
The Distal Promoter of the B438L Gene of African Swine Fever Virus Is Responsible for the Transcription of the Alternatively Spliced B169L

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
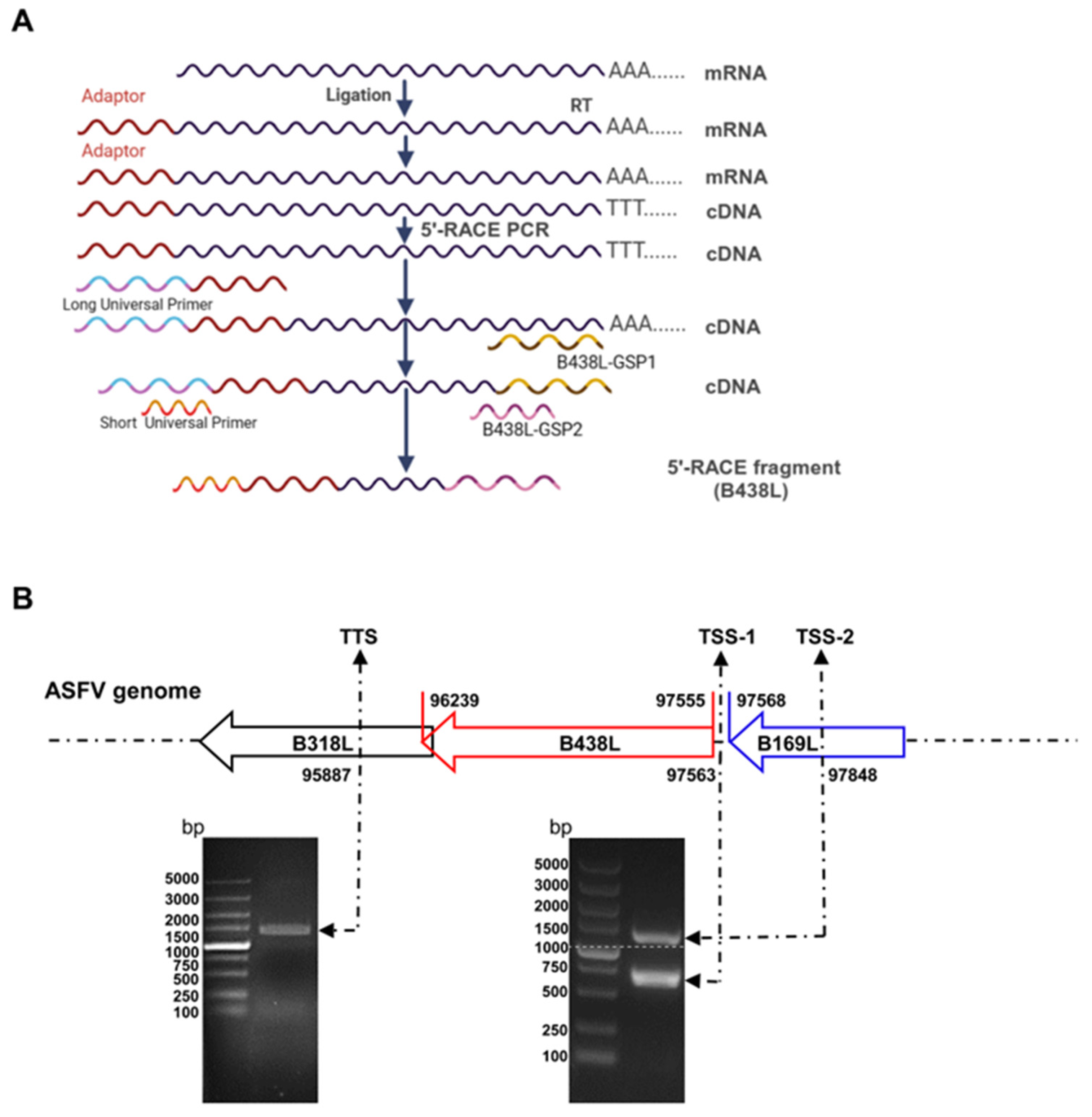
2.2. 5′ and 3′ Rapid Amplification of cDNA Ends (RACE) Assay
2.3. Construction of Plasmids
2.4. Luciferase Reporter Assay
2.5. Western Blotting Analysis
2.6. RNA Extraction and RT-qPCR
2.7. Statistical Analysis
3. Results
3.1. Two Distinct Initiation Sites for B438L Transcription
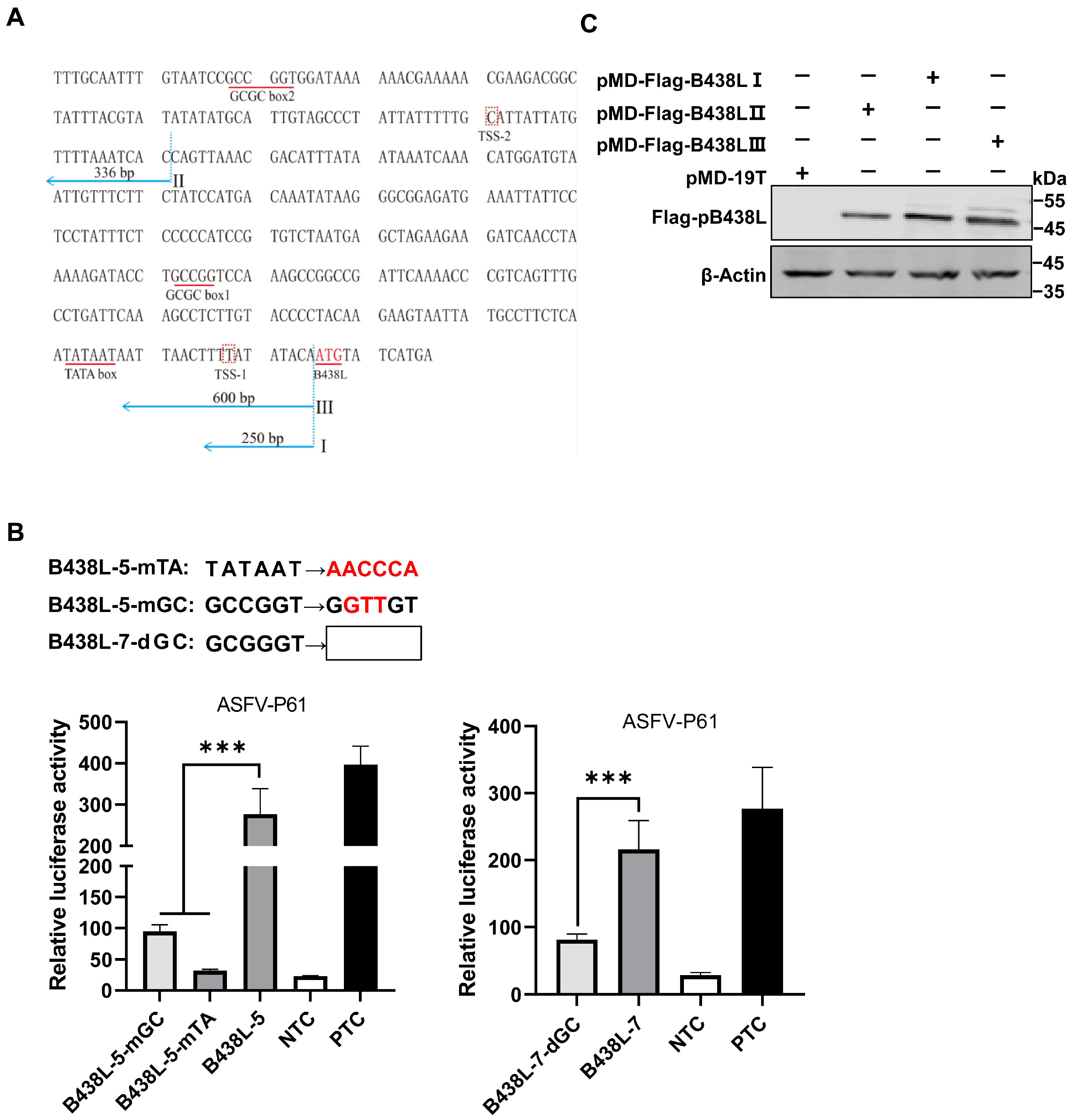
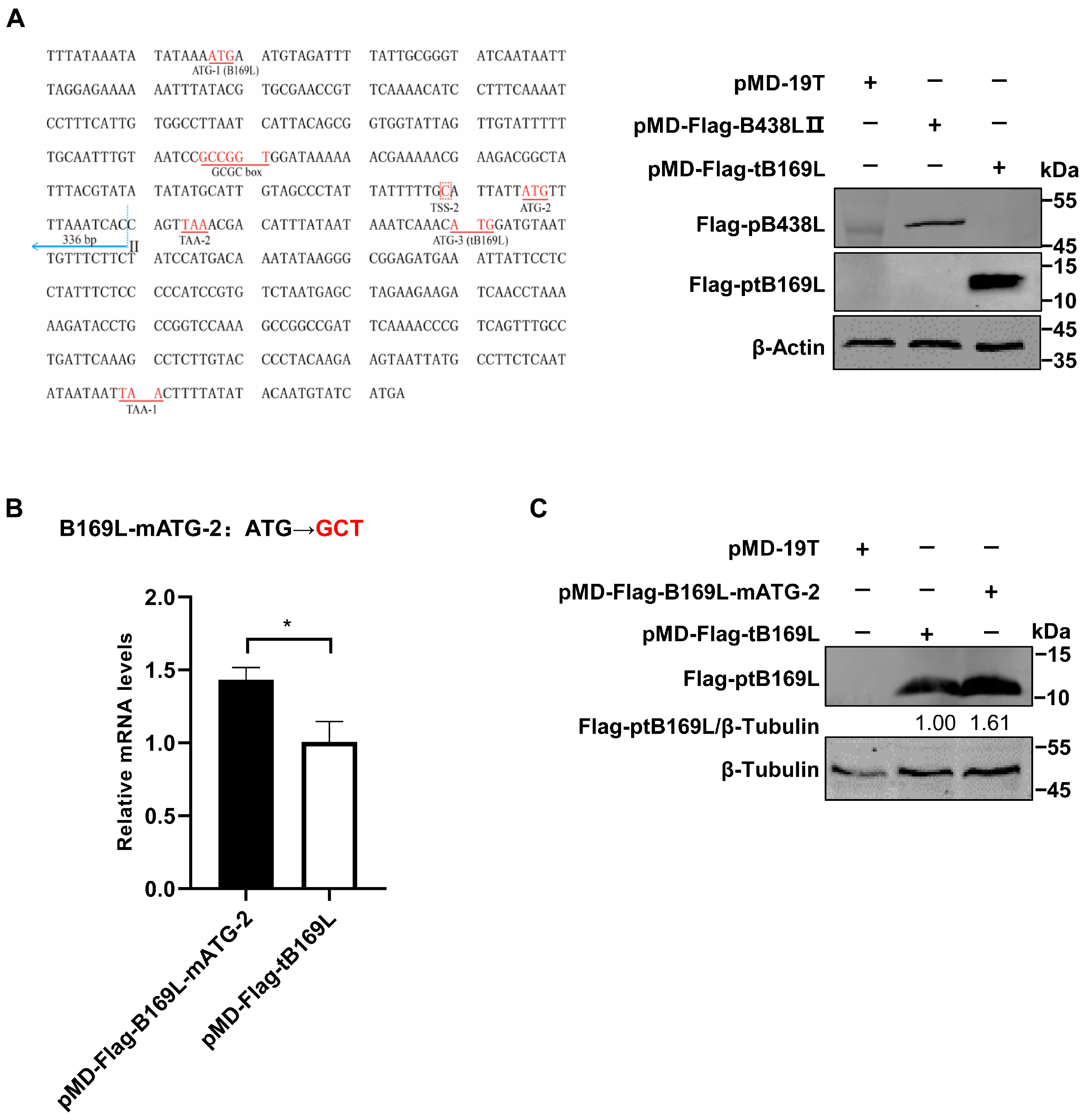
3.2. Mapping of the B438L Promoter Region Located in the B169L Gene
3.3. Identification of the TATA and GC Boxes in the B438L Promoter
3.4. Identification of the B438L Distal Promoter That Initiates Transcription of B169L-2
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Iyer, L.M.; Aravind, L.; Koonin, E.V. Common origin of four diverse families of large eukaryotic DNA viruses. J. Virol. 2001, 75, 11720–11734. [Google Scholar] [CrossRef] [PubMed]
- Alejo, A.; Andrés, G.; Salas, M.L. African swine fever virus proteinase is essential for core maturation and infectivity. J. Virol. 2003, 77, 5571–5577. [Google Scholar] [CrossRef] [PubMed]
- Carrascosa, A.L.; Bustos, M.J.; de Leon, P. Methods for growing and titrating African swine fever virus: Field and laboratory samples. Curr. Protoc. Cell Biol. 2011, 26, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhao, D.; He, X.; Liu, R.; Wang, Z.; Zhang, X.; Li, F.; Shan, D.; Chen, H.; Zhang, J.; et al. A seven-gene-deleted African swine fever virus is safe and effective as a live attenuated vaccine in pigs. Sci. China Life Sci. 2020, 63, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Dixon, L.K.; Chapman, D.A.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef]
- Cackett, G.; Matelska, D.; Sýkora, M.; Portugal, R.; Malecki, M.; Bähler, J.; Dixon, L.; Werner, F. The African swine fever virus transcriptome. J. Virol. 2020, 94, e00119-20. [Google Scholar] [CrossRef] [PubMed]
- Epifano, C.; Krijnse-Locker, J.; Salas, M.L.; Salas, J.; Rodríguez, J.M. Generation of filamentous instead of icosahedral particles by repression of African swine fever virus structural protein pB438L. J. Virol. 2006, 80, 11456–11466. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Zhang, L.H.; Lin, Y.; Cai, Y.F.; Zou, Y.W.; Hao, Z.Y.; Luo, Z.H.; Wang, N.D.; Deng, Z.B.; Yang, Y.; et al. Development and preliminary testing of a probe-based duplex real-time PCR assay for the detection of African swine fever virus. Mol. Cell. Probes 2021, 59, 101764. [Google Scholar] [CrossRef]
- Cackett, G.; Sýkora, M.; Werner, F. Transcriptome view of a killer: African swine fever virus. Biochem. Soc. Trans. 2020, 48, 1569–1581. [Google Scholar] [CrossRef]
- Zhao, D.; Liu, R.; Zhang, X.; Li, F.; Wang, J.; Zhang, J.; Liu, X.; Wang, L.; Zhang, J.; Wu, X.; et al. Replication and virulence in pigs of the first African swine fever virus isolated in China. Emerg. Microbes Infect. 2019, 8, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Li, L.F.; Zhang, K.; Wang, B.; Tang, L.; Li, M.; Wang, T.; Sun, Y.; Li, S.; Qiu, H.J. Deletion of the H240R gene of African swine fever virus decreases infectious progeny virus production due to aberrant virion morphogenesis and enhances inflammatory cytokine expression in porcine macrophages. J. Virol. 2022, 96, e0166721. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, L.; Han, Y.; Pan, L.; Yang, J.; Sun, M.; Zhou, P.; Sun, Y.; Bi, Y.; Qiu, H.J. Adaptation of African swine fever virus to HEK293T cells. Transbound. Emerg. Dis. 2021, 68, 2853–2866. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Chang, W.C.; Hsu, J.B.; Chang, T.H.; Shien, D.M. GPMiner: An integrated system for mining combinatorial cis-regulatory elements in mammalian gene group. BMC. Genomics 2012, 13 (Suppl. S1), S3. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Bhanja, C.J.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus upregulates beclin1 for induction of autophagy and activates mTOR signaling. J. Virol. 2012, 86, 8705–8712. [Google Scholar] [CrossRef]
- Smale, S.T.; Kadonaga, J.T. The RNA polymerase II core promoter. Annu. Rev. Biochem. 2003, 72, 449–479. [Google Scholar] [CrossRef]
- Wasserman, W.W.; Sandelin, A. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 2004, 5, 276–287. [Google Scholar] [CrossRef]
- Rodríguez, J.M.; Salas, M.L. African swine fever virus transcription. Virus Res. 2013, 173, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.B.; Amaral, P.P.; Schlesinger, F.J.; Dinger, M.E.; Taft, R.J.; Rinn, J.L.; Ponting, C.P.; Stadler, P.F.; Morris, K.V.; Morillon, A.; et al. The reality of pervasive transcription. PLoS Biol. 2011, 9, e1000625. [Google Scholar] [CrossRef]
- García-Escudero, R.; Viñuela, E. Structure of African swine fever virus late promoters: Requirement of a TATA sequence at the initiation region. J. Virol. 2000, 74, 8176–8182. [Google Scholar] [CrossRef]
- Yang, Z.; Martens, C.A.; Bruno, D.P.; Porcella, S.F.; Moss, B. Pervasive initiation and 3′-end formation of poxvirus postreplicative RNAs. J. Biol. Chem. 2012, 287, 31050–31060. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Yutin, N. Origin and evolution of eukaryotic large nucleo-cytoplasmic DNA viruses. Intervirology 2010, 53, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Reteno, D.G.; Benamar, S.; Khalil, J.B.; Andreani, J.; Armstrong, N.; Klose, T.; Rossmann, M.; Colson, P.; Raoult, D.; Lascola, B. Faustovirus, an Asfarvirus-related new lineage of giant viruses infecting amoebae. J. Virol. 2015, 89, 6585–6594. [Google Scholar] [CrossRef] [PubMed]
- Alejo, A.; Matamoros, T.; Guerra, M.; Andrés, G. A proteomic atlas of the African swine fever virus particle. J. Virol. 2018, 92, e01293-18. [Google Scholar] [CrossRef] [PubMed]
- Salas, M.L.; Kuznar, J.; Viñuela, E. Polyadenylation, methylation, and capping of the RNA synthesized in vitro by African swine fever virus. Virology 1981, 113, 484–491. [Google Scholar] [CrossRef]
- Furtado, M.R.; Balachandran, R.; Gupta, P.; Wolinsky, S.M. Analysis of alternatively spliced human immunodeficiency virus type-1 mRNA species, one of which encodes a novel Tat-Env fusion protein. Virology 1991, 185, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Telwatte, S.; Lee, S.; Somsouk, M.; Hatano, H.; Baker, C.; Kaiser, P.; Kim, P.; Chen, T.H.; Milush, J.; Hunt, P.W.; et al. Gut and blood differ in constitutive blocks to HIV transcription, suggesting tissue-specific differences in the mechanisms that govern HIV latency. PLoS Pathog. 2018, 14, e1007357. [Google Scholar] [CrossRef]
- Shimoda, A.; Sugata, F.; Chen, H.S.; Miller, R.H.; Purcell, R.H. Evidence for a bidirectional promoter complex within the X gene of woodchuck hepatitis virus. Virus Res. 1998, 56, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Slagle, B.L.; Bouchard, M.J. Hepatitis B virus X and regulation of viral gene expression. Cold Spring Harb. Perspect. Med. 2016, 6, a021402. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Tanaka, M.; Yokoymama, A.; Matsuda, G.; Kato, K.; Kagawa, H.; Hirai, K.; Roizman, B. Herpes simplex virus 1 alpha regulatory protein ICP0 functionally interacts with cellular transcription factor BMAL1. Proc. Natl. Acad. Sci. USA 2001, 98, 1877–1882. [Google Scholar] [CrossRef]
- Wijesekera, N.; Hazell, N.; Jones, C. Independent cis-regulatory modules within the herpes simplex virus 1 infected cell protein 0 (ICP0) promoter are transactivated by Krüppel-like factor 15 and glucocorticoid receptor. Viruses 2022, 14, 1284. [Google Scholar] [CrossRef] [PubMed]
- Jeang, K.T.; Rawlins, D.R.; Rosenfeld, P.J.; Shero, J.H.; Kelly, T.J.; Hayward, G.S. Multiple tandemly repeated binding sites for cellular nuclear factor 1 that surround the major immediate-early promoters of simian and human cytomegalovirus. J. Virol. 1987, 61, 1559–1570. [Google Scholar] [CrossRef] [PubMed]
- Stinski, M.F.; Isomura, H. Role of the cytomegalovirus major immediate early enhancer in acute infection and reactivation from latency. Med. Microbiol. Immunol. 2008, 197, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Pai, S.; Khanna, R. Role of LMP1 in immune control of EBV infection. Semin. Cancer Biol. 2001, 11, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.L.; Morgan, D.R.; Dominguez, R.L.; Thorne, L.B.; Elmore, S.H.; Mino-Kenudson, M.; Lauwers, G.Y.; Booker, J.K.; Gulley, M.L. High levels of Epstein-Barr virus DNA in latently infected gastric adenocarcinoma. Lab. Investig. 2009, 89, 80–90. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Sequences (5′–3′) | Description |
|---|---|---|
| Long Universal Primer | CTAATACGACTCACTATAGGGCAAGCAGTGGTATCAACGCAGAGT | B438L |
| Short Universal Primer | CTAATACGACTCACTATAGGGC | B438L |
| GSP1 | GATTACGCCAAGCTTAGCATCGGCACGTCCGTGTAGGTAC | B438L |
| GSP2 | GATTACGCCAAGCTTGTGTGCTCGCTGAACCTCGTAGAAG | B438L |
| B438L-1.F | CCGCTCGAGTGTATATAAAAGTTAATTATTATATTG | |
| B438L-1.R | GGGGTACCTGCGACTACATTAGCAATCTGGGCACC | B438L-1 |
| B438L-2.F | GGGGTACCCCATTAGATAACTATCCCGTGCCAC | B438L-2 |
| B438L-3.F | GGGGTACCAGTCGGGCCTTTTTGAAGAATCTTC | B438L-3 |
| B438L-4.F | GGGGTACCTAGATTTTATTGCGGGTATCAATAA | B438L-4 |
| B438L-5.F | GGGGTACCGCCCTATTATTTTTGCATTATTATG | B438L-5 |
| B438L-6.F | GGGGTACCATAAGGGCGGAGATGAAATTATTCC | B438L-6 |
| B438L-7.F | GGGGTACCTGCGACTACATTAGCAATCTGGGCACC | B438L-7 |
| B438L-7.R | CCGCTCGAGTACAATGCATATATATACGTAAATAGC | |
| B438L-5-mTA.F | CCTTCTCAAAACCAAAATAATTAAC | B438L-5-mTA |
| B438L-5-mTA.R | GTTAATTATTTTGGTTTTGAGAAGG | |
| B438L-5-mGC.F | ATTATGCCTTCTCAAAATTAACTTT | B438L-5-mGC |
| B438L-5-mGC.R | AAAGTTAATTTTGAGAAGGCATAAT | |
| B438L-7-dGC.F | AATGTAGATTTTATTATCAATAATTTAGGA | B438L-7-dGC. |
| B438L-7-d.GC.R | TCCTAAATTATTGATAATAAAATCTACATT | |
| pMD-Flag-B438L-I.F | CGGATCCTATAATAAATCAAACATGGATGTA | pMD-Flag-B438L-I |
| pMD-Flag-B438L-I.R | TAAGAACTACTTATCGTCGTCATCCTTGTAATCCAATGATGGAGATATAGATG | |
| pMD-Flag-B438L-II.1F | CGGATCCGTCGGGCCTTTTTGAAGAATCTTCA | pMD-Flag-B438L-II |
| pMD-Flag-B438L-II.1R | CATAATCATGATACATTGATTTAAAACATAA | |
| pMD-Flag-B438L-II.2F | TTATGTTTTAAATCAATGTATCATGATTATG | |
| pMD-Flag-B438L-II.2R | TAAGAACTACTTATCGTCGTCATCCTTGTAATCCAATGATGGAGATATAGATG | |
| pMD-Flag-B438L-III.F | CGGATCCGTCGGGCCTTTTTGAAGAATCTTCA | pMD-Flag-B438L-III |
| pMD-Flag-B438L-III.R | TAAGAACTACTTATCGTCGTCATCCTTGTAATCCAATGATGGAGATATAGATG | |
| pMD-Flag-tB169L.F | CGGATCCTCCTTTCAAAATCCTTTCATTGTGGC | pMD-Flag-tB169L |
| pMD-Flag-tB169L.R | GGAATTCTTACTTGTCGTCATCGTCTTTGTAGTCATTATTATATTGAGAAGGC | |
| pMD-Flag-B169-mATG-2.F | TATTATTTTTGCATTATTGCTTTTTAAATCACCAGTTAA | pMD-Flag-B169-mATG-2 |
| pMD-Flag-tB169-mATG-2.R | TTAACTGGTGATTTAAAACATAATAATGCAAAAATAATA | |
| qB169L-2-F | TTTGCAATTTGTAATCCGCCGGTG | RT-qPCR for B169L-mRNA2 |
| qB169L-2-R | ACTTCTTGTAGGGGTACAAGAGG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, H.; Deng, H.; Wang, Y.; Liu, D.; Li, L.; Li, M.; Peng, D.; Dai, J.; Li, J.; Qiu, H.; et al. The Distal Promoter of the B438L Gene of African Swine Fever Virus Is Responsible for the Transcription of the Alternatively Spliced B169L. Viruses 2024, 16, 1058. https://doi.org/10.3390/v16071058
Cao H, Deng H, Wang Y, Liu D, Li L, Li M, Peng D, Dai J, Li J, Qiu H, et al. The Distal Promoter of the B438L Gene of African Swine Fever Virus Is Responsible for the Transcription of the Alternatively Spliced B169L. Viruses. 2024; 16(7):1058. https://doi.org/10.3390/v16071058
Chicago/Turabian StyleCao, Hongwei, Hao Deng, Yanjin Wang, Diqiu Liu, Lianfeng Li, Meilin Li, Dingkun Peng, Jingwen Dai, Jiaqi Li, Huaji Qiu, and et al. 2024. "The Distal Promoter of the B438L Gene of African Swine Fever Virus Is Responsible for the Transcription of the Alternatively Spliced B169L" Viruses 16, no. 7: 1058. https://doi.org/10.3390/v16071058
APA StyleCao, H., Deng, H., Wang, Y., Liu, D., Li, L., Li, M., Peng, D., Dai, J., Li, J., Qiu, H., & Li, S. (2024). The Distal Promoter of the B438L Gene of African Swine Fever Virus Is Responsible for the Transcription of the Alternatively Spliced B169L. Viruses, 16(7), 1058. https://doi.org/10.3390/v16071058