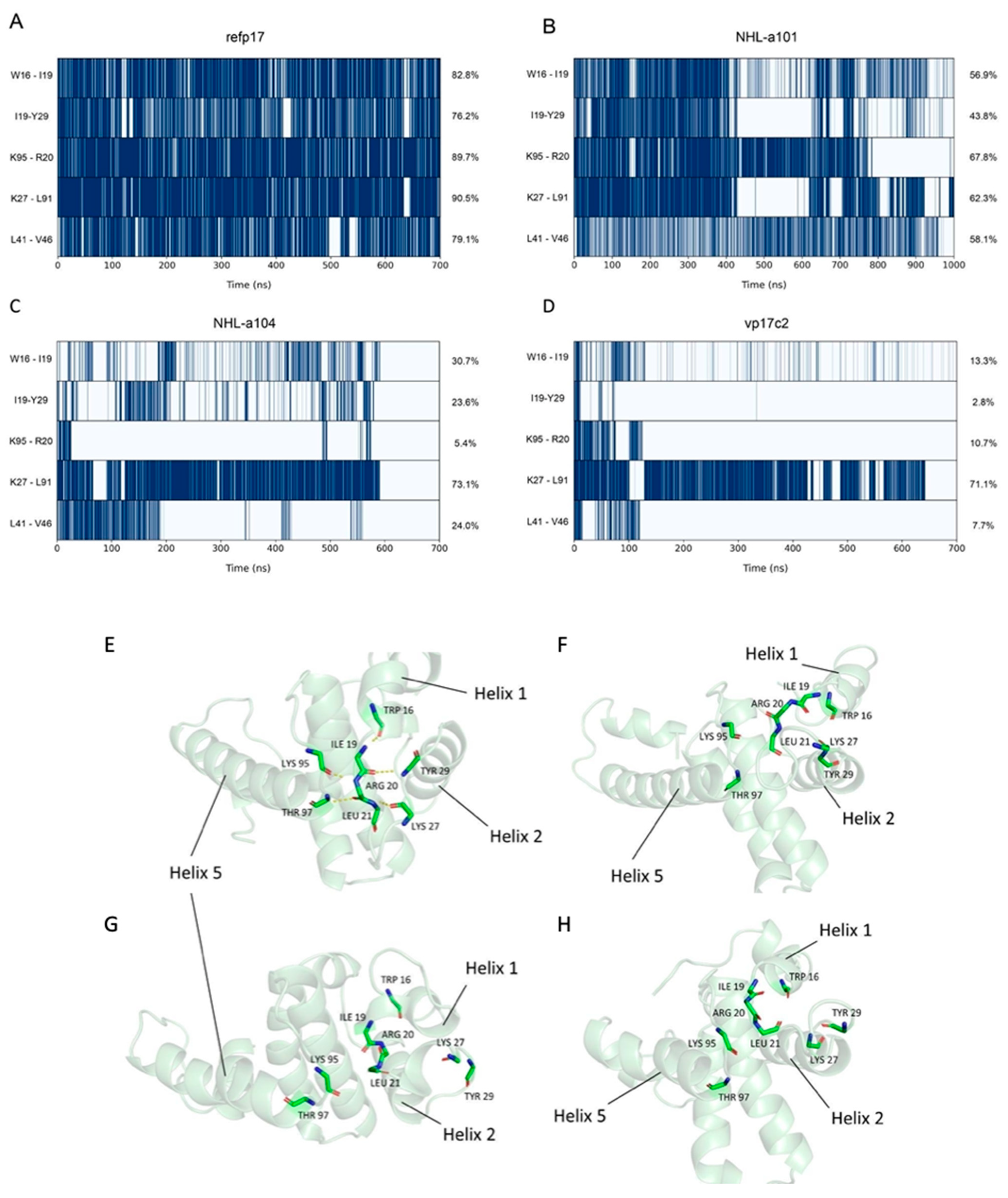
3.3. Vp17s Loss of Interaction
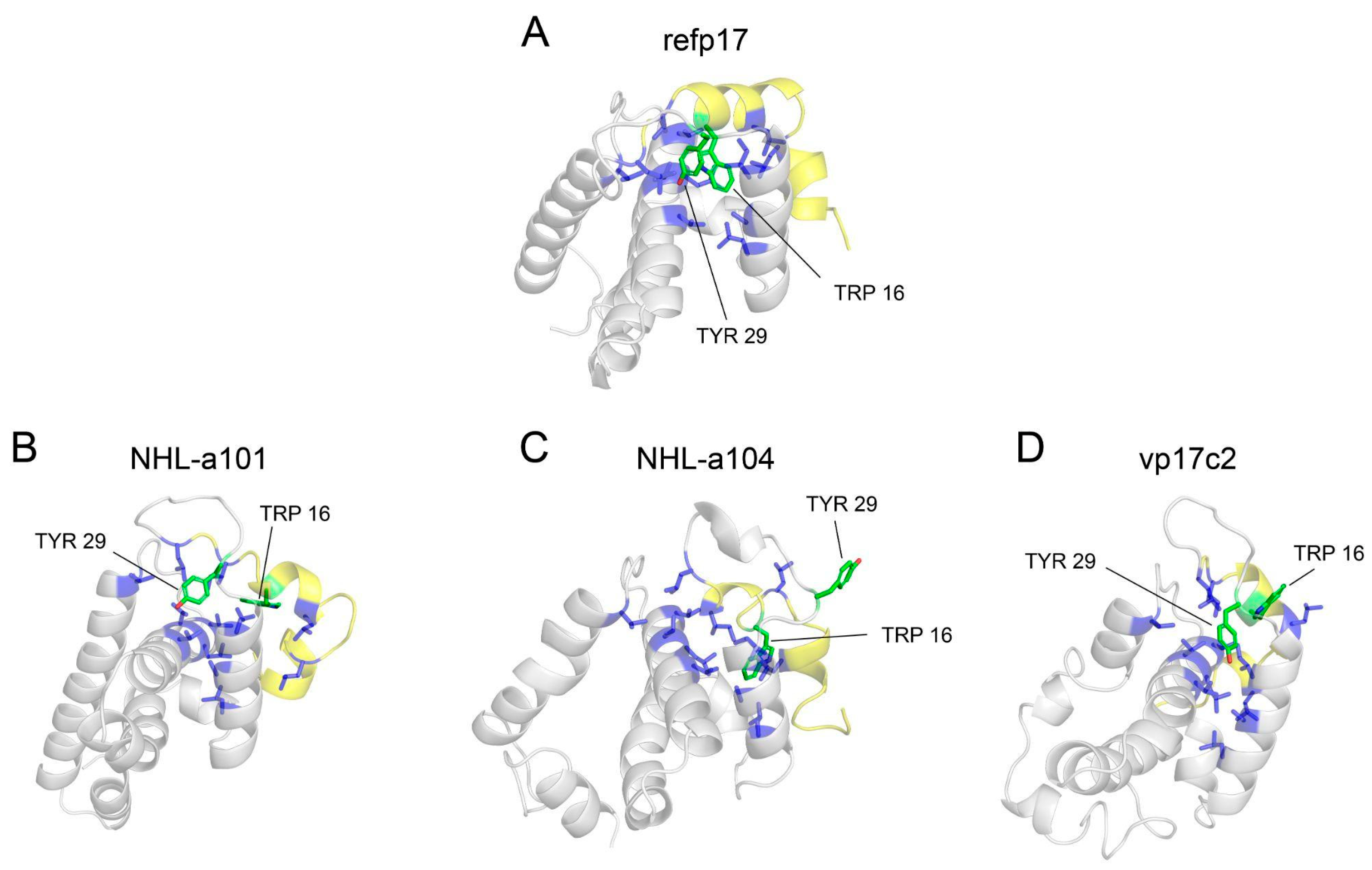
3.4. Trp16 and Tyr29 Play a Key Role in Maintaining the Stability of HC-AT20
3.5. Conformational Changes in the AT20 Region of vp17s
3.6. Trp16 and Tyr29 Are Fundamental Core Residues for the Structural Maintenance of the HC-AT20
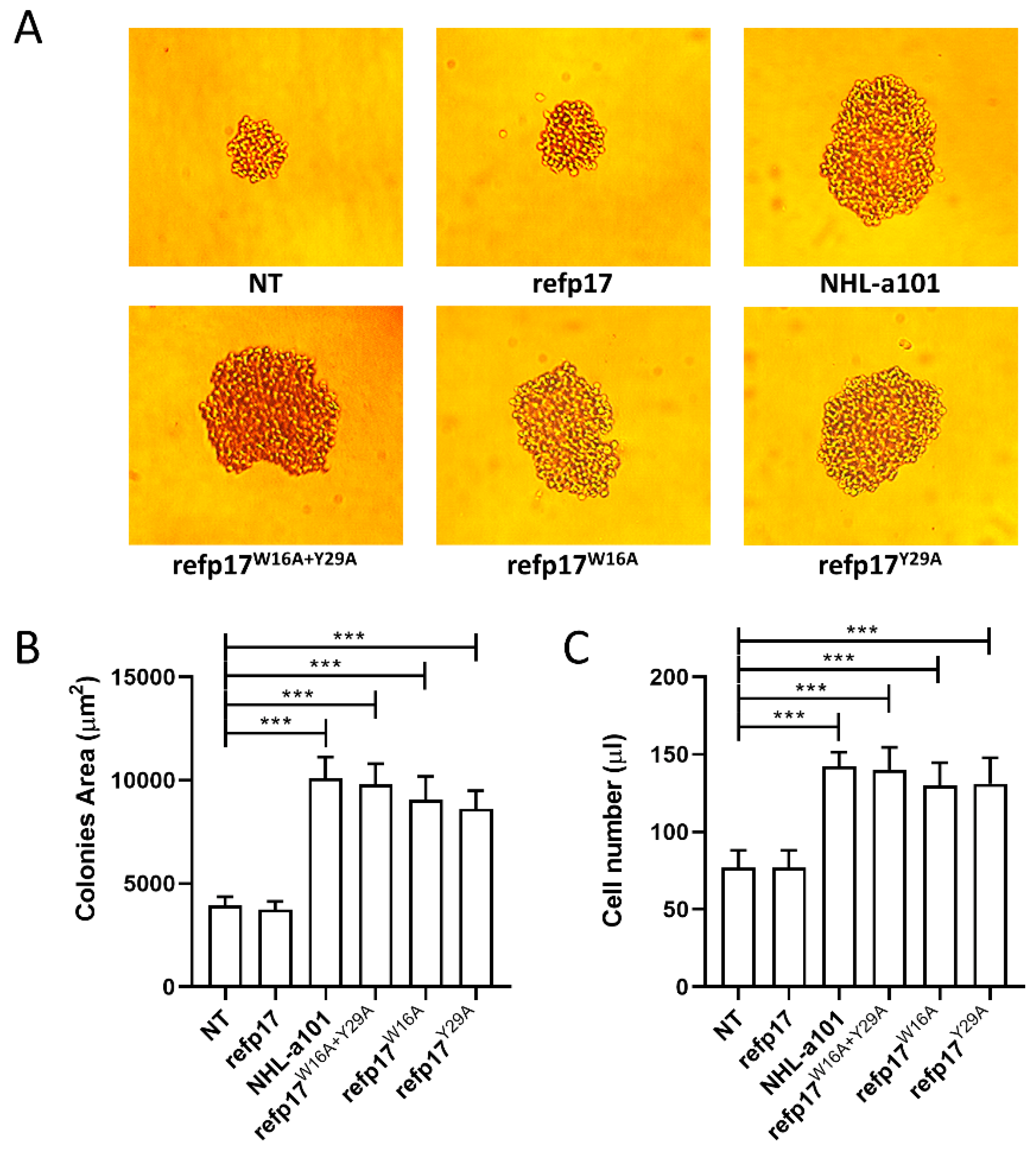
3.7. Replacement of Trp16 and Tyr29 with Alanine on the refp17 Backbone Confers B Cell Clonogenic Activity to the Viral Protein
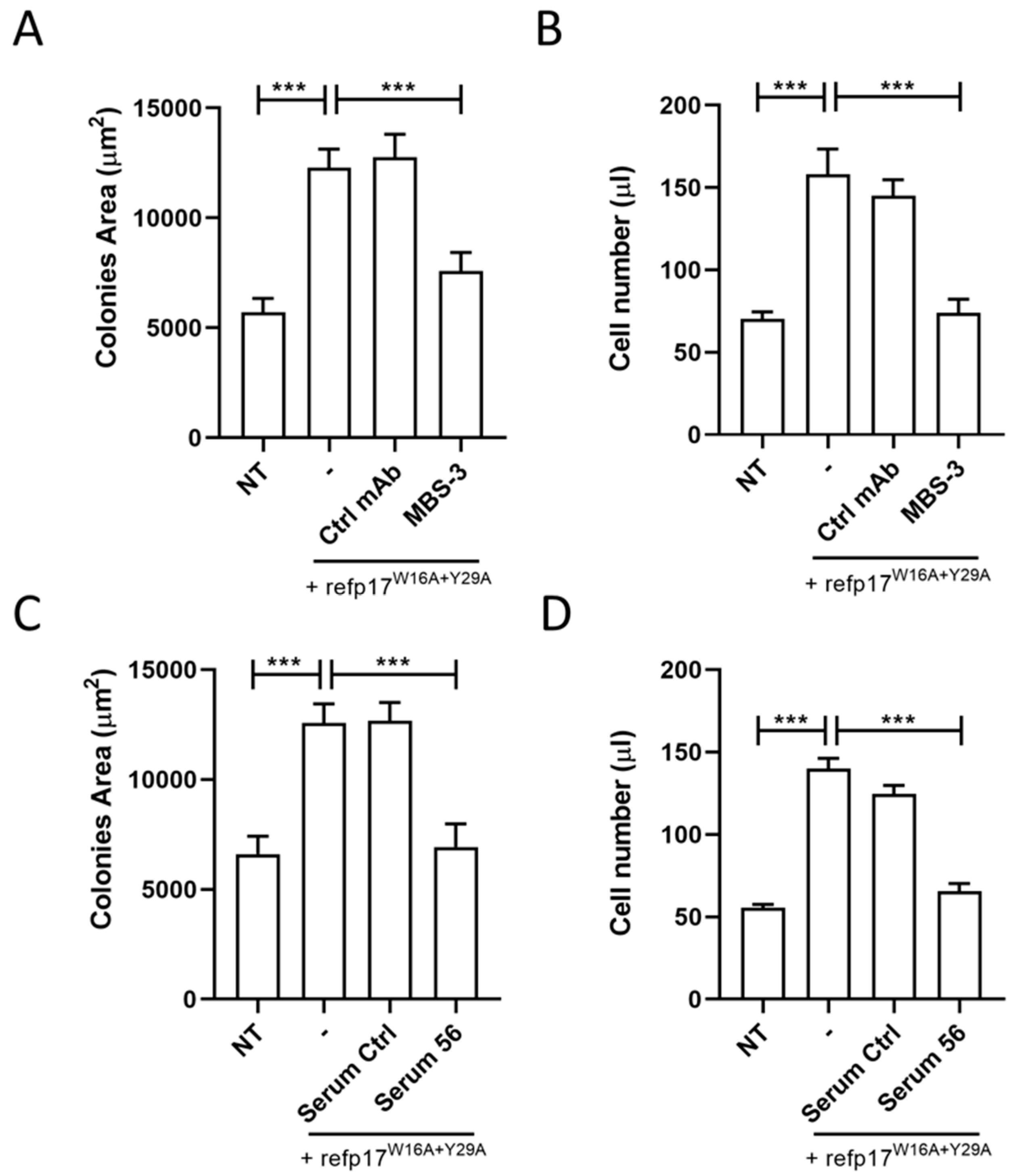
3.8. The p17-Neutralizing mAb MBS-3 Impairs the refp17W16A+Y29A Cell-Growth-Promoting Activity
3.9. Serum Derived from an HIV+ Patient Immunized with AT20-KLH Suppressed the refp17W16A+Y29A Clonogenic Activity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fiorentini, S.; Marini, E.; Caracciolo, S.; Caruso, A. Functions of the HIV-1 matrix protein p17. New Microbiol. 2006, 29, 1–10. [Google Scholar] [PubMed]
- Mazzuca, P.; Caruso, A.; Caccuri, F. HIV-1 infection, microenvironment and endothelial cell dysfunction. New Microbiol. 2016, 39, 163–173. [Google Scholar] [PubMed]
- Popovic, M.; Tenner-Racz, K.; Pelser, C.; Stellbrink, H.J.; van Lunzen, J.; Lewis, G.; Kalyanaraman, V.S.; Gallo, R.C.; Racz, P. Persistence of HIV-1 structural proteins and glycoproteins in lymph nodes of patients under highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2005, 102, 14807–14812. [Google Scholar] [CrossRef] [PubMed]
- Caccuri, F.; Iaria, M.L.; Campilongo, F.; Varney, K.; Rossi, A.; Mitola, S.; Schiarea, S.; Bugatti, A.; Mazzuca, P.; Giagulli, C.; et al. Cellular aspartyl proteases promote the unconventional secretion of biologically active HIV-1 matrix protein p17. Sci. Rep. 2016, 6, 38027. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, S.; Giagulli, C.; Caccuri, F.; Magiera, A.K.; Caruso, A. HIV-1 matrix protein p17: A candidate antigen for therapeutic vaccines against AIDS. Pharmacol. Ther. 2010, 128, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.P.; Worthylake, D.; Bancroft, D.P.; Christensen, A.M.; Sundquist, W.I. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: Implications for membrane association and assembly. Proc. Natl. Acad. Sci. USA 1996, 93, 3099–3104. [Google Scholar] [CrossRef]
- Dolcetti, R.; Giagulli, C.; He, W.; Selleri, M.; Caccuri, F.; Eyzaguirre, L.M.; Mazzuca, P.; Corbellini, S.; Campilongo, F.; Marsico, S.; et al. Role of HIV-1 matrix protein p17 variants in lymphoma pathogenesis. Proc. Natl. Acad. Sci. USA 2015, 112, 14331–14336. [Google Scholar] [CrossRef] [PubMed]
- Giagulli, C.; Marsico, S.; Magiera, A.K.; Bruno, R.; Caccuri, F.; Barone, I.; Fiorentini, S.; Andò, S.; Caruso, A. Opposite effects of HIV-1 p17 variants on PTEN activation and cell growth in B cells. PLoS ONE 2011, 6, e17831. [Google Scholar] [CrossRef] [PubMed]
- Caccuri, F.; Messali, S.; Zani, A.; Campisi, G.; Giovanetti, M.; Zanussi, S.; Vaccher, E.; Fabris, S.; Bugatti, A.; Focà, E.; et al. HIV-1 mutants expressing B cell clonogenic matrix protein p17 variants are increasing their prevalence worldwide. Proc. Natl. Acad. Sci. USA 2022, 119, e2122050119. [Google Scholar] [CrossRef]
- Dolcetti, R.; Gloghini, A.; Caruso, A.; Carbone, A. A lymphomagenic role for HIV beyond immune suppression? Blood 2016, 127, 1403–1409. [Google Scholar] [CrossRef]
- Giagulli, C.; Caccuri, F.; Zorzan, S.; Bugatti, A.; Zani, A.; Filippini, F.; Manocha, E.; D’Ursi, P.; Orro, A.; Dolcetti, R.; et al. B-cell clonogenic activity of HIV-1 p17 variants is driven by PAR1-mediated EGF transactivation. Cancer Gene Ther. 2021, 28, 649–666. [Google Scholar] [CrossRef]
- Giagulli, C.; D’Ursi, P.; He, W.; Zorzan, S.; Caccuri, F.; Varney, K.; Orro, A.; Marsico, S.; Otjacques, B.; Laudanna, C.; et al. A single amino acid substitution confers B-cell clonogenic activity to the HIV-1 matrix protein p17. Sci. Rep. 2017, 7, 6555. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Mazzuca, P.; Yuan, W.; Varney, K.; Bugatti, A.; Cagnotto, A.; Giagulli, C.; Rusnati, M.; Marsico, S.; Diomede, L.; et al. Identification of amino acid residues critical for the B cell growth-promoting activity of HIV-1 matrix protein p17 variants. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 13–24. [Google Scholar] [CrossRef]
- Miotto, M.; Olimpieri, P.P.; Di Rienzo, L.; Ambrosetti, F.; Corsi, P.; Lepore, R.; Tartaglia, G.G.; Milanetti, E. Insights on protein thermal stability: A graph representation of molecular interactions. Bioinformatics 2019, 35, 2569–2577. [Google Scholar] [CrossRef]
- Priyakumar, U.D. Role of hydrophobic core on the thermal stability of proteins—Molecular dynamics simulations on a single point mutant of Sso7d abstract. J. Biomol. Struct. Dyn. 2012, 29, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Van den Burg, B.; Dijkstra, B.W.; Vriend, G.; Van der Vinne, B.; Venema, G.; Eijsink, V.G. Protein stabilization by hydrophobic interactions at the surface. Eur. J. Biochem. 1994, 220, 981–985. [Google Scholar] [CrossRef]
- Banach, M.; Fabian, P.; Stapor, K.; Konieczny, L.; Roterman, A.I. Structure of the Hydrophobic Core Determines the 3D Protein Structure-Verification by Single Mutation Proteins. Biomolecules 2020, 10, 767. [Google Scholar] [CrossRef] [PubMed]
- Fabian, P.; Stapor, K.; Banach, M.; Ptak-Kaczor, M.; Konieczny, L.; Roterman, I. Alternative Hydrophobic Core in Proteins—The Effect of Specific Synergy. Symmetry 2020, 12, 273. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Sali, A.A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK—A program to check the stereochemical quality of protein structures. J. App. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo-distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham III, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Machado, M.R.; Pantano, S. Split the Charge Difference in Two! A Rule of Thumb for Adding Proper Amounts of Ions in MD Simulations. J. Chem. Theory Comput. 2020, 16, 1367–1372. [Google Scholar] [CrossRef]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef]
- Miao, Y.; Feixas, F.; Eun, C.; McCammon, J.A. Accelerated molecular dynamics simulations of protein folding. J. Comput. Chem. 2015, 36, 1536–1549. [Google Scholar] [CrossRef]
- Pierce, L.C.; Salomon-Ferrer, R.; Augusto F de Oliveira, C.; McCammon, J.A.; Walker, R.C. Routine Access to Millisecond Time Scale Events with Accelerated Molecular Dynamics. J. Chem. Theory Comput. 2012, 8, 2997–3002. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. HBonanza: A computer algorithm for molecular-dynamics-trajectory hydrogen-bond analysis. J. Mol. Graph. Model. 2011, 31, 5–9. [Google Scholar] [CrossRef]
- Tina, K.G.; Bhadra, R.; Srinivasan, N. PIC: Protein Interactions Calculator. Nucleic Acids Res. 2007, 35, W473–W476. [Google Scholar] [CrossRef]
- De Francesco, M.A.; Baronio, M.; Fiorentini, S.; Signorini, C.; Bonfanti, C.; Poiesi, C.; Popovic, M.; Grassi, M.; Garrafa, E.; Bozzo, L.; et al. HIV-1 matrix protein p17 increases the production of proinflammatory cytokines and counteracts IL-4 activity by binding to a cellular receptor. Proc. Natl. Acad. Sci. USA 2002, 99, 9972–9977. [Google Scholar] [CrossRef]
- Caccuri, F.; Giagulli, C.; Bugatti, A.; Benetti, A.; Alessandri, G.; Ribatti, D.; Marsico, S.; Apostoli, P.; Slevin, M.A.; Rusnati, M.; et al. HIV-1 matrix protein p17 promotes angiogenesis via chemokine receptors CXCR1 and CXCR2. Proc. Natl. Acad. Sci. USA 2012, 109, 14580–14585. [Google Scholar] [CrossRef]
- Kathuria, S.V.; Chan, Y.H.; Nobrega, R.P.; Özen, A.; Matthews, C.R. Clusters of isoleucine, leucine, and valine side chains define cores of stability in high-energy states of globular proteins: Sequence determinants of structure and stability. Protein Sci. 2016, 25, 662–675. [Google Scholar] [CrossRef]
- Yue, P.; Li, Z.; Moult, J. Loss of protein structure stability as a major causative factor in monogenic disease. J. Mol. Biol. 2005, 353, 459–473. [Google Scholar] [CrossRef]
- Yue, P.; Melamud, E.; Moult, J. SNPs3D: Candidate gene and SNP selection for association studies. BMC Bioinform. 2006, 7, 166. [Google Scholar] [CrossRef]
- Feichtinger, H.; Putkonen, P.; Parravicini, C.; Li, S.L.; Kaaya, E.E.; Böttiger, D.; Biberfeld, G.; Biberfeld, P. Malignant lymphomas in cynomolgus monkeys infected with simian immunodeficiency virus. Am. J. Pathol. 1990, 137, 1311–1315. [Google Scholar]
- Rao, Z.; Belyaev, A.S.; Fry, E.; Roy, P.; Jones, I.M.; Stuart, D.I. Crystal structure of SIV matrix antigen and implications for virus assembly. Nature 1995, 378, 743–747. [Google Scholar] [CrossRef]
- Massiah, M.A.; Starich, M.R.; Paschall, C.; Summers, M.F.; Christensen, A.M.; Sundquist, W.I. Three-dimensional structure of the human immunodeficiency virus type 1 matrix protein. J. Mol. Biol. 1994, 244, 198–223. [Google Scholar] [CrossRef]
- Matthews, S.; Barlow, P.; Clark, N.; Kingsman, S.; Kingsman, A.; Campbell, I. Refined solution structure of p17, the HIV matrix protein. Biochem. Soc. Trans. 1995, 23, 725–729. [Google Scholar] [CrossRef]
- Galindo-Murillo, R.; Roe, D.R.; Cheatham, T.E., 3rd. Convergence and reproducibility in molecular dynamics simulations of the DNA duplex d(GCACGAACGAACGAACGC). Biochim. Biophys. Acta 2015, 1850, 1041–1058. [Google Scholar] [CrossRef] [PubMed]
- Iaria, M.L.; Fiorentini, S.; Focà, E.; Zicari, S.; Giagulli, C.; Caccuri, F.; Francisci, D.; Di Perri, G.; Castelli, F.; Baldelli, F.; et al. Synthetic HIV-1 matrix protein p17-based AT20-KLH therapeutic immunization in HIV-1-infected patients receiving antiretroviral treatment: A phase I safety and immunogenicity study. Vaccine 2014, 32, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Focà, E.; Iaria, M.L.; Caccuri, F.; Fiorentini, S.; Motta, D.; Giagulli, C.; Castelli, F.; Caruso, A. Long-lasting humoral immune response induced in HIV-1-infected patients by a synthetic peptide (AT20) derived from the HIV-1 matrix protein p17 functional epitope. HIV Clin. Trials 2015, 16, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Hoang, T.X.; Trovato, A.; Seno, F.; Banavar, J.R.; Maritan, A. Geometry and symmetry presculpt the free-energy landscape of proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 7960–7964. [Google Scholar] [CrossRef]
- Venkat, A.; Tehrani, D.; Taujale, R.; Yeung, W.; Gravel, N.; Moremen, K.W.; Kannan, N. Modularity of the hydrophobic core and evolution of functional diversity in fold A glycosyltransferases. J. Biol. Chem. 2022, 298, 102212. [Google Scholar] [CrossRef]
- Wu, Z.; Alexandratos, J.; Ericksen, B.; Lubkowski, J.; Gallo, R.C.; Lu, W. Total chemical synthesis of N-myristoylated HIV-1 matrix protein p17: Structural and mechanistic implications of p17 myristoylation. Proc. Natl. Acad. Sci. USA 2004, 101, 11587–11592. [Google Scholar] [CrossRef] [PubMed]
- Saad, J.S.; Miller, J.; Tai, J.; Kim, A.; Ghanam, R.H.; Summers, M.F. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. USA 2006, 103, 11364–11369. [Google Scholar] [CrossRef]
- Bugatti, A.; Caccuri, F.; Filippini, F.; Ravelli, C.; Caruso, A. Binding to PI(4,5)P2 is indispensable for secretion of B-cell clonogenic HIV-1 matrix protein p17 variants. J. Biol. Chem. 2021, 297, 100934. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, A.K.; Campbell, N.; Panganiban, A.; Ratner, L. Characterization of replication defects induced by mutations in the basic domain and C terminus of HIV-1 matrix. Virology 2007, 369, 47–54. [Google Scholar] [CrossRef]
- Caccuri, F.; Rueckert, C.; Giagulli, C.; Schulze, K.; Basta, D.; Zicari, S.; Marsico, S.; Cervi, E.; Fiorentini, S.M.; Slevin, C.A.; et al. HIV-1 matrix protein p17 promotes lymphangiogenesis and activates the endothelin-1/endothelin B receptor axis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 846–856. [Google Scholar] [CrossRef]
- Paduch, R. The role of lymphangiogenesis and angiogenesis in tumor metastasis. Cell. Oncol. 2016, 39, 397–410. [Google Scholar] [CrossRef]
- Fiorentini, S.; Marini, E.; Bozzo, L.; Trainini, L.; Saadoune, L.; Avolio, M.; Pontillo, A.; Bonfanti, C.; Sarmientos, P.; Caruso, A. Preclinical studies on immunogenicity of the HIV-1 p17-based synthetic peptide AT20-KLH. Biopolymers 2004, 76, 334–343. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Ursi, P.; Rondina, A.; Zani, A.; Uggeri, M.; Messali, S.; Caruso, A.; Caccuri, F. Molecular Mechanisms Involved in the B Cell Growth and Clonogenic Activity of HIV-1 Matrix Protein p17 Variants. Viruses 2024, 16, 1048. https://doi.org/10.3390/v16071048
D’Ursi P, Rondina A, Zani A, Uggeri M, Messali S, Caruso A, Caccuri F. Molecular Mechanisms Involved in the B Cell Growth and Clonogenic Activity of HIV-1 Matrix Protein p17 Variants. Viruses. 2024; 16(7):1048. https://doi.org/10.3390/v16071048
Chicago/Turabian StyleD’Ursi, Pasqualina, Alessandro Rondina, Alberto Zani, Matteo Uggeri, Serena Messali, Arnaldo Caruso, and Francesca Caccuri. 2024. "Molecular Mechanisms Involved in the B Cell Growth and Clonogenic Activity of HIV-1 Matrix Protein p17 Variants" Viruses 16, no. 7: 1048. https://doi.org/10.3390/v16071048