
Regulation of Mitochondria-Derived Immune Activation by ‘Antiviral’ TRIM Proteins

Abstract
:1. Introduction
2. Stimulation of “Antiviral” Responses by Mitochondria
3. Mitochondria Participate in Antiviral Responses
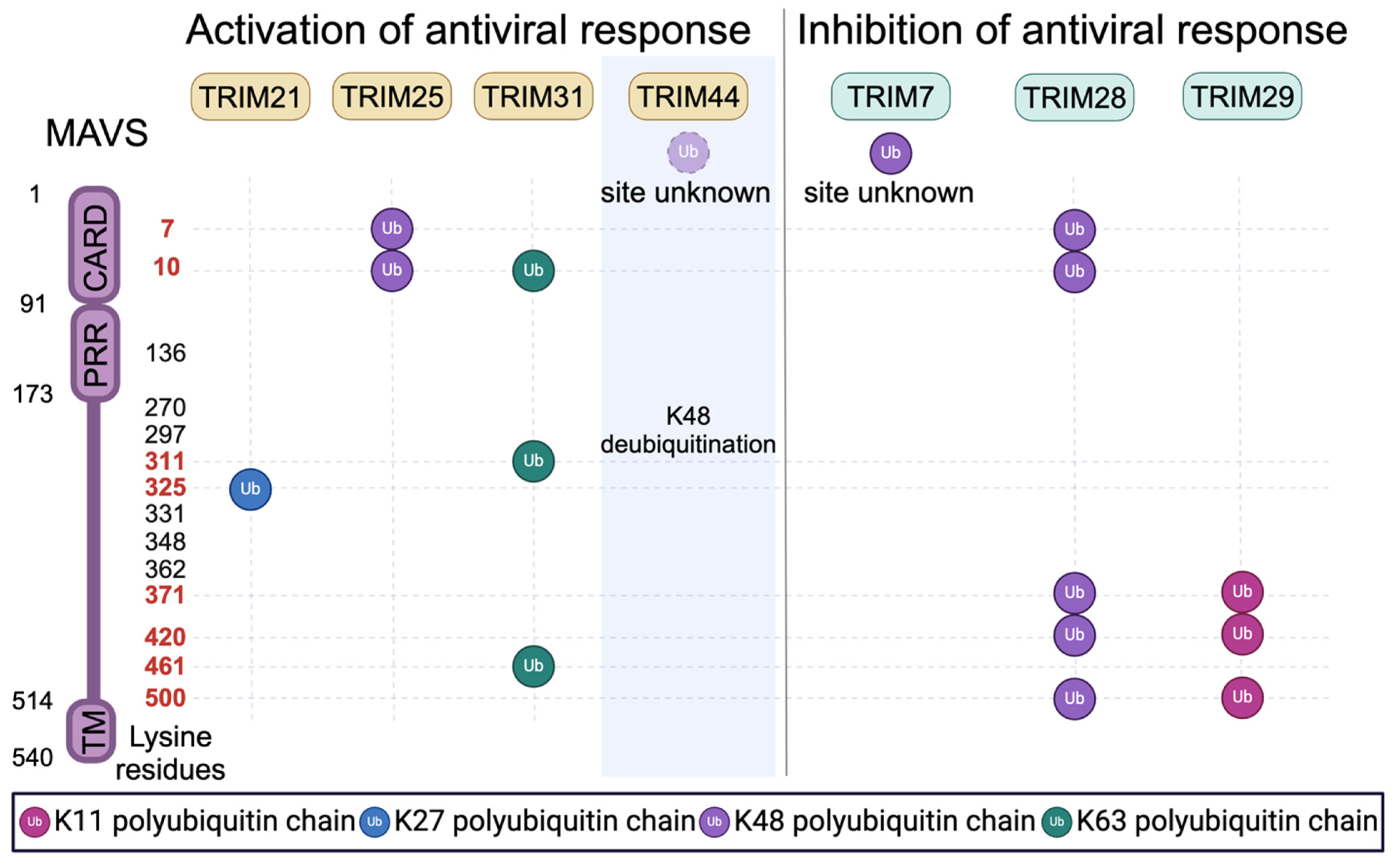
4. Regulation of Innate Immune Signaling by TRIMs
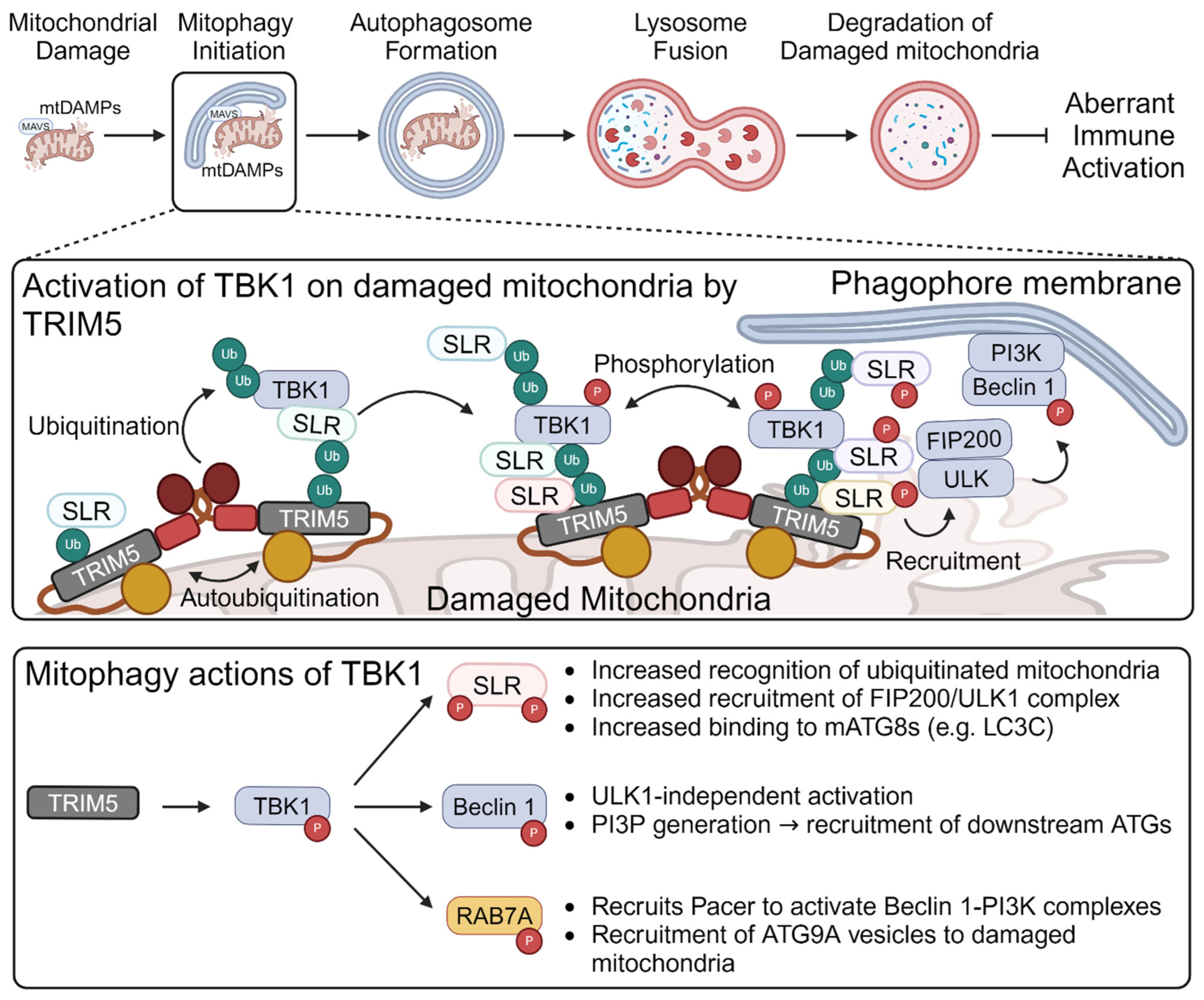
5. TRIM-Mediated Mitophagy Regulates Mitochondrial “Antiviral-like” Signaling
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koepke, L.; Gack, M.U.; Sparrer, K.M. The antiviral activities of TRIM proteins. Curr. Opin. Microbiol. 2021, 59, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.; Kumar, S.; Jain, A.; Ponpuak, M.; Mudd, M.H.; Kimura, T.; Choi, S.W.; Peters, R.; Mandell, M.; Bruun, J.A.; et al. TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-direct Autophagy in Endomembrane Damage Homeostasis. Dev. Cell 2016, 39, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, A.; Di Rienzo, M.; Petruccioli, E.; Fusco, C.; Palucci, I.; Micale, L.; Mazza, T.; Delogu, G.; Merla, G.; Goletti, D.; et al. The ubiquitin ligase TRIM32 promotes the autophagic response to Mycobacterium tuberculosis infection in macrophages. Cell Death Dis. 2023, 14, 505. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Li, Y.; Chen, T.; Liu, F.; Zheng, Y.; Liu, B.; Zhao, W.; Qi, X.; Sun, W.; Gao, C. TRIM26 alleviates fatal immunopathology by regulating inflammatory neutrophil infiltration during Candida infection. PLoS Pathog. 2024, 20, e1011902. [Google Scholar] [CrossRef] [PubMed]
- Hoffpauir, C.T.; Bell, S.L.; West, K.O.; Jing, T.; Wagner, A.R.; Torres-Odio, S.; Cox, J.S.; West, A.P.; Li, P.; Patrick, K.L.; et al. TRIM14 Is a Key Regulator of the Type I IFN Response during Mycobacterium tuberculosis Infection. J. Immunol. 2020, 205, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C., III. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Langevin, C.; Levraud, J.P.; Boudinot, P. Fish antiviral tripartite motif (TRIM) proteins. Fish Shellfish Immunol. 2019, 86, 724–733. [Google Scholar] [CrossRef]
- Carthagena, L.; Bergamaschi, A.; Luna, J.M.; David, A.; Uchil, P.D.; Margottin-Goguet, F.; Mothes, W.; Hazan, U.; Transy, C.; Pancino, G.; et al. Human TRIM gene expression in response to interferons. PLoS ONE 2009, 4, e4894. [Google Scholar] [CrossRef]
- Versteeg, G.A.; Rajsbaum, R.; Sánchez-Aparicio, M.T.; Maestre, A.M.; Valdiviezo, J.; Shi, M.; Inn, K.S.; Fernandez-Sesma, A.; Jung, J.; García-Sastre, A. The E3-Ligase TRIM Family of Proteins Regulates Signaling Pathways Triggered by Innate Immune Pattern-Recognition Receptors. Immunity 2013, 38, 384–398. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Pornillos, O. Restriction of HIV-1 and other retroviruses by TRIM5. Nat. Rev. Microbiol. 2019, 17, 546–556. [Google Scholar] [CrossRef]
- Becker, Y.L.C.; Duvvuri, B.; Fortin, P.R.; Lood, C.; Boilard, E. The role of mitochondria in rheumatic diseases. Nat. Rev. Rheumatol. 2022, 18, 621–640. [Google Scholar] [CrossRef] [PubMed]
- Moehlman, A.T.; Youle, R.J. Mitochondrial Quality Control and Restraining Innate Immunity. Annu. Rev. Cell Dev. Biol. 2020, 36, 265–289. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A.; Nunnari, J. Mitochondria at the crossroads of health and disease. Cell 2024, 187, 2601–2627. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef]
- Cosentino, K.; Hertlein, V.; Jenner, A.; Dellmann, T.; Gojkovic, M.; Pena-Blanco, A.; Dadsena, S.; Wajngarten, N.; Danial, J.S.H.; Thevathasan, J.V.; et al. The interplay between BAX and BAK tunes apoptotic pore growth to control mitochondrial-DNA-mediated inflammation. Mol. Cell 2022, 82, 933–949.e9. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 2018, 37, e99238. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Rai, P.; Janardhan, K.S.; Meacham, J.; Madenspacher, J.H.; Lin, W.C.; Karmaus, P.W.F.; Martinez, J.; Li, Q.Z.; Yan, M.; Zeng, J.; et al. IRGM1 links mitochondrial quality control to autoimmunity. Nat. Immunol. 2021, 22, 312–321. [Google Scholar] [CrossRef]
- Andreeva, L.; Hiller, B.; Kostrewa, D.; Lassig, C.; de Oliveira Mann, C.C.; Jan Drexler, D.; Maiser, A.; Gaidt, M.; Leonhardt, H.; Hornung, V.; et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 2017, 549, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Gulen, M.F.; Samson, N.; Keller, A.; Schwabenland, M.; Liu, C.; Gluck, S.; Thacker, V.V.; Favre, L.; Mangeat, B.; Kroese, L.J.; et al. cGAS-STING drives ageing-related inflammation and neurodegeneration. Nature 2023, 620, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Moehlman, A.T.; Kanfer, G.; Youle, R.J. Loss of STING in parkin mutant flies suppresses muscle defects and mitochondria damage. PLoS Genet. 2023, 19, e1010828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Dhir, A.; Dhir, S.; Borowski, L.S.; Jimenez, L.; Teitell, M.; Rotig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018, 560, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Ponia, S.S.; Robertson, S.J.; McNally, K.L.; Subramanian, G.; Sturdevant, G.L.; Lewis, M.; Jessop, F.; Kendall, C.; Gallegos, D.; Hay, A.; et al. Mitophagy antagonism by ZIKV reveals Ajuba as a regulator of PINK1 signaling, PKR-dependent inflammation, and viral invasion of tissues. Cell Rep. 2021, 37, 109888. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Liu, Q.; Wu, Y.; Ma, L.; Zhang, Z.; Liu, T.; Jin, S.; She, Y.; Li, Y.P.; Cui, J. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1-caspase-1 axis. EMBO J. 2018, 37, e99347. [Google Scholar] [CrossRef] [PubMed]
- Webb, L.G.; Veloz, J.; Pintado-Silva, J.; Zhu, T.; Rangel, M.V.; Mutetwa, T.; Zhang, L.; Bernal-Rubio, D.; Figueroa, D.; Carrau, L.; et al. Chikungunya virus antagonizes cGAS-STING mediated type-I interferon responses by degrading cGAS. PLoS Pathog. 2020, 16, e1008999. [Google Scholar] [CrossRef]
- Moriyama, M.; Koshiba, T.; Ichinohe, T. Influenza A virus M2 protein triggers mitochondrial DNA-mediated antiviral immune responses. Nat. Commun. 2019, 10, 4624. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, Z.; Xue, Q.; Yang, F.; Li, Z.; Xue, Z.; Cao, W.; He, J.; Guo, J.; Liu, X.; et al. Innate sensing of picornavirus infection involves cGAS-STING-mediated antiviral responses triggered by mitochondrial DNA release. PLoS Pathog. 2023, 19, e1011132. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Sundstrom, K.B.; Chew, J.J.; Bist, P.; Gan, E.S.; Tan, H.C.; Goh, K.C.; Chawla, T.; Tang, C.K.; Ooi, E.E. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci. Rep. 2017, 7, 3594. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Hoshi, M.; Ikeda, F.; Fujiyuki, T.; Yoneda, M.; Kai, C. Downregulation of mitochondrial biogenesis by virus infection triggers antiviral responses by cyclic GMP-AMP synthase. PLoS Pathog. 2021, 17, e1009841. [Google Scholar] [CrossRef] [PubMed]
- Domizio, J.D.; Gulen, M.F.; Saidoune, F.; Thacker, V.V.; Yatim, A.; Sharma, K.; Nass, T.; Guenova, E.; Schaller, M.; Conrad, C.; et al. The cGAS-STING pathway drives type I IFN immunopathology in COVID-19. Nature 2022, 603, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Aarreberg, L.D.; Esser-Nobis, K.; Driscoll, C.; Shuvarikov, A.; Roby, J.A.; Gale, M., Jr. Interleukin-1beta Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol. Cell 2019, 74, 801–815.e6. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA virus recognition. Virology 2015, 479, 110–121. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Refolo, G.; Vescovo, T.; Piacentini, M.; Fimia, G.M.; Ciccosanti, F. Mitochondrial Interactome: A Focus on Antiviral Signaling Pathways. Front. Cell Dev. Biol. 2020, 8, 8. [Google Scholar] [CrossRef]
- Harding, O.; Holzer, E.; Riley, J.F.; Martens, S.; Holzbaur, E.L.F. Damaged mitochondria recruit the effector NEMO to activate NF-kappaB signaling. Mol. Cell 2023, 83, 3188–3204.e7. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Berlemann, L.A.; Bader, V.; Sehr, D.A.; Dawin, E.; Covallero, A.; Meschede, J.; Angersbach, L.; Showkat, C.; Michaelis, J.B.; et al. LUBAC assembles a ubiquitin signaling platform at mitochondria for signal amplification and transport of NF-kappaB to the nucleus. EMBO J. 2022, 41, e112006. [Google Scholar] [CrossRef] [PubMed]
- Courtois, G.; Israel, A. IKK Regulation and Human Genetics. Curr. Top. Microbiol. Immunol. 2011, 349, 73–95. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Wei, L.; Yu, Z.B.; Yao, Z.Y.; Cheng, J.; Wang, Y.T.; Song, X.T.; Li, M. The Roles of TRIMs in Antiviral Innate Immune Signaling. Front. Cell. Infect. Microbiol. 2021, 11, 628275. [Google Scholar] [CrossRef] [PubMed]
- van Gent, M.; Sparrer, K.M.J.; Gack, M.U. TRIM Proteins and Their Roles in Antiviral Host Defenses. Annu. Rev. Virol. 2018, 5, 385–405. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, C.; Wynne, C.; Higgs, R. Antiviral TRIMs: Friend or foe in autoimmune and autoinflammatory disease? Nat. Rev. Immunol. 2011, 11, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Pertel, T.; Hausmann, S.; Morger, D.; Zuger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef]
- Lang, X.; Tang, T.; Jin, T.; Ding, C.; Zhou, R.; Jiang, W. TRIM65-catalized ubiquitination is essential for MDA5-mediated antiviral innate immunity. J. Exp. Med. 2017, 214, 459–473. [Google Scholar] [CrossRef]
- Tan, P.; He, L.; Cui, J.; Qian, C.; Cao, X.; Lin, M.; Zhu, Q.; Li, Y.; Xing, C.; Yu, X.; et al. Assembly of the WHIP-TRIM14-PPP6C Mitochondrial Complex Promotes RIG-I-Mediated Antiviral Signaling. Mol. Cell 2017, 68, 293–307.e5. [Google Scholar] [CrossRef]
- Chen, M.; Meng, Q.; Qin, Y.; Liang, P.; Tan, P.; He, L.; Zhou, Y.; Chen, Y.; Huang, J.; Wang, R.F.; et al. TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor p62 to Promote Innate Immune Responses. Mol. Cell 2016, 64, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Jain, A.; Choi, S.W.; Mandell, M.A.; Johansen, T.; Deretic, V. TRIM-Directed Selective Autophagy Regulates Immune Activation. Autophagy 2016, 13, 989–990. [Google Scholar] [CrossRef]
- Zhao, C.; Jia, M.; Song, H.; Yu, Z.; Wang, W.; Li, Q.; Zhang, L.; Zhao, W.; Cao, X. The E3 Ubiquitin Ligase TRIM40 Attenuates Antiviral Immune Responses by Targeting MDA5 and RIG-I. Cell Rep. 2017, 21, 1613–1623. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhao, W.; Zhao, K.; Zhang, L.; Gao, C. TRIM26 negatively regulates interferon-beta production and antiviral response through polyubiquitination and degradation of nuclear IRF3. PLoS Pathog. 2015, 11, e1004726. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Deng, W.; Bi, E.; Mao, K.; Ji, Y.; Lin, G.; Wu, X.; Tao, Z.; Li, Z.; Cai, X.; et al. TRIM30 alpha negatively regulates TLR-mediated NF-kappa B activation by targeting TAB2 and TAB3 for degradation. Nat. Immunol. 2008, 9, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Ran, X.H.; Zhu, J.W.; Ni, R.Z.; Zheng, Y.T.; Chen, Y.Y.; Zheng, W.H.; Mu, D. TRIM5alpha recruits HDAC1 to p50 and Sp1 and promotes H3K9 deacetylation at the HIV-1 LTR. Nat. Commun. 2023, 14, 3343. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, S.; Ohbayashi, N.; Ikeda, O.; Togi, S.; Muromoto, R.; Sekine, Y.; Ohta, K.; Ishiyama, H.; Matsuda, T. KAP1 regulates type I interferon/STAT1-mediated IRF-1 gene expression. Biochem. Biophys. Res. Commun. 2008, 370, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Tisserand, J.; Khetchoumian, K.; Thibault, C.; Dembele, D.; Chambon, P.; Losson, R. Tripartite motif 24 (Trim24/Tif1alpha) tumor suppressor protein is a novel negative regulator of interferon (IFN)/signal transducers and activators of transcription (STAT) signaling pathway acting through retinoic acid receptor alpha (Raralpha) inhibition. J. Biol. Chem. 2011, 286, 33369–33379. [Google Scholar] [CrossRef]
- Portilho, D.M.; Fernandez, J.; Ringeard, M.; Machado, A.K.; Boulay, A.; Mayer, M.; Muller-Trutwin, M.; Beignon, A.S.; Kirchhoff, F.; Nisole, S.; et al. Endogenous TRIM5alpha Function Is Regulated by SUMOylation and Nuclear Sequestration for Efficient Innate Sensing in Dendritic Cells. Cell Rep. 2016, 14, 355–369. [Google Scholar] [CrossRef]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.X.; Chen, Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, M.; Chu, H.; Zhang, H.; Wu, H.; Song, G.; Wang, P.; Zhao, K.; Hou, J.; Wang, X.; et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol. 2017, 18, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Yang, X. SUMO E3 ligase activity of TRIM proteins. Oncogene 2011, 30, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Zhang, D.E. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J. Biol. Chem. 2006, 281, 3989–3994. [Google Scholar] [CrossRef] [PubMed]
- Akizuki, Y.; Kaypee, S.; Ohtake, F.; Ikeda, F. The emerging roles of non-canonical ubiquitination in proteostasis and beyond. J. Cell Biol. 2024, 223, e202311171. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Duong, D.M.; Seyfried, N.T.; Cheng, D.; Xie, Y.; Robert, J.; Rush, J.; Hochstrasser, M.; Finley, D.; Peng, J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 2009, 137, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, C.; Novack, J.P.; Reed, J.C.; Matsuzawa, S.I. K63 ubiquitination in immune signaling. Trends Immunol. 2022, 43, 148–162. [Google Scholar] [CrossRef]
- Liu, F.; Zhuang, W.; Song, B.; Yang, Y.; Liu, J.; Zheng, Y.; Liu, B.; Zheng, J.; Zhao, W.; Gao, C. MAVS-loaded unanchored Lys63-linked polyubiquitin chains activate the RIG-I-MAVS signaling cascade. Cell. Mol. Immunol. 2023, 20, 1186–1202. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jia, X.; Xue, Q.; Dou, Z.; Ma, Y.; Zhao, Z.; Jiang, Z.; He, B.; Jin, Q.; Wang, J. TRIM14 is a mitochondrial adaptor that facilitates retinoic acid-inducible gene-I-like receptor-mediated innate immune response. Proc. Natl. Acad. Sci. USA 2014, 111, E245–E254. [Google Scholar] [CrossRef]
- Xue, B.; Li, H.; Guo, M.; Wang, J.; Xu, Y.; Zou, X.; Deng, R.; Li, G.; Zhu, H. TRIM21 Promotes Innate Immune Response to RNA Viral Infection through Lys27-Linked Polyubiquitination of MAVS. J. Virol. 2018, 92, e00321-18. [Google Scholar] [CrossRef]
- Yang, B.; Zhang, G.; Qin, X.; Huang, Y.; Ren, X.; Sun, J.; Ma, S.; Liu, Y.; Song, D.; Liu, Y.; et al. Negative Regulation of RNF90 on RNA Virus-Triggered Antiviral Immune Responses Targeting MAVS. Front. Immunol. 2021, 12, 730483. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Ran, X.H.; Ni, R.Z.; Mu, D. TRIM28 negatively regulates the RLR signaling pathway by targeting MAVS for degradation via K48-linked polyubiquitination. J. Biol. Chem. 2023, 299, 104660. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Zhang, A.; Minze, L.J.; Li, X.C.; Zhang, Z. TRIM29 Negatively Regulates the Type I IFN Production in Response to RNA Virus. J. Immunol. 2018, 201, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Castanier, C.; Zemirli, N.; Portier, A.; Garcin, D.; Bidere, N.; Vazquez, A.; Arnoult, D. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Orchard, R.C.; Sullender, M.E.; Dunlap, B.F.; Balce, D.R.; Doench, J.G.; Virgin, H.W. Identification of Antinorovirus Genes in Human Cells Using Genome-Wide CRISPR Activation Screening. J. Virol. 2019, 93, e01324-18. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, M.I.; Xia, H.; Aguilera-Aguirre, L.; Hage, A.; van Tol, S.; Shan, C.; Xie, X.; Sturdevant, G.L.; Robertson, S.J.; McNally, K.L.; et al. Envelope protein ubiquitination drives entry and pathogenesis of Zika virus. Nature 2020, 585, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Mar, K.B.; Sari, L.; Gaszek, I.K.; Cheng, Q.; Evers, B.M.; Shelton, J.M.; Wight-Carter, M.; Siegwart, D.J.; Lin, M.M.; et al. TRIM7 inhibits enterovirus replication and promotes emergence of a viral variant with increased pathogenicity. Cell 2021, 184, 3410–3425.e17. [Google Scholar] [CrossRef]
- Gonzalez-Orozco, M.; Tseng, H.C.; Hage, A.; Xia, H.; Behera, P.; Afreen, K.; Penaflor-Tellez, Y.; Giraldo, M.I.; Huante, M.; Puebla-Clark, L.; et al. TRIM7 ubiquitinates SARS-CoV-2 membrane protein to limit apoptosis and viral replication. bioRxiv 2024. [Google Scholar] [CrossRef]
- Yang, B.; Wang, J.; Wang, Y.; Zhou, H.; Wu, X.; Tian, Z.; Sun, B. Novel function of Trim44 promotes an antiviral response by stabilizing VISA. J. Immunol. 2013, 190, 3613–3619. [Google Scholar] [CrossRef] [PubMed]
- Urano, T.; Usui, T.; Takeda, S.; Ikeda, K.; Okada, A.; Ishida, Y.; Iwayanagi, T.; Otomo, J.; Ouchi, Y.; Inoue, S. TRIM44 interacts with and stabilizes terf, a TRIM ubiquitin E3 ligase. Biochem. Biophys. Res. Commun. 2009, 383, 263–268. [Google Scholar] [CrossRef]
- Lyu, L.; Chen, Z.; McCarty, N. TRIM44 links the UPS to SQSTM1/p62-dependent aggrephagy and removing misfolded proteins. Autophagy 2022, 18, 783–798. [Google Scholar] [CrossRef]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef] [PubMed]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shen, J.; Ran, Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 2020, 16, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, J.; Berge, A.K.M.; Overa, K.S.; Larsen, K.B.; Bhujabal, Z.; Brech, A.; Abudu, Y.P.; Lamark, T.; Johansen, T.; Sjottem, E. TRIM27 is an autophagy substrate facilitating mitochondria clustering and mitophagy via phosphorylated TBK1. FEBS J. 2023, 290, 1096–1116. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Salemi, M.; Williams, G.L.; Oh, S.; Paffett, M.L.; Phinney, B.; Mandell, M.A. Interactomic analysis reveals a homeostatic role for the HIV restriction factor TRIM5alpha in mitophagy. Cell Rep. 2022, 39, 110797. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Olsvik, H.; Williams, G.L.; Oh, S.; Evjen, G.; Sjøttem, E.; Mandell, M.A. TBK1 is ubiquitinated by TRIM5α to assemble mitophagy machinery. Cell Rep. 2024, 43, 114294. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Chiramel, A.I.; Meyerson, N.R.; McNally, K.L.; Broeckel, R.M.; Montoya, V.R.; Mendez-Solis, O.; Robertson, S.J.; Sturdevant, G.L.; Lubick, K.J.; Nair, V.; et al. TRIM5alpha Restricts Flavivirus Replication by Targeting the Viral Protease for Proteasomal Degradation. Cell Rep. 2019, 27, 3269–3283.e6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lu, Y.; Richardson, S.; Sreekumar, M.; Albarnaz, J.D.; Smith, G.L. TRIM5alpha restricts poxviruses and is antagonized by CypA and the viral protein C6. Nature 2023, 620, 873–880. [Google Scholar] [CrossRef]
- Yamada, Y.; Yasukochi, Y.; Kato, K.; Oguri, M.; Horibe, H.; Fujimaki, T.; Takeuchi, I.; Sakuma, J. Identification of 26 novel loci that confer susceptibility to early-onset coronary artery disease in a Japanese population. Biomed. Rep. 2018, 9, 383–404. [Google Scholar] [CrossRef]
- van der Harst, P.; Verweij, N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ. Res. 2018, 122, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Kato, K.; Oguri, M.; Horibe, H.; Fujimaki, T.; Yasukochi, Y.; Takeuchi, I.; Sakuma, J. Identification of 13 novel susceptibility loci for early-onset myocardial infarction, hypertension, or chronic kidney disease. Int. J. Mol. Med. 2018, 42, 2415–2436. [Google Scholar] [CrossRef] [PubMed]
- Aragam, K.G.; Jiang, T.; Goel, A.; Kanoni, S.; Wolford, B.N.; Atri, D.S.; Weeks, E.M.; Wang, M.; Hindy, G.; Zhou, W.; et al. Discovery and systematic characterization of risk variants and genes for coronary artery disease in over a million participants. Nat. Genet. 2022, 54, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.E.; Clarke, S.L.; Wu, K.H.; Kanoni, S.; Zajac, G.J.M.; Ramdas, S.; Surakka, I.; Ntalla, I.; Vedantam, S.; Winkler, T.W.; et al. The power of genetic diversity in genome-wide association studies of lipids. Nature 2021, 600, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Klimentidis, Y.C.; Arora, A.; Newell, M.; Zhou, J.; Ordovas, J.M.; Renquist, B.J.; Wood, A.C. Phenotypic and Genetic Characterization of Lower LDL Cholesterol and Increased Type 2 Diabetes Risk in the UK Biobank. Diabetes 2020, 69, 2194–2205. [Google Scholar] [CrossRef] [PubMed]
- Richardson, T.G.; Sanderson, E.; Palmer, T.M.; Ala-Korpela, M.; Ference, B.A.; Davey Smith, G.; Holmes, M.V. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: A multivariable Mendelian randomisation analysis. PLoS Med. 2020, 17, e1003062. [Google Scholar] [CrossRef] [PubMed]
- Ripatti, P.; Ramo, J.T.; Mars, N.J.; Fu, Y.; Lin, J.; Soderlund, S.; Benner, C.; Surakka, I.; Kiiskinen, T.; Havulinna, A.S.; et al. Polygenic Hyperlipidemias and Coronary Artery Disease Risk. Circ. Genom. Precis. Med. 2020, 13, e002725. [Google Scholar] [CrossRef] [PubMed]
- Richardson, T.G.; Leyden, G.M.; Wang, Q.; Bell, J.A.; Elsworth, B.; Davey Smith, G.; Holmes, M.V. Characterising metabolomic signatures of lipid-modifying therapies through drug target mendelian randomisation. PLoS Biol. 2022, 20, e3001547. [Google Scholar] [CrossRef] [PubMed]
- Nexo, B.A.; Christensen, T.; Frederiksen, J.; Moller-Larsen, A.; Oturai, A.B.; Villesen, P.; Hansen, B.; Nissen, K.K.; Laska, M.J.; Petersen, T.S.; et al. The etiology of multiple sclerosis: Genetic evidence for the involvement of the human endogenous retrovirus HERV-Fc1. PLoS ONE 2011, 6, e16652. [Google Scholar] [CrossRef]
- Mandell, M.A.; Jain, A.; Arko-Mensah, J.; Chauhan, S.; Kimura, T.; Dinkins, C.; Silvestri, G.; Munch, J.; Kirchhoff, F.; Simonsen, A.; et al. TRIM proteins regulate autophagy and can target autophagic substrates by direct recognition. Dev. Cell 2014, 30, 394–409. [Google Scholar] [CrossRef]
- Ribeiro, C.M.; Sarrami-Forooshani, R.; Setiawan, L.C.; Zijlstra-Willems, E.M.; van Hamme, J.L.; Tigchelaar, W.; van der Wel, N.N.; Kootstra, N.A.; Gringhuis, S.I.; Geijtenbeek, T.B. Receptor usage dictates HIV-1 restriction by human TRIM5alpha in dendritic cell subsets. Nature 2016, 540, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Chisholm, D.; Kell, A.M.; Mandell, M.A. A non-canonical role for the autophagy machinery in anti-retroviral signaling mediated by TRIM5alpha. PLoS Pathog. 2020, 16, e1009017. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Simonsen, A. Diversity of mitophagy pathways at a glance. J. Cell Sci. 2022, 135, jcs259748. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [PubMed]
- Vargas, J.N.S.; Wang, C.; Bunker, E.; Hao, L.; Maric, D.; Schiavo, G.; Randow, F.; Youle, R.J. Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol. Cell 2019, 74, 347–362.e6. [Google Scholar] [CrossRef]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Zachari, M.; Gudmundsson, S.R.; Li, Z.; Manifava, M.; Cugliandolo, F.; Shah, R.; Smith, M.; Stronge, J.; Karanasios, E.; Piunti, C.; et al. Selective Autophagy of Mitochondria on a Ubiquitin-Endoplasmic-Reticulum Platform. Dev. Cell 2019, 50, 627–643.e5. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef]
- Heo, J.M.; Ordureau, A.; Swarup, S.; Paulo, J.A.; Shen, K.; Sabatini, D.M.; Harper, J.W. RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK-PARKIN pathway. Sci. Adv. 2018, 4, eaav0443. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Sawa-Makarska, J.; Khuu, G.; Lam, W.K.; Adriaenssens, E.; Fracchiolla, D.; Shoebridge, S.; Bernklau, D.; Padman, B.S.; Skulsuppaisarn, M.; et al. Unconventional initiation of PINK1/Parkin mitophagy by Optineurin. Mol. Cell 2023, 83, 1693–1709.e9. [Google Scholar] [CrossRef] [PubMed]
- Tudorica, D.A.; Basak, B.; Puerta Cordova, A.S.; Khuu, G.; Rose, K.; Lazarou, M.; Holzbaur, E.L.F.; Hurley, J.H. A RAB7A phosphoswitch coordinates Rubicon Homology protein regulation of Parkin-dependent mitophagy. J. Cell Biol. 2024, 223, e202309015. [Google Scholar] [CrossRef] [PubMed]
- Zurek, B.; Schoultz, I.; Neerincx, A.; Napolitano, L.M.; Birkner, K.; Bennek, E.; Sellge, G.; Lerm, M.; Meroni, G.; Soderholm, J.D.; et al. TRIM27 negatively regulates NOD2 by ubiquitination and proteasomal degradation. PLoS ONE 2012, 7, e41255. [Google Scholar] [CrossRef]
- Zheng, Q.; Hou, J.; Zhou, Y.; Yang, Y.; Xie, B.; Cao, X. Siglec1 suppresses antiviral innate immune response by inducing TBK1 degradation via the ubiquitin ligase TRIM27. Cell Res. 2015, 25, 1121–1136. [Google Scholar] [CrossRef]
- Qin, F.; Cai, B.; Cao, R.; Bai, X.; Yuan, J.; Zhang, Y.; Liu, Y.; Chen, T.; Liu, F.; Sun, W.; et al. Listerin promotes cGAS protein degradation through the ESCRT pathway to negatively regulate cGAS-mediated immune response. Proc. Natl. Acad. Sci. USA 2023, 120, e2308853120. [Google Scholar] [CrossRef]
- Barde, I.; Rauwel, B.; Marin-Florez, R.M.; Corsinotti, A.; Laurenti, E.; Verp, S.; Offner, S.; Marquis, J.; Kapopoulou, A.; Vanicek, J.; et al. A KRAB/KAP1-miRNA cascade regulates erythropoiesis through stage-specific control of mitophagy. Science 2013, 340, 350–353. [Google Scholar] [CrossRef]
- Liu, H.; Zhou, Z.; Deng, H.; Tian, Z.; Wu, Z.; Liu, X.; Ren, Z.; Jiang, Z. Trim65 attenuates isoproterenol-induced cardiac hypertrophy by promoting autophagy and ameliorating mitochondrial dysfunction via the Jak1/Stat1 signaling pathway. Eur. J. Pharmacol. 2023, 949, 175735. [Google Scholar] [CrossRef] [PubMed]
- Tsoupri, E.; Kostavasili, I.; Kloukina, I.; Tsikitis, M.; Miliou, D.; Vasilaki, E.; Varela, A.; Nakos-Bimpos, M.; Davos, C.; Mavroidis, M.; et al. Myospryn deficiency leads to impaired cardiac structure and function and schizophrenia-associated symptoms. Cell Tissue Res. 2021, 385, 675–696. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, S.; Liu, M.; Wei, Y.; Wang, Q.; Shen, W.; Lei, C.Q.; Zhu, Q. The nucleoprotein of influenza A virus inhibits the innate immune response by inducing mitophagy. Autophagy 2023, 19, 1916–1933. [Google Scholar] [CrossRef]
- Li, X.; Hou, P.; Ma, W.; Wang, X.; Wang, H.; Yu, Z.; Chang, H.; Wang, T.; Jin, S.; Wang, X.; et al. SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell. Mol. Immunol. 2022, 19, 67–78. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, Y.; Ren, C.; Yang, S.; Tian, S.; Chen, H.; Jin, M.; Zhou, H. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy 2021, 17, 496–511. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Yu, J.W.; Ahn, J.H.; Choi, S.T.; Park, H.; Yun, J.; Shin, O.S. Varicella zoster virus glycoprotein E facilitates PINK1/Parkin-mediated mitophagy to evade STING and MAVS-mediated antiviral innate immunity. Cell Death Dis. 2024, 15, 16. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Gonzalez, P.; Li, C.; Meng, G.; Jiang, A.; Wang, H.; Gao, Q.; Debatin, K.M.; Beltinger, C.; Wei, J. Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J. Virol. 2014, 88, 5152–5164. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ma, H.; Liu, H.; Ye, W.; Li, Z.; Cheng, L.; Zhang, L.; Lei, Y.; Shen, L.; Zhang, F. The Glycoprotein and Nucleocapsid Protein of Hantaviruses Manipulate Autophagy Flux to Restrain Host Innate Immune Responses. Cell Rep. 2019, 27, 2075–2091.e5. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Wang, Y.; Yin, L.; Liang, W.; Zhang, J.; Ma, C.; Zhang, Y.; Liu, B.; Wang, J.; Zhao, W.; et al. The nonstructural protein 1 of respiratory syncytial virus hijacks host mitophagy as a novel mitophagy receptor to evade the type I IFN response in HEp-2 cells. mBio 2023, 14, e0148023. [Google Scholar] [CrossRef]
- Tomar, D.; Prajapati, P.; Lavie, J.; Singh, K.; Lakshmi, S.; Bhatelia, K.; Roy, M.; Singh, R.; Benard, G.; Singh, R. TRIM4; a novel mitochondrial interacting RING E3 ligase, sensitizes the cells to hydrogen peroxide (H2O2) induced cell death. Free Radic. Biol. Med. 2015, 89, 1036–1048. [Google Scholar] [CrossRef]
- Wang, J.; Qin, X.; Huang, Y.; Zhang, Q.; Pei, J.; Wang, Y.; Goren, I.; Ma, S.; Song, Z.; Liu, Y.; et al. TRIM7/RNF90 promotes autophagy via regulation of ATG7 ubiquitination during L. monocytogenes infection. Autophagy 2023, 19, 1844–1862. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.Y.; Rahmanto, A.S.; Tan, O.; Norris, M.D.; Haber, M.; Marshall, G.M.; Cheung, B.B. TRIM16 overexpression induces apoptosis through activation of caspase-2 in cancer cells. Apoptosis 2013, 18, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Magiera, M.M.; Mora, S.; Mojsa, B.; Robbins, I.; Lassot, I.; Desagher, S. Trim17-mediated ubiquitination and degradation of Mcl-1 initiate apoptosis in neurons. Cell Death Differ. 2013, 20, 281–292. [Google Scholar] [CrossRef]
- Lionnard, L.; Duc, P.; Brennan, M.S.; Kueh, A.J.; Pal, M.; Guardia, F.; Mojsa, B.; Damiano, M.A.; Mora, S.; Lassot, I.; et al. TRIM17 and TRIM28 antagonistically regulate the ubiquitination and anti-apoptotic activity of BCL2A1. Cell Death Differ. 2019, 26, 902–917. [Google Scholar] [CrossRef]
- Shen, J.; Yang, H.; Qiao, X.; Chen, Y.; Zheng, L.; Lin, J.; Lang, J.; Yu, Q.; Wang, Z. The E3 ubiquitin ligase TRIM17 promotes gastric cancer survival and progression via controlling BAX stability and antagonizing apoptosis. Cell Death Differ. 2023, 30, 2322–2335. [Google Scholar] [CrossRef] [PubMed]
- Gentric, G.; Kieffer, Y.; Mieulet, V.; Goundiam, O.; Bonneau, C.; Nemati, F.; Hurbain, I.; Raposo, G.; Popova, T.; Stern, M.H.; et al. PML-Regulated Mitochondrial Metabolism Enhances Chemosensitivity in Human Ovarian Cancers. Cell Metab. 2019, 29, 156–173.e10. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Ito, K.; Lin, H.K.; Santangelo, C.; Wieckowski, M.R.; Lebiedzinska, M.; Bononi, A.; Bonora, M.; Duszynski, J.; Bernardi, R.; et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 2010, 330, 1247–1251. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Perrone, M.; Gafa, R.; Nicoli, F.; Bonora, M.; Morciano, G.; Boncompagni, C.; Marchi, S.; Lebiedzinska-Arciszewska, M.; Vezzani, B.; et al. PML at mitochondria-associated membranes governs a trimeric complex with NLRP3 and P2X7R that modulates the tumor immune microenvironment. Cell Death Differ. 2023, 30, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, M.; Song, Y.; Xu, W. TRIM21 Restricts Coxsackievirus B3 Replication, Cardiac and Pancreatic Injury via Interacting With MAVS and Positively Regulating IRF3-Mediated Type-I Interferon Production. Front. Immunol. 2018, 9, 2479. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Yu, T.; Gan, S.; Wang, Y.; Pei, Y.; Zhao, Q.; Pei, S.; Hao, S.; Yuan, J.; Xu, J.; et al. TRIM24 facilitates antiviral immunity through mediating K63-linked TRAF3 ubiquitination. J. Exp. Med. 2020, 217, e20192083. [Google Scholar] [CrossRef]
- Cheng, C.T.; Kuo, C.Y.; Ouyang, C.; Li, C.F.; Chung, Y.; Chan, D.C.; Kung, H.J.; Ann, D.K. Metabolic Stress-Induced Phosphorylation of KAP1 Ser473 Blocks Mitochondrial Fusion in Breast Cancer Cells. Cancer Res. 2016, 76, 5006–5018. [Google Scholar] [CrossRef]
- Zhao, Z.; Song, X.; Wang, Y.; Yu, L.; Huang, G.; Li, Y.; Zong, R.; Liu, T.; Ji, Q.; Zheng, Y.; et al. E3 ubiquitin ligase TRIM31 alleviates dopaminergic neurodegeneration by promoting proteasomal degradation of VDAC1 in Parkinson’s Disease model. Cell Death Differ. 2024. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, P.; Gohel, D.; Shinde, A.; Roy, M.; Singh, K.; Singh, R. TRIM32 regulates mitochondrial mediated ROS levels and sensitizes the oxidative stress induced cell death. Cell. Signal. 2020, 76, 109777. [Google Scholar] [CrossRef]
- Goyani, S.; Shinde, A.; Shukla, S.; Saranga, M.V.; Currim, F.; Mane, M.; Singh, J.; Roy, M.; Gohel, D.; Chandak, N.; et al. Enhanced translocation of TRIM32 to mitochondria sensitizes dopaminergic neuronal cells to apoptosis during stress conditions in Parkinson’s disease. FEBS J. 2024, 291, 2636–2655. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, M.M.; Wang, Y.Y.; Shu, H.B. TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Ji, B.; Sun, D. TRIM34 localizes to the mitochondria and mediates apoptosis through the mitochondrial pathway in HEK293T cells. Heliyon 2020, 6, e03115. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.J.; Zhang, L.; Tang, W.; Chen, C.; Yang, C.S.; Kornbluth, S. The Trim39 ubiquitin ligase inhibits APC/CCdh1-mediated degradation of the Bax activator MOAP-1. J. Cell Biol. 2012, 197, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Hata, S.; Witt, C.C.; Ono, Y.; Lerche, S.; Ojima, K.; Chiba, T.; Doi, N.; Kitamura, F.; Tanaka, K.; et al. Muscle RING-finger protein-1 (MuRF1) as a connector of muscle energy metabolism and protein synthesis. J. Mol. Biol. 2008, 376, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Mattox, T.A.; Young, M.E.; Rubel, C.E.; Spaniel, C.; Rodriguez, J.E.; Grevengoed, T.J.; Gautel, M.; Xu, Z.; Anderson, E.J.; Willis, M.S. MuRF1 activity is present in cardiac mitochondria and regulates reactive oxygen species production in vivo. J. Bioenerg. Biomembr. 2014, 46, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Witt, S.H.; Granzier, H.; Witt, C.C.; Labeit, S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: Towards understanding MURF-dependent muscle ubiquitination. J. Mol. Biol. 2005, 350, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Demirdizen, E.; Al-Ali, R.; Narayanan, A.; Sun, X.; Varga, J.P.; Steffl, B.; Brom, M.; Krunic, D.; Schmidt, C.; Schmidt, G.; et al. TRIM67 drives tumorigenesis in oligodendrogliomas through Rho GTPase-dependent membrane blebbing. Neuro Oncol. 2023, 25, 1031–1043. [Google Scholar] [CrossRef]
- Wu, M.; Li, H.; He, J.; Liang, J.; Liu, Y.; Zhang, W. TRIM72 Alleviates Muscle Inflammation in mdx Mice via Promoting Mitophagy-Mediated NLRP3 Inflammasome Inactivation. Oxidative Med. Cell Longev. 2023, 2023, 8408574. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| TRIMs | Synonym | Mitochondrial Functions |
|---|---|---|
| TRIM4 | Regulates mitochondrial membrane potential and ROS production in response to oxidative stress [126]. | |
| TRIM5 | Regulates mitophagy and inflammation [85,86]. | |
| TRIM7 | RNF90/GNIP | Promotes MAVS degradation through K48 ubiquitination [127]. |
| TRIM14 | Enhancing RLR signaling via interacting with NEMO and RIG-I [50,68]. | |
| TRIM16 | EBBP | Regulates mitochondrial membrane potential and apoptosis [128]. |
| TRIM17 | TERF | Regulates apoptosis via regulating the stability of apoptotic proteins [129,130,131]. |
| TRIM19 | PML | Regulates mitochondrial metabolism, induces apoptosis, inhibits inflammation [132,133,134]. |
| TRIM21 | SSA/RO52 | Induces K27 ubiquitination of MAVS enhancing TBK1-IRF3 activation [69,135]. |
| TRIM24 | TIF1α | Regulates antiviral response via TRAF3 ubiquitination enhancing MAVS interaction [136]. |
| TRIM25 | EFP | Promotes MAVS degradation through K48 ubiquitination [44,73]. |
| TRIM27 | RFP | Induces mitophagy [84]. |
| TRIM28 | TIF1β/KAP1/KRIP-1 | Promotes MAVS degradation through K48 ubiquitination, regulates apoptosis, mitophagy, and mitochondrial fusion [71,116,137]. |
| TRIM29 | ATDC | Promotes MAVS degradation through K11 ubiquitination [72]. |
| TRIM31 | RING | Induce MAVS aggregation and activation; proteasomal degradation of VDAC1. [61,67,138]. |
| TRIM32 | BBS11/HT2A | Regulates mitochondrial membrane potential and ROS production in response to oxidative stress. Regulates apoptosis and antiviral response [139,140,141]. |
| TRIM34 | IFP1 | Regulates apoptosis [142]. |
| TRIM39 | TFP | Regulates apoptosis [143]. |
| TRIM40 | RNF35 | Interacts with MAVS [53]. |
| TRIM44 | Stabilizes MAVS by deubiquitylation [78]. | |
| TRIM63 | MURF1 | Regulates mitochondrial metabolism [144,145,146]. |
| TRIM65 | May regulate mitophagy/apoptosis [117]. | |
| TRIM67 | TNL | Interacts with MAVS [147]. |
| TRIM72 | MG53 | Regulates mitophagy and inflammation [148]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, S.; Mandell, M.A. Regulation of Mitochondria-Derived Immune Activation by ‘Antiviral’ TRIM Proteins. Viruses 2024, 16, 1161. https://doi.org/10.3390/v16071161
Oh S, Mandell MA. Regulation of Mitochondria-Derived Immune Activation by ‘Antiviral’ TRIM Proteins. Viruses. 2024; 16(7):1161. https://doi.org/10.3390/v16071161
Chicago/Turabian StyleOh, Seeun, and Michael A. Mandell. 2024. "Regulation of Mitochondria-Derived Immune Activation by ‘Antiviral’ TRIM Proteins" Viruses 16, no. 7: 1161. https://doi.org/10.3390/v16071161
APA StyleOh, S., & Mandell, M. A. (2024). Regulation of Mitochondria-Derived Immune Activation by ‘Antiviral’ TRIM Proteins. Viruses, 16(7), 1161. https://doi.org/10.3390/v16071161