
HIV-1 Intasomes Assembled with Excess Integrase C-Terminal Domain Protein Facilitate Structural Studies by Cryo-EM and Reveal the Role of the Integrase C-Terminal Tail in HIV-1 Integration

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Substrates, Peptides, and Other Reagents
2.2. Protein Preparation
2.3. In Vitro Concerted Integration Assay
2.4. Intasome Assembly and Electrophoretic Mobility Shift Assays
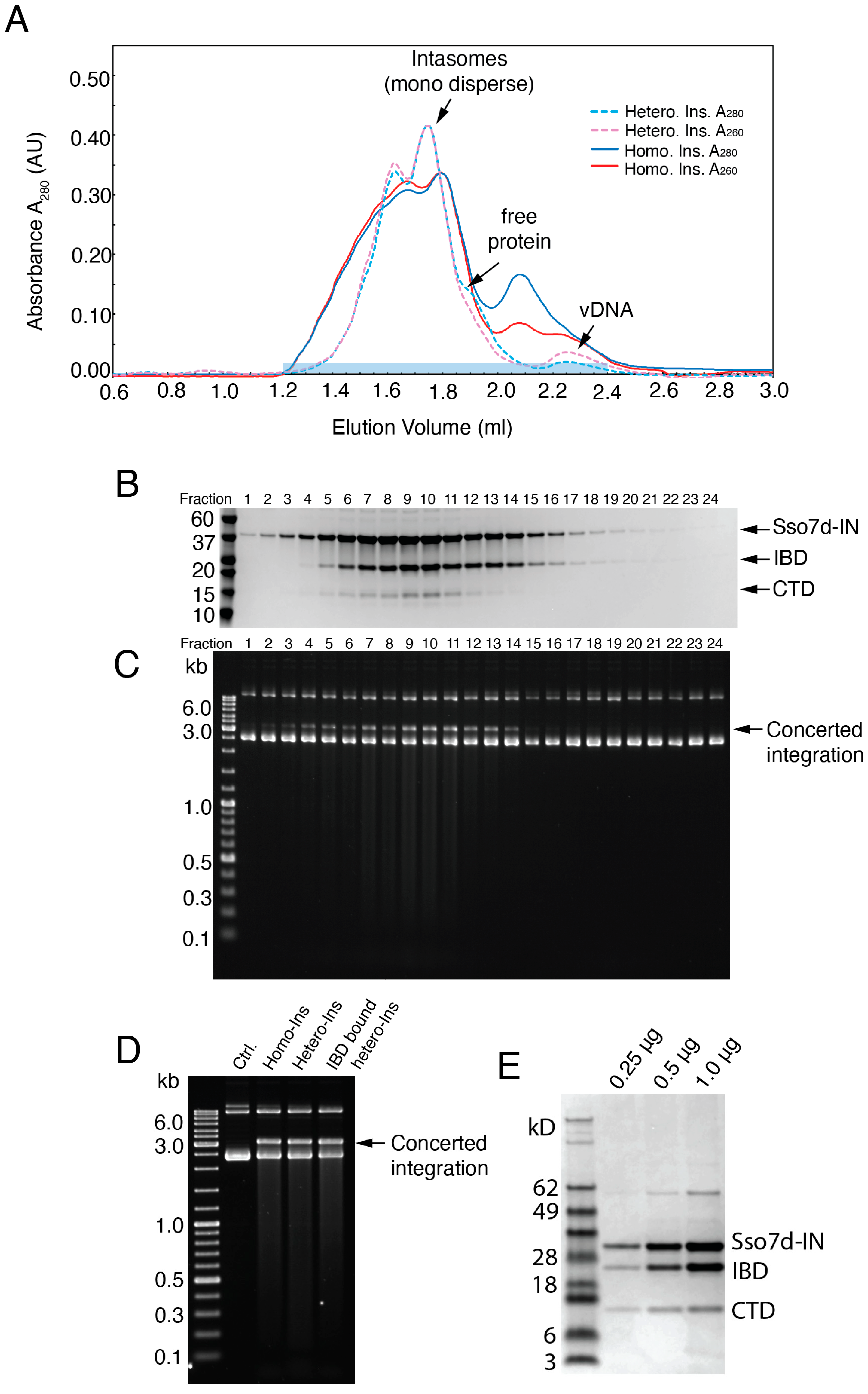
2.5. Hetero-Intasome Assembly and Purification for Cryo-EM
2.6. Cryo-EM Grid Preparation and Data Acquisition
2.7. Structure Determination and Refinement
2.8. Virus-Based Work
2.8.1. HIV-1 Production and Infection
2.8.2. Quantitative PCR Assays
3. Results
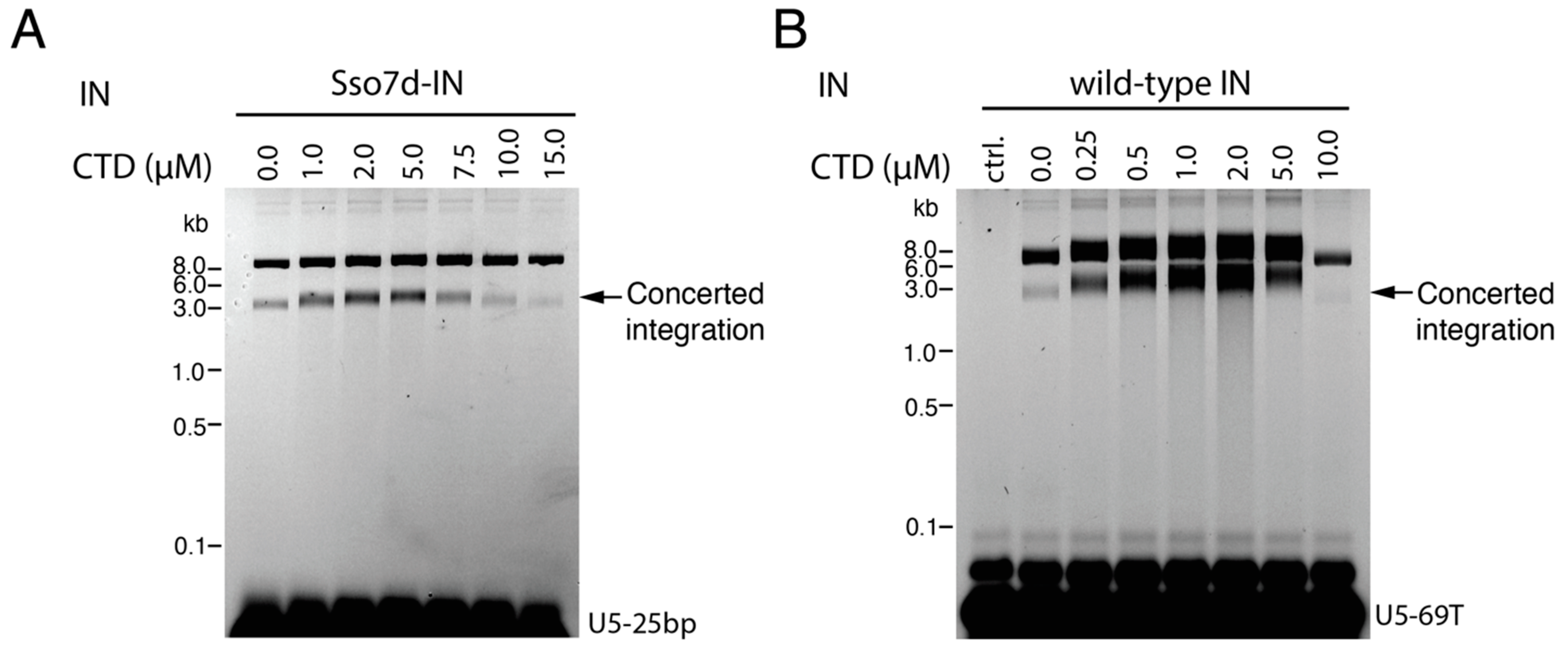
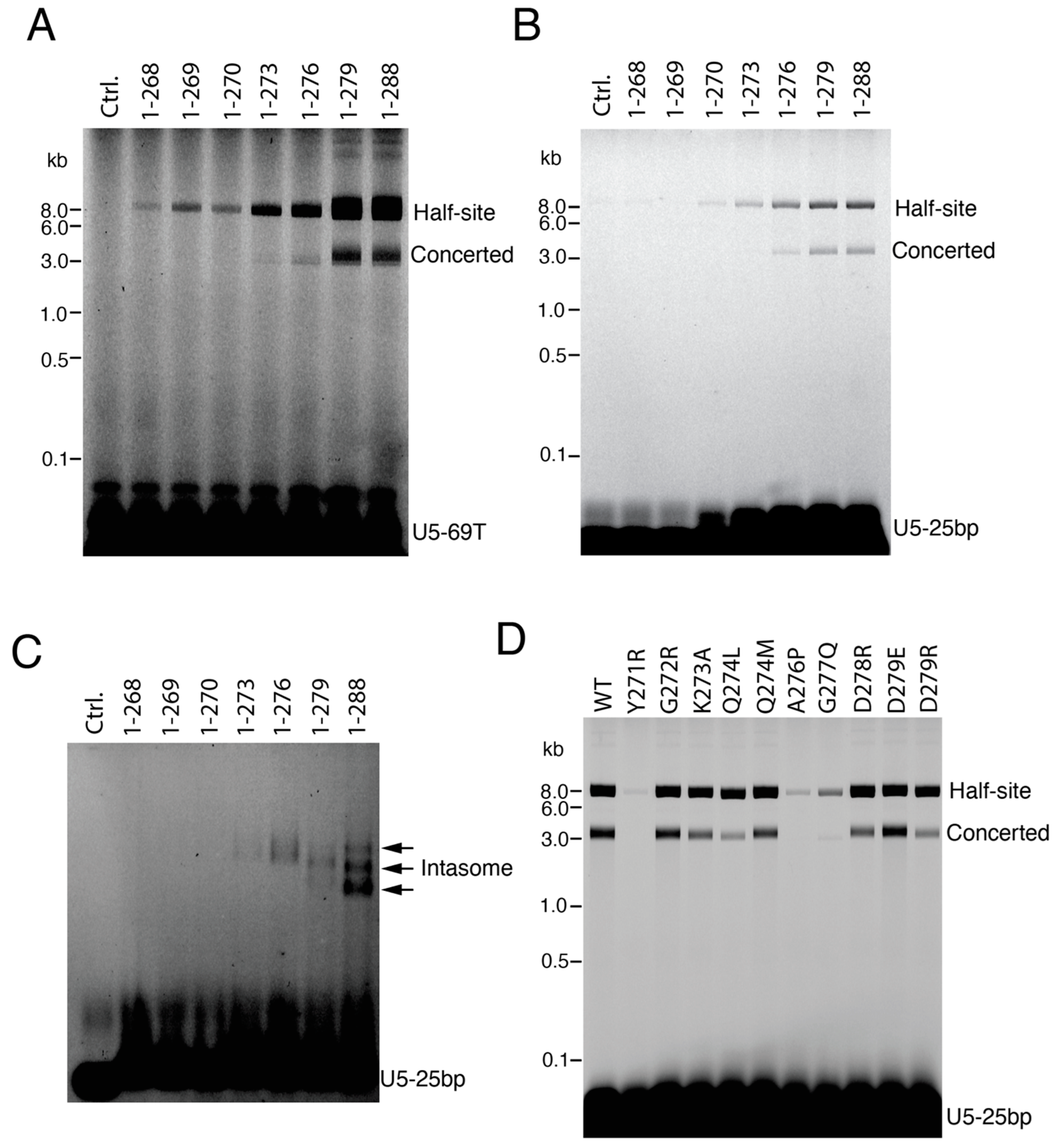
3.1. CTD Stimulates the Full Length IN Integration Activity
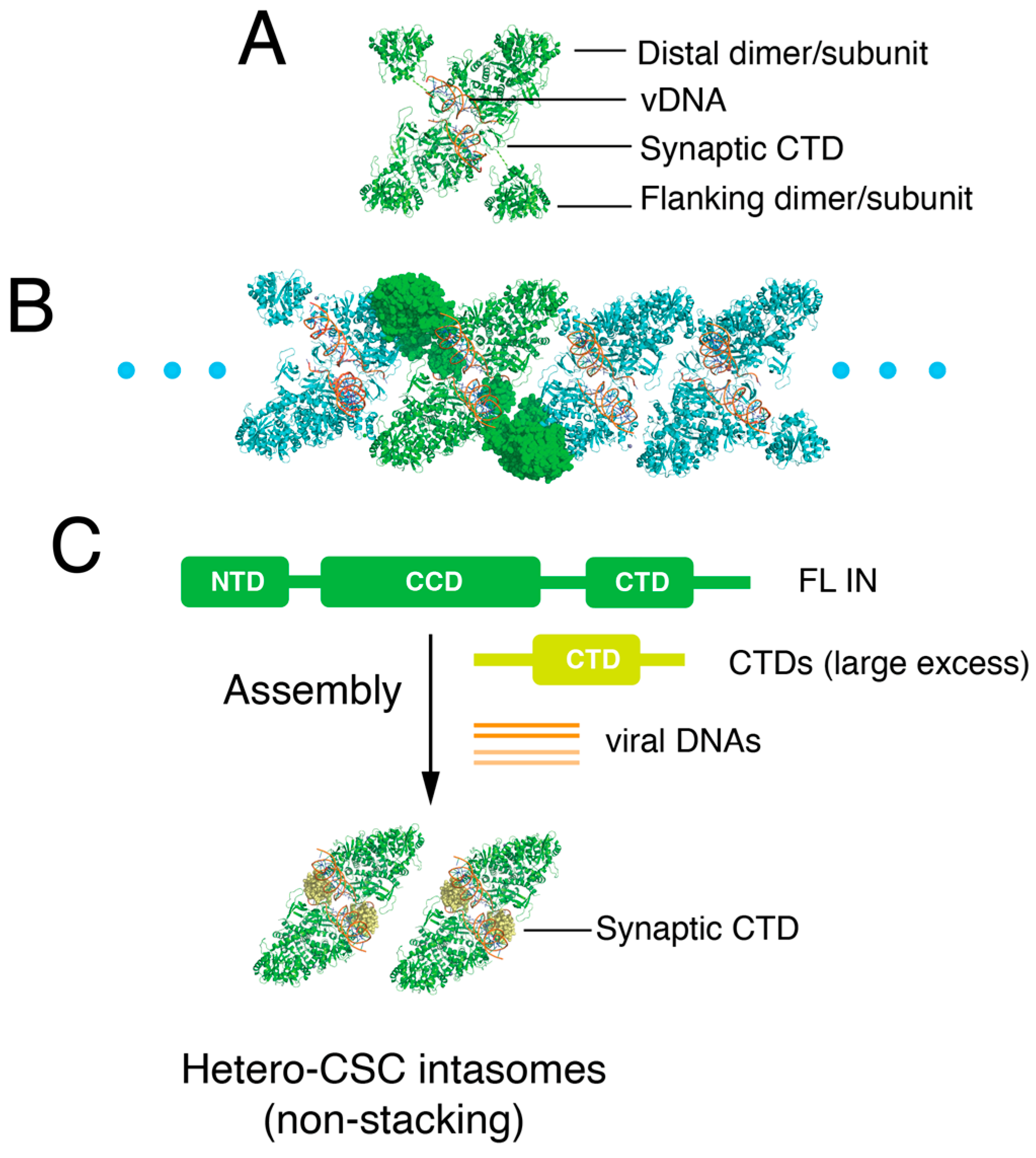
3.2. Exogenous CTD Can Be Incorporated into Intasomes
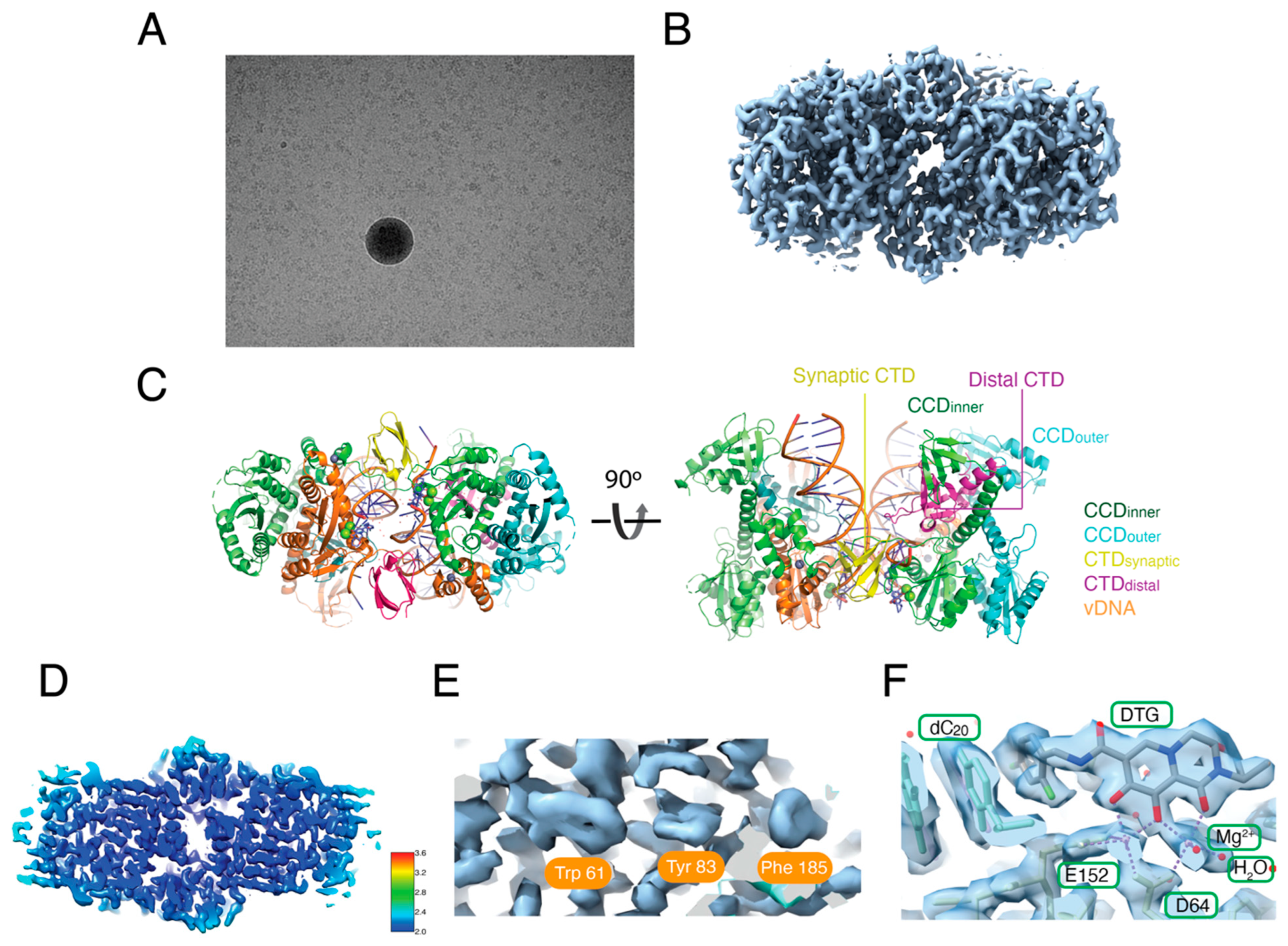
3.3. Reconstruction of the Hetero-CSC Intasome Core by Cryo-EM
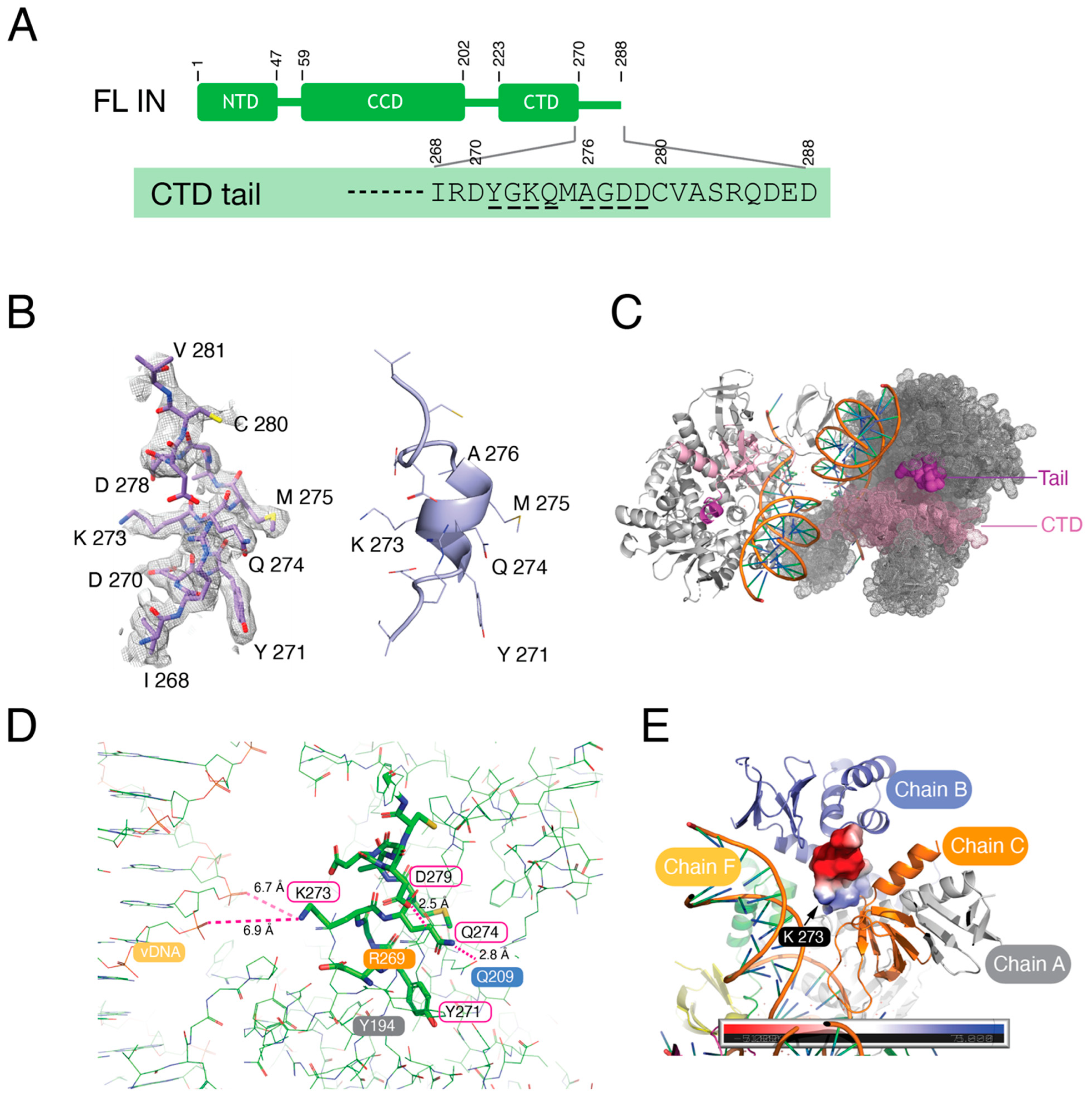
3.4. The C-Terminal Tail of HIV-1 Integrase Stabilizes the Intasome Structure
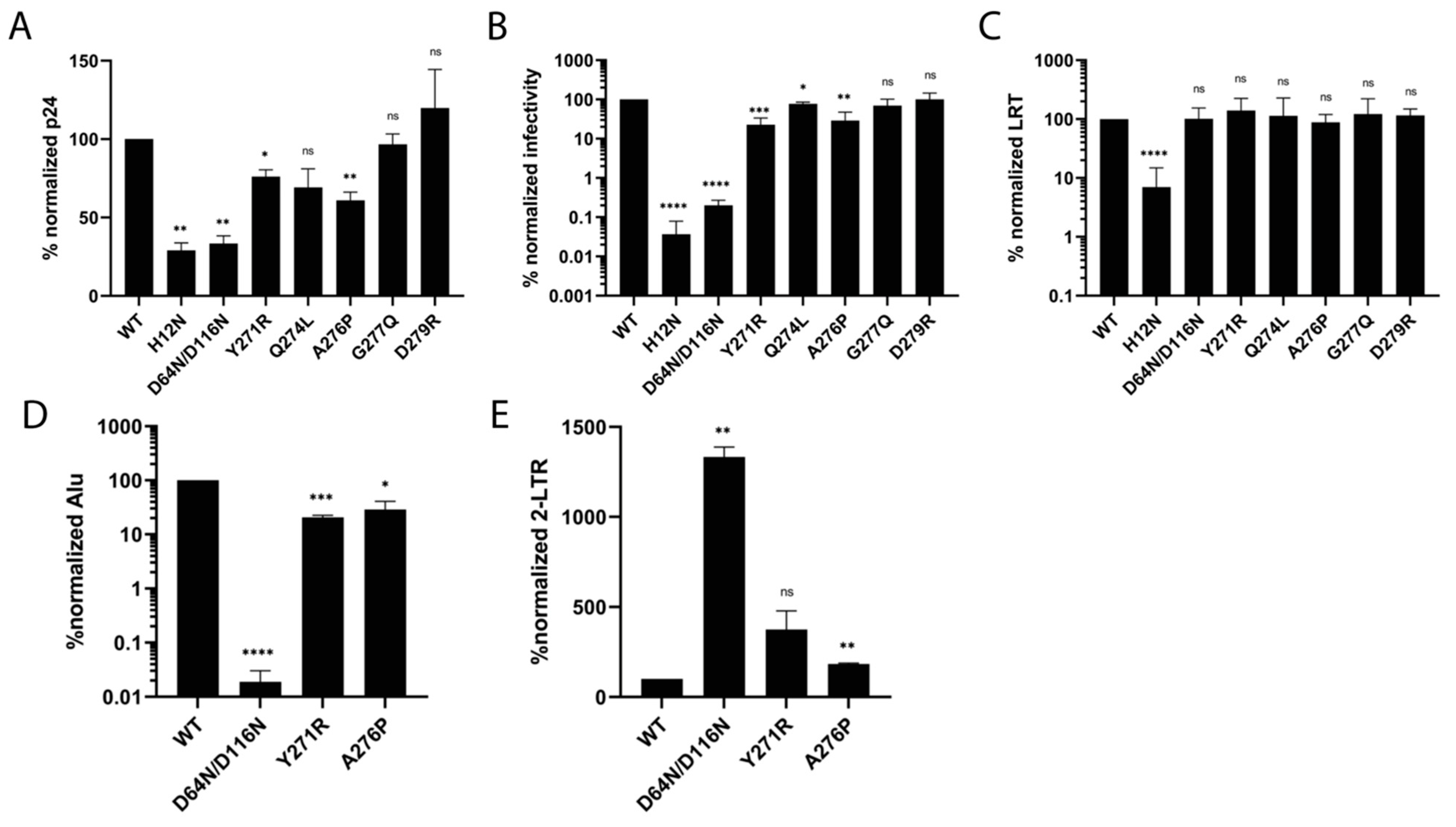
3.5. IN Tail Mutants Y271R and A276P Are Defective for Integration during HIV-1 Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Engelman, A.; Mizuuchi, K.; Craigie, R. HIV-1 DNA integration: Mechanism of viral DNA cleavage and DNA strand transfer. Cell 1991, 67, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Lesbats, P.; Engelman, A.N.; Cherepanov, P. Retroviral DNA integration. Chem. Rev. 2016, 116, 12730–12757. [Google Scholar] [CrossRef] [PubMed]
- Craigie, R. Nucleoprotein intermediates in HIV-1 DNA integration: Structure and function of HIV-1 intasomes. In Virus Protein and Nucleoprotein Complexes; Harris, J.R., Bhella, D., Eds.; Springer: Berlin/Heidelberg, Germany, 2018; Volume 88, pp. 189–210. [Google Scholar]
- Hare, S.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010, 464, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Ballandras-Colas, A.; Browne, M.; Cook, N.J.; Dewdney, T.G.; Domeler, B.; Cherepanov, P.; Lyumkis, D.; Engelman, A.N. Cryo-EM reveals a novel octameric integrase structure for betaretroviral intasome function. Nature 2016, 530, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Ballandras-Colas, A.; Maskell, D.P.; Serrao, E.; Locke, J.; Swuec, P.; Jonsson, S.R.; Kotecha, A.; Cook, N.J.; Pye, V.E.; Taylor, I.A.; et al. A supramolecular assembly mediates lentiviral DNA integration. Science 2017, 355, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Maertens, G.N.; Engelman, A.N.; Cherepanov, P. Structure and function of retroviral integrase. Nat. Rev. Microbiol. 2022, 20, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Passos, D.O.; Li, M.; Yang, R.B.; Rebensburg, S.V.; Ghirlando, R.; Jeon, Y.; Shkriabai, N.; Kvaratskhelia, M.; Craigie, R.; Lyumkis, D. Cryo-EM structures and atomic model of the HIV-1 strand transfer complex intasome. Science 2017, 355, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Passos, D.O.; Li, M.; Jozwik, I.K.; Zhao, X.Z.; Santos-Martins, D.; Yang, R.; Smith, S.J.; Jeon, Y.; Forli, S.; Hughes, S.H.; et al. Structural basis for strand-transfer inhibitor binding to HIV intasomes. Science 2020, 367, 810–814. [Google Scholar] [CrossRef]
- Li, M.; Passos, D.O.; Shan, Z.L.; Smith, S.J.; Sun, Q.F.; Biswas, A.; Choudhuri, I.; Strutzenberg, T.S.; Haldane, A.; Deng, N.J.; et al. Mechanisms of HIV-1 integrase resistance to dolutegravir and potent inhibition of drug-resistant variants. Sci. Adv. 2023, 9, eadg5953. [Google Scholar] [CrossRef]
- Li, M.; Yang, R.B.; Chen, X.M.; Wang, H.B.; Ghirlando, R.; Dimitriadis, E.K.; Craigie, R. HIV-1 Integrase Assembles Multiple Species of Stable Synaptic Complex Intasomes That Are Active for Concerted DNA Integration In vitro. J. Mol. Biol. 2024, 436, 168557. [Google Scholar] [CrossRef]
- Cai, M.; Zheng, R.; Caffrey, M.; Craigie, R.; Clore, G.M.; Gronenborn, A.M. Solution structure of the N-terminal zinc binding domain of HIV-1 integrase. Nat. Struct. Biol. 1997, 4, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Dyda, F.; Hickman, A.B.; Jenkins, T.M.; Engelman, A.; Craigie, R.; Davies, D.R. Crystal structure of the catalytic domain of HIV-1 integrase: Similarity to other polynucleotidyl transferases. Science 1994, 266, 1981–1986. [Google Scholar] [CrossRef] [PubMed]
- Lodi, P.J.; Ernst, J.A.; Kuszewski, J.; Hickman, A.B.; Engelman, A.; Craigie, R.; Clore, G.M.; Gronenborn, A.M. Solution structure of the DNA binding domain of HIV-1 integrase. Biochemistry 1995, 34, 9826–9833. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenboom, A.P.; Lutzke, R.A.; Boelens, R.; Plasterk, R.H.; Kaptein, R.; Hard, K. The DNA-binding domain of HIV-1 integrase has an SH3-like fold. Nat. Struct. Biol. 1995, 2, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Shi, K.; Banerjee, S.; Pandey, K.K.; Bera, S.; Grandgenett, D.P.; Aihara, H. Crystal structure of the Rous sarcoma virus intasome. Nature 2016, 530, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.J.; Li, W.; Berta, D.; Badaoui, M.; Ballandras-Colase, A.; Nans, A.; Kotecha, A.; Rosta, E.; Engelman, A.N.; Cherepanov, P. Structural basis of second-generation HIV integrase inhibitor action and viral resistance. Science 2020, 367, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Dar, M.J.; Monel, B.; Krishnan, L.; Shun, M.C.; Di Nunzio, F.; Helland, D.E.; Engelman, A. Biochemical and virological analysis of the 18-residue C-terminal tail of HIV-1 integrase. Retrovirology 2009, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jurado, K.A.; Lin, S.; Engelman, A.; Craigie, R. Engineered hyperactive integrase for concerted HIV-1 DNA integration. PLoS ONE 2014, 9, e105078. [Google Scholar] [CrossRef] [PubMed]
- Schorb, M.; Haberbosch, I.; Hagen, W.J.H.; Schwab, Y.; Mastronarde, D.N. Software tools for automated transmission electron microscopy. Nat. Methods 2019, 16, 471–477. [Google Scholar] [CrossRef]
- Zheng, S.Q.; Palovcak, E.; Armache, J.P.; Verba, K.A.; Cheng, Y.F.; Agard, D.A. MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 2017, 14, 331–332. [Google Scholar] [CrossRef]
- Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 2016, 193, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Leiro, R.; Scheres, S.H.W. A pipeline approach to single-particle processing in RELION. Acta Crystallogr. Sect. D-Struct. Biol. 2017, 73, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Garcia, R.; Gomez-Blanco, J.; Cuervo, A.; Carazo, J.M.; Sorzano, C.O.S.; Vargas, J. DeepEMhancer: A deep learning solution for cryo-EM volume post-processing. Commun. Biol. 2021, 4, 874. [Google Scholar] [CrossRef] [PubMed]
- Swint-Kruse, L.; Brown, C.S. Resmap: Automated representation of macromolecular interfaces as two-dimensional networks. Bioinformatics 2005, 21, 3327–3328. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Brunger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. Sect. D-Biol. Crystallogr. 1998, 54, 905–921. [Google Scholar]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D-Struct. Biol. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Limon, A.; Devroe, E.; Silver, P.A.; Cherepanov, P.; Engelman, A. Class II integrase mutants with changes in putative nuclear localization signals are primarily blocked at a postnuclear entry step of human immunodeficiency virus type 1 replication. J. Virol. 2004, 78, 12735–12746. [Google Scholar] [CrossRef]
- Li, W.; Singh, P.K.; Sowd, G.A.; Bedwell, G.J.; Jang, S.; Achuthan, V.; Oleru, A.V.; Wong, D.; Fadel, H.J.; Lee, K.; et al. CPSF6-Dependent Targeting of Speckle-Associated Domains Distinguishes Primate from Nonprimate Lentiviral Integration. Mbio 2020, 11, e02254-20. [Google Scholar] [CrossRef]
- Jurado, K.A.; Wang, H.; Slaughter, A.; Feng, L.; Kessl, J.J.; Koh, Y.; Wang, W.; Ballandras-Colas, A.; Patel, P.A.; Fuchs, J.R.; et al. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. USA 2013, 110, 8690–8695. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, X.M.; Wang, H.B.; Jurado, K.A.; Engelman, A.N.; Craigie, R. A Peptide Derived from Lens Epithelium-Derived Growth Factor Stimulates HIV-1 DNA Integration and Facilitates Intasome Structural Studies. J. Mol. Biol. 2020, 432, 2055–2066. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, K.D.; Topper, M.B.; Muesing, M.A. Sequential Deletion of the Integrase (Gag-Pol) Carboxyl Terminus Reveals Distinct Phenotypic Classes of Defective HIV-1. J. Virol. 2011, 85, 4654–4666. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Turkki, V.; Sherrill-Mix, S.; Hwang, Y.; Eilers, G.; Taylor, L.; McDanal, C.; Wang, P.; Temelkoff, D.; Nolte, R.T.; et al. Structural Basis for Inhibitor-Induced Aggregation of HIV Integrase. PLoS Biol. 2016, 14, e1002584. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A. In vivo analysis of retroviral integrase structure and function. Adv. Virus Res. 1999, 52, 411–426. [Google Scholar] [PubMed]
- Engelman, A.; Englund, G.; Orenstein, J.M.; Martin, M.A.; Craigie, R. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J. Virol. 1995, 69, 2729–2736. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.L.; Eschbach, J.E.; Koneru, P.C.; Li, W.; Puray-Chavez, M.; Townsend, D.; Lawson, D.Q.; Engelman, A.N.; Kvaratskhelia, M.; Kutluay, S.B. Integrase-RNA interactions underscore the critical role of integrase in HIV-1 virion morphogenesis. Elife 2020, 9, e54311. [Google Scholar] [CrossRef] [PubMed]
- Butler, S.L.; Hansen, M.S.T.; Bushman, F.D. A quantitative assay for HIV DNA integration in vivo. Nat. Med. 2001, 7, 631–634. [Google Scholar] [CrossRef] [PubMed]
- Rocchi, C.; Gouet, P.; Parissi, V.; Fiorini, F. The C-Terminal Domain of HIV-1 Integrase: A Swiss Army Knife for the Virus? Viruses 2022, 14, 1397. [Google Scholar] [CrossRef]
- Gandhi, R.T.; Bedimo, R.; Hoy, J.F.; Landovitz, R.J.; Smith, D.M.; Eaton, E.F.; Lehmann, C.; Springer, S.A.; Sax, P.E.; Thompson, M.A.; et al. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults 2022 Recommendations of the International Antiviral Society-USA Panel. Jama-J. Am. Med. Assoc. 2023, 329, 63–84. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Li, Z.; Chen, X.; Cui, Y.; Engelman, A.N.; Craigie, R. HIV-1 Intasomes Assembled with Excess Integrase C-Terminal Domain Protein Facilitate Structural Studies by Cryo-EM and Reveal the Role of the Integrase C-Terminal Tail in HIV-1 Integration. Viruses 2024, 16, 1166. https://doi.org/10.3390/v16071166
Li M, Li Z, Chen X, Cui Y, Engelman AN, Craigie R. HIV-1 Intasomes Assembled with Excess Integrase C-Terminal Domain Protein Facilitate Structural Studies by Cryo-EM and Reveal the Role of the Integrase C-Terminal Tail in HIV-1 Integration. Viruses. 2024; 16(7):1166. https://doi.org/10.3390/v16071166
Chicago/Turabian StyleLi, Min, Zhen Li, Xuemin Chen, Yanxiang Cui, Alan N. Engelman, and Robert Craigie. 2024. "HIV-1 Intasomes Assembled with Excess Integrase C-Terminal Domain Protein Facilitate Structural Studies by Cryo-EM and Reveal the Role of the Integrase C-Terminal Tail in HIV-1 Integration" Viruses 16, no. 7: 1166. https://doi.org/10.3390/v16071166