
Exploring Canine Picornavirus Diversity in the USA Using Wastewater Surveillance: From High-Throughput Genomic Sequencing to Immuno-Informatics and Capsid Structure Modeling

 , ,
, ,  , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
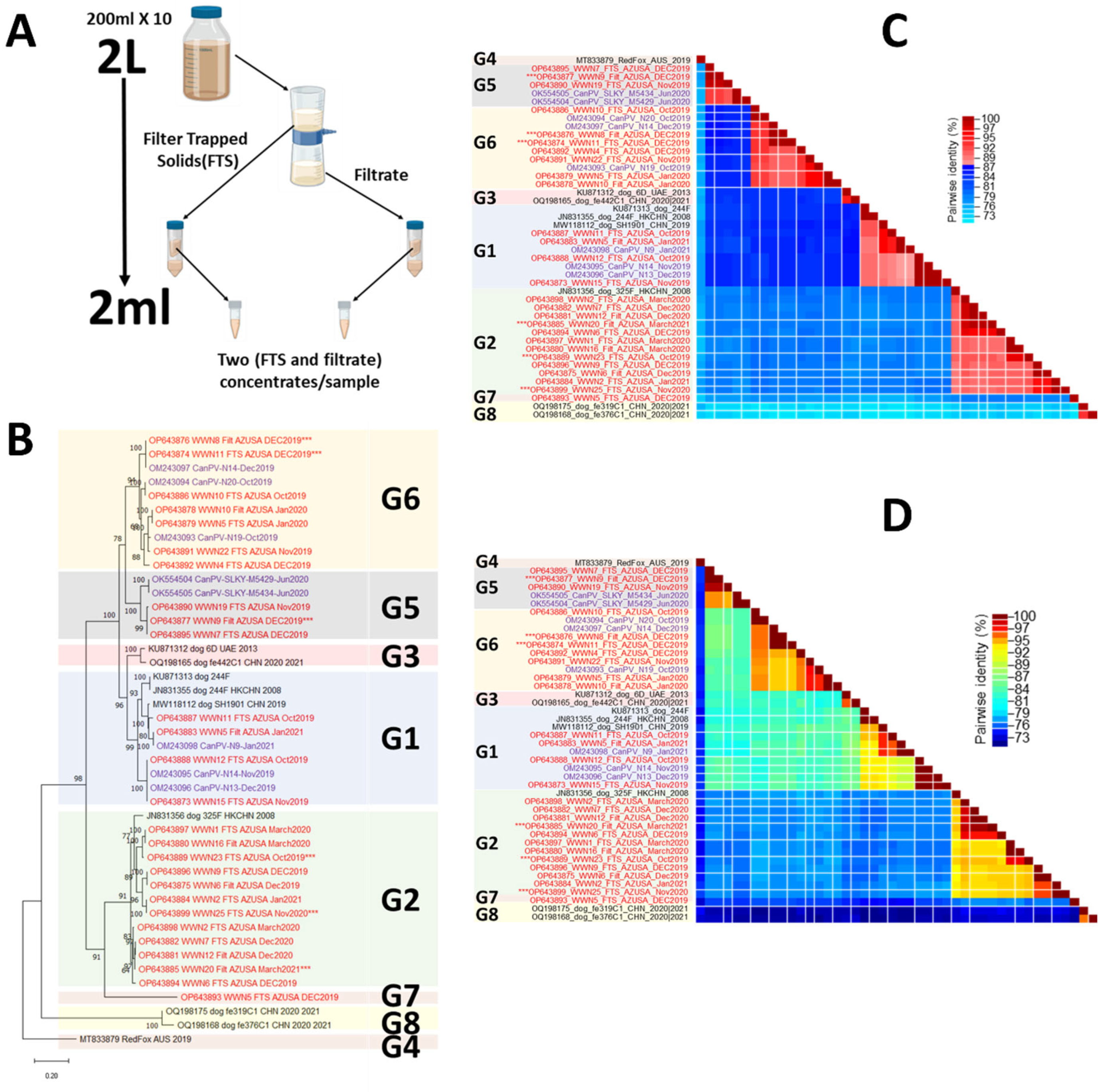
2.1. Sample Collection and Processing
2.2. Nucleic Acid Extraction and Polymerase Chain Reaction (PCR)
2.3. Sequencing
2.4. Reads Processing
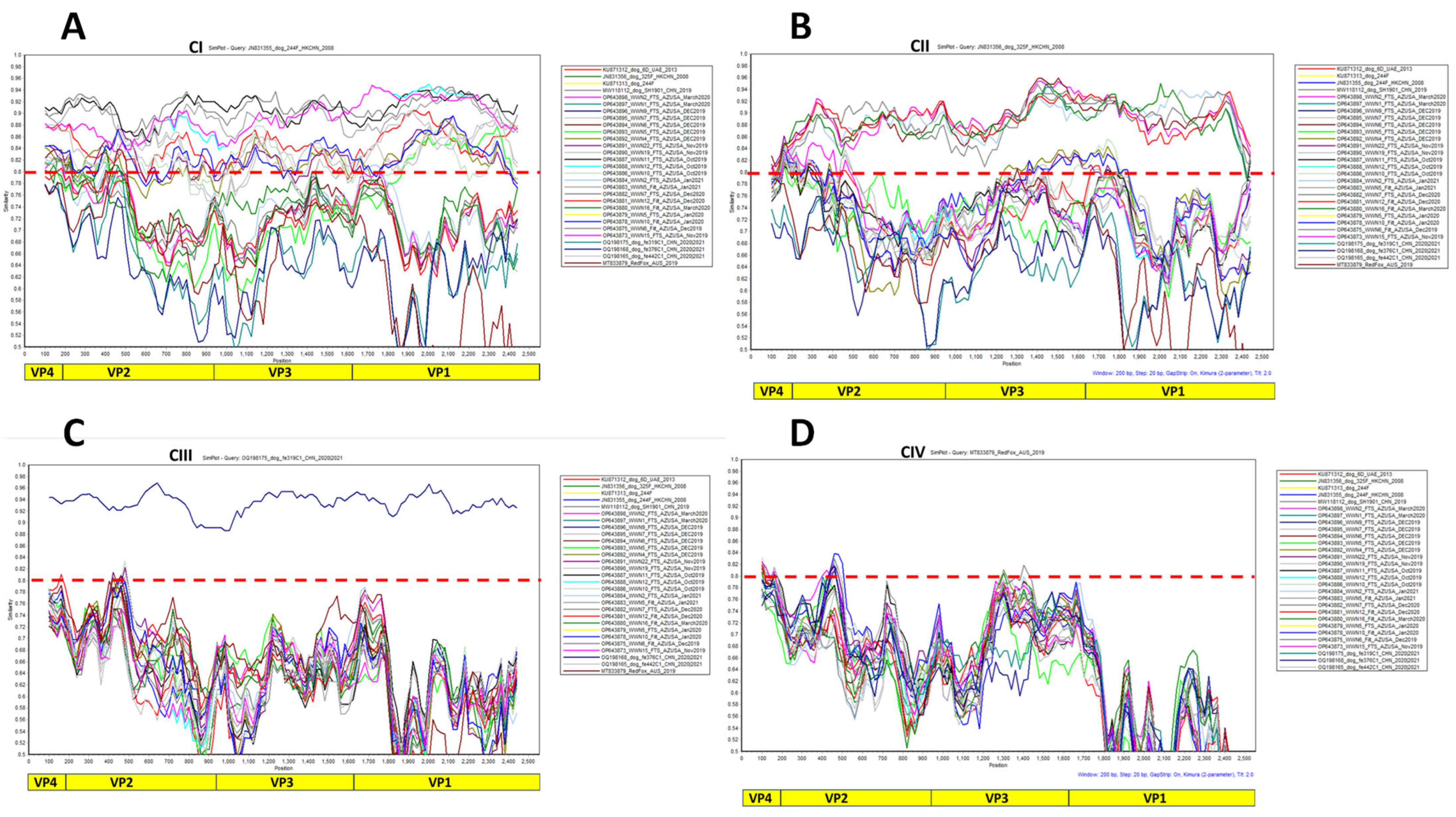
2.5. Virus Typing and Phylogenetic Analysis
2.6. Immunoinformatics and CanPV Capsid Structure Prediction
3. Results
3.1. Nested PCR Results
3.2. Reads and Contigs Obtained
3.3. Virus Typing
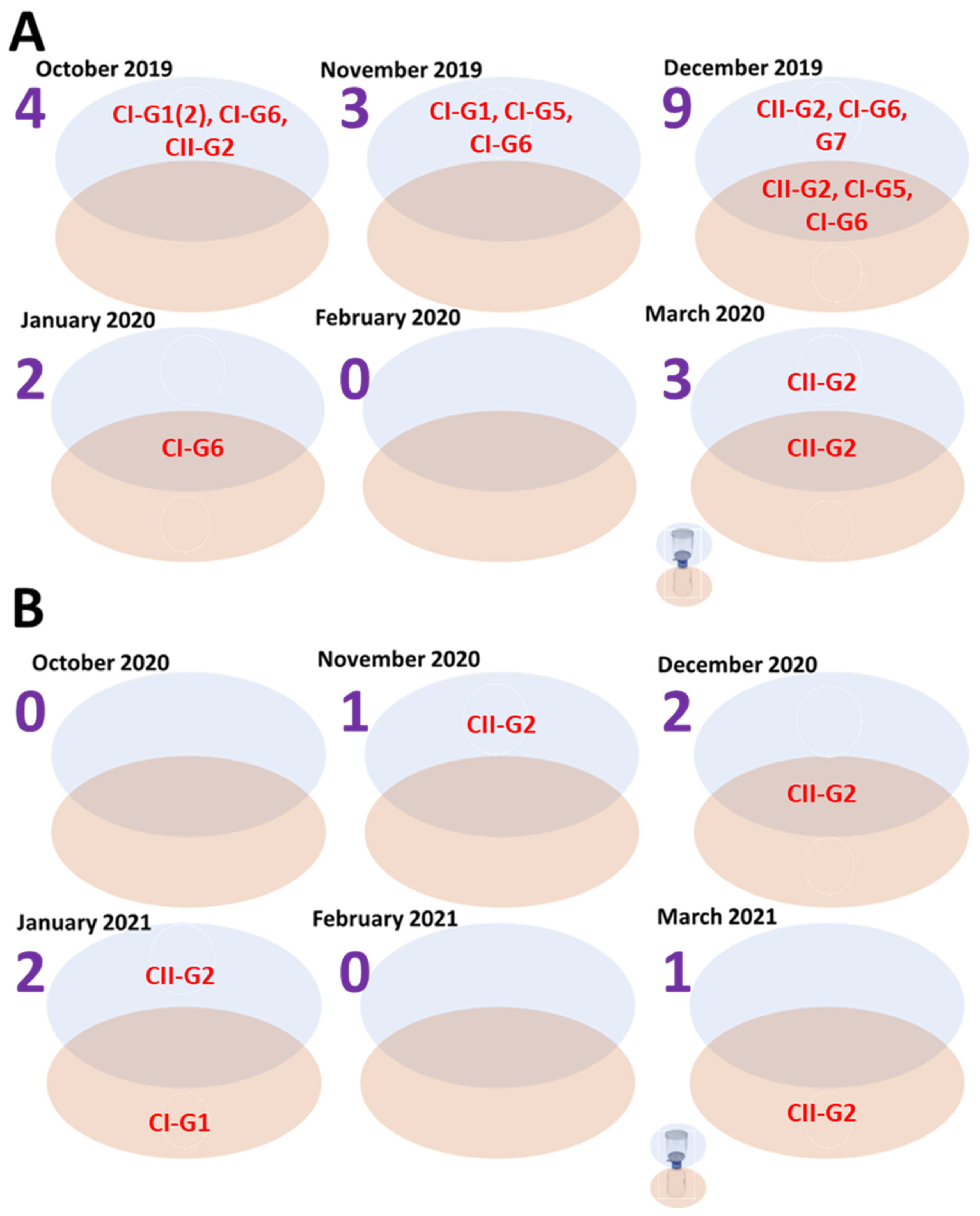
3.4. Diversity by Fraction and Season
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hill, V.; Githinji, G.; Vogels, C.B.F.; Bento, A.I.; Chaguza, C.; Carrington, C.V.F.; Grubaugh, N.D. Toward a global virus genomic surveillance network. Cell Host Microbe 2023, 31, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.J.; Ashley, W.; Gil-Fernandez, M.; Newsome, T.M.; Di Giallonardo, F.; Ortiz-Baez, A.S.; Mahar, J.E.; Towerton, A.L.; Gillings, M.; Holmes, E.C.; et al. Red fox viromes in urban and rural landscapes. Virus Evol. 2020, 6, veaa065. [Google Scholar] [CrossRef]
- Faleye, T.O.C.; Driver, E.M.; Bowes, D.A.; Holm, R.H.; Talley, D.; Yeager, R.; Bhatnagar, A.; Smith, T.; Varsani, A.; Halden, R.U.; et al. Detection of human, porcine and canine picornaviruses in municipal sewage sludge using pan-enterovirus amplicon-based long-read Illumina sequencing. Emerg. Microbes Infect. 2022, 11, 1339–1342. [Google Scholar] [CrossRef]
- Faleye, T.O.C.; Skidmore, P.T.; Elyaderani, A.; Smith, A.; Kaiser, N.; Adhikari, S.; Yanez, A.; Perleberg, T.; Driver, E.M.; Halden, R.U.; et al. Canine picornaviruses detected in wastewater in Arizona, USA 2019 and 2021. Infect. Genet. Evol. 2022, 103, 105315. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, Y.; Li, H.; Chen, Z.; Zhou, J.; Liu, G.; Wang, Y. Genomic characterization and phylogenetic analysis of a new canine picornavirus variant in the mainland of China. Virus Res. 2021, 296, 198351. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.; Lau, S.K.; Choi, G.K.; Yip, C.C.; Huang, Y.; Tsoi, H.W.; Yuen, K.Y. Complete genome sequence of a novel picornavirus, canine picornavirus, discovered in dogs. J. Virol. 2012, 86, 3402–3403. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Choi, G.K.Y.; Huang, Y.; Sivakumar, S.; Tsoi, H.W.; Yip, C.C.Y.; Jose, S.V.; Bai, R.; Wong, E.Y.M.; et al. Molecular epidemiology of canine picornavirus in Hong Kong and Dubai and proposal of a novel genus in Picornaviridae. Infect. Genet. Evol. 2016, 41, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Wasik, B.R.; Voorhees, I.E.H.; Parrish, C.R. Canine and Feline Influenza. Cold Spring Harb. Perspect. Med. 2021, 11, a038562. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2021, 49, D92–D96. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef]
- Mirdita, M.; Schutze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Montiel-Garcia, D.; Santoyo-Rivera, N.; Ho, P.; Carrillo-Tripp, M.; Iii, C.L.B.; Johnson, J.E.; Reddy, V.S. VIPERdb v3.0: A structure-based data analytics platform for viral capsids. Nucleic Acids Res. 2021, 49, D809–D816. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef]
- Wang, K.; Zheng, B.; Zhang, L.; Cui, L.; Su, X.; Zhang, Q.; Guo, Z.; Guo, Y.; Zhang, W.; Zhu, L.; et al. Serotype specific epitopes identified by neutralizing antibodies underpin immunogenic differences in Enterovirus B. Nat. Commun. 2020, 11, 4419. [Google Scholar] [CrossRef]
- McIntyre, C.L.; Knowles, N.J.; Simmonds, P. Proposals for the classification of human rhinovirus species A, B and C into genotypically assigned types. J. Gen. Virol. 2013, 94, 1791–1806. [Google Scholar] [CrossRef]
- Oberste, M.S.; Nix, W.A.; Maher, K.; Pallansch, M.A. Improved molecular identification of enteroviruses by RT-PCR and amplicon sequencing. J. Clin. Virol. 2003, 26, 375–377. [Google Scholar] [CrossRef] [PubMed]
- Bessaud, M.; Razafindratsimandresy, R.; Nougairede, A.; Joffret, M.L.; Deshpande, J.M.; Dubot-Peres, A.; Heraud, J.M.; de Lamballerie, X.; Delpeyroux, F.; Bailly, J.L. Molecular comparison and evolutionary analyses of VP1 nucleotide sequences of new African human enterovirus 71 isolates reveal a wide genetic diversity. PLoS ONE 2014, 9, e90624. [Google Scholar] [CrossRef]
- Benschop, K.S.M.; Broberg, E.K.; Hodcroft, E.; Schmitz, D.; Albert, J.; Baicus, A.; Bailly, J.L.; Baldvinsdottir, G.; Berginc, N.; Blomqvist, S.; et al. Molecular Epidemiology and Evolutionary Trajectory of Emerging Echovirus 30, Europe. Emerg. Infect. Dis. 2021, 27, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Lema, C.; Torres, C.; Van der Sanden, S.; Cisterna, D.; Freire, M.C.; Gomez, R.M. Global phylodynamics of Echovirus 30 revealed differential behavior among viral lineages. Virology 2019, 531, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Miranda, C.; Thompson, G. Canine parvovirus: The worldwide occurrence of antigenic variants. J. Gen. Virol. 2016, 97, 2043–2057. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, W.; Bertsch, P.M.; Bivins, A.; Bibby, K.; Farkas, K.; Gathercole, A.; Haramoto, E.; Gyawali, P.; Korajkic, A.; McMinn, B.R.; et al. Comparison of virus concentration methods for the RT-qPCR-based recovery of murine hepatitis virus, a surrogate for SARS-CoV-2 from untreated wastewater. Sci. Total Environ. 2020, 739, 139960. [Google Scholar] [CrossRef] [PubMed]
- Farkas, K.; Pellett, C.; Alex-Sanders, N.; Bridgman, M.T.P.; Corbishley, A.; Grimsley, J.M.S.; Kasprzyk-Hordern, B.; Kevill, J.L.; Pantea, I.; Richardson-O’Neill, I.S.; et al. Comparative Assessment of Filtration- and Precipitation-Based Methods for the Concentration of SARS-CoV-2 and Other Viruses from Wastewater. Microbiol. Spectr. 2022, 10, e0110222. [Google Scholar] [CrossRef] [PubMed]
- Fores, E.; Bofill-Mas, S.; Itarte, M.; Martinez-Puchol, S.; Hundesa, A.; Calvo, M.; Borrego, C.M.; Corominas, L.L.; Girones, R.; Rusinol, M. Evaluation of two rapid ultrafiltration-based methods for SARS-CoV-2 concentration from wastewater. Sci. Total Environ. 2021, 768, 144786. [Google Scholar] [CrossRef]
- Petala, M.; Dafou, D.; Kostoglou, M.; Karapantsios, T.; Kanata, E.; Chatziefstathiou, A.; Sakaveli, F.; Kotoulas, K.; Arsenakis, M.; Roilides, E.; et al. A physicochemical model for rationalizing SARS-CoV-2 concentration in sewage. Case study: The city of Thessaloniki in Greece. Sci. Total Environ. 2021, 755, 142855. [Google Scholar] [CrossRef]
- Ye, Y.; Ellenberg, R.M.; Graham, K.E.; Wigginton, K.R. Survivability, Partitioning, and Recovery of Enveloped Viruses in Untreated Municipal Wastewater. Environ. Sci. Technol. 2016, 50, 5077–5085. [Google Scholar] [CrossRef] [PubMed]
- Erickson, A.K.; Jesudhasan, P.R.; Mayer, M.J.; Narbad, A.; Winter, S.E.; Pfeiffer, J.K. Bacteria Facilitate Enteric Virus Co-infection of Mammalian Cells and Promote Genetic Recombination. Cell Host Microbe 2018, 23, 77–88.e75. [Google Scholar] [CrossRef]
- Lu, H.; Lehrman, M.A.; Pfeiffer, J.K. Use of a Glycan Library Reveals a New Model for Enteric Virus Oligosaccharide Binding and Virion Stabilization. J. Virol. 2020, 94, e01894-19. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Jesudhasan, P.R.; Pfeiffer, J.K. Bacterial lipopolysaccharide binding enhances virion stability and promotes environmental fitness of an enteric virus. Cell Host Microbe 2014, 15, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Faleye, T.O.C.; Driver, E.M.; Wright, J.M.; Halden, R.U.; Varsani, A.; Scotch, M. Direct detection of canine picornavirus complete coding sequence in wastewater using long-range reverse-transcriptase polymerase chain reaction and long-read sequencing. Infect. Genet. Evol. 2024, 118, 105550. [Google Scholar] [CrossRef] [PubMed]
- Oberste, M.S. Progress of polio eradication and containment requirements after eradication. Transfusion 2018, 58 (Suppl. S3), 3078–3083. [Google Scholar] [CrossRef]
- Previsani, N.; Tangermann, R.H.; Tallis, G.; Jafari, H.S. World Health Organization Guidelines for Containment of Poliovirus Following Type-Specific Polio Eradication-Worldwide, 2015. MMWR Morb. Mortal. Wkly. Rep. 2015, 64, 913–917. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Month | Filtrate | FTS | ||
|---|---|---|---|---|
| Conc ID | CanPV | Conc ID | CanPV | |
| October 2019 | 1 | 13 | + | |
| November 2019 | 2 | 14 | + | |
| December 2019 | 3 | + | 15 | + |
| January 2020 | 4 | + | 16 | + |
| February 2020 | 5 | 17 | ||
| March 2020 | 6 | + | 18 | + |
| Season 1 | 3/6 | 5/6 | ||
| October 2020 | 7 | 19 | ||
| November 2020 | 8 | 20 | + | |
| December 2020 | 9 | + | 21 | + |
| January 2021 | 10 | + | 22 | + |
| February 2021 | 11 | 23 | ||
| March 2021 | 12 | + | 24 | |
| Season 2 | 3/6 | 3/6 | ||
| Total | 6/12 | 8/12 | ||
| VP2 | VP3 | VP1 | |||
|---|---|---|---|---|---|
| Phylogenetic Clusters | South-Wall Rim (aa 194–217) | South-Wall Rim (aa 56–62) | North-Wall Rim (aa 143–161) | South-Wall Rim (aa 219–228) | North-Wall Rim (aa 246–254) |
| C-I | CQSSSGERS/LS/NTIFELA/V/TD/EN/T/I/YDFR/GDYP | SN/SD/EGNV | DPVSRARN/DQV/IPNISD | QNFS/NPS/TAA/VGY | SYRTNDGVS |
| C-II | CQSSSGERSN/DSIFQ/PLQQERDFA/SDYP | SD/ENTNTF | DPASRSRNNVPNLTD | RNFNPEA/TAGY | NFRSSDNTS |
| C-III | CQAS/PQARTSSIFELQDTDFQDYP | SGEVT/ATP | DPS/TSRNRVNGS/NA/TTIPNNSD | QNFQPQTSGY | SYKDNNTS |
| C-IV | CQSSSGERSASIFQLTEQDFADYP | GESATP | DPSSTGLSNSTTISD | ANFQPQAAQY | ARKDNTGTS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faleye, T.O.C.; Skidmore, P.; Elyaderani, A.; Adhikari, S.; Kaiser, N.; Smith, A.; Yanez, A.; Perleberg, T.; Driver, E.M.; Halden, R.U.; et al. Exploring Canine Picornavirus Diversity in the USA Using Wastewater Surveillance: From High-Throughput Genomic Sequencing to Immuno-Informatics and Capsid Structure Modeling. Viruses 2024, 16, 1188. https://doi.org/10.3390/v16081188
Faleye TOC, Skidmore P, Elyaderani A, Adhikari S, Kaiser N, Smith A, Yanez A, Perleberg T, Driver EM, Halden RU, et al. Exploring Canine Picornavirus Diversity in the USA Using Wastewater Surveillance: From High-Throughput Genomic Sequencing to Immuno-Informatics and Capsid Structure Modeling. Viruses. 2024; 16(8):1188. https://doi.org/10.3390/v16081188
Chicago/Turabian StyleFaleye, Temitope O. C., Peter Skidmore, Amir Elyaderani, Sangeet Adhikari, Nicole Kaiser, Abriana Smith, Allan Yanez, Tyler Perleberg, Erin M. Driver, Rolf U. Halden, and et al. 2024. "Exploring Canine Picornavirus Diversity in the USA Using Wastewater Surveillance: From High-Throughput Genomic Sequencing to Immuno-Informatics and Capsid Structure Modeling" Viruses 16, no. 8: 1188. https://doi.org/10.3390/v16081188
APA StyleFaleye, T. O. C., Skidmore, P., Elyaderani, A., Adhikari, S., Kaiser, N., Smith, A., Yanez, A., Perleberg, T., Driver, E. M., Halden, R. U., Varsani, A., & Scotch, M. (2024). Exploring Canine Picornavirus Diversity in the USA Using Wastewater Surveillance: From High-Throughput Genomic Sequencing to Immuno-Informatics and Capsid Structure Modeling. Viruses, 16(8), 1188. https://doi.org/10.3390/v16081188