
CavitOmiX Drug Discovery: Engineering Antivirals with Enhanced Spectrum and Reduced Side Effects for Arboviral Diseases

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mutation Analysis of Viral Genome Sequences
2.2. Generation of Structural Models and Point-Cloud Representations of Druggable Sites
2.3. Comparison and Clustering of Binding Site Cavities
2.4. Selection of Small-Molecule Candidates for Refinement
2.5. Refinement of Molecules with a Genetic Algorithm
2.6. Visualizations
3. Results
3.1. Approved Drugs for Off-Label Use against CHIKV Disease
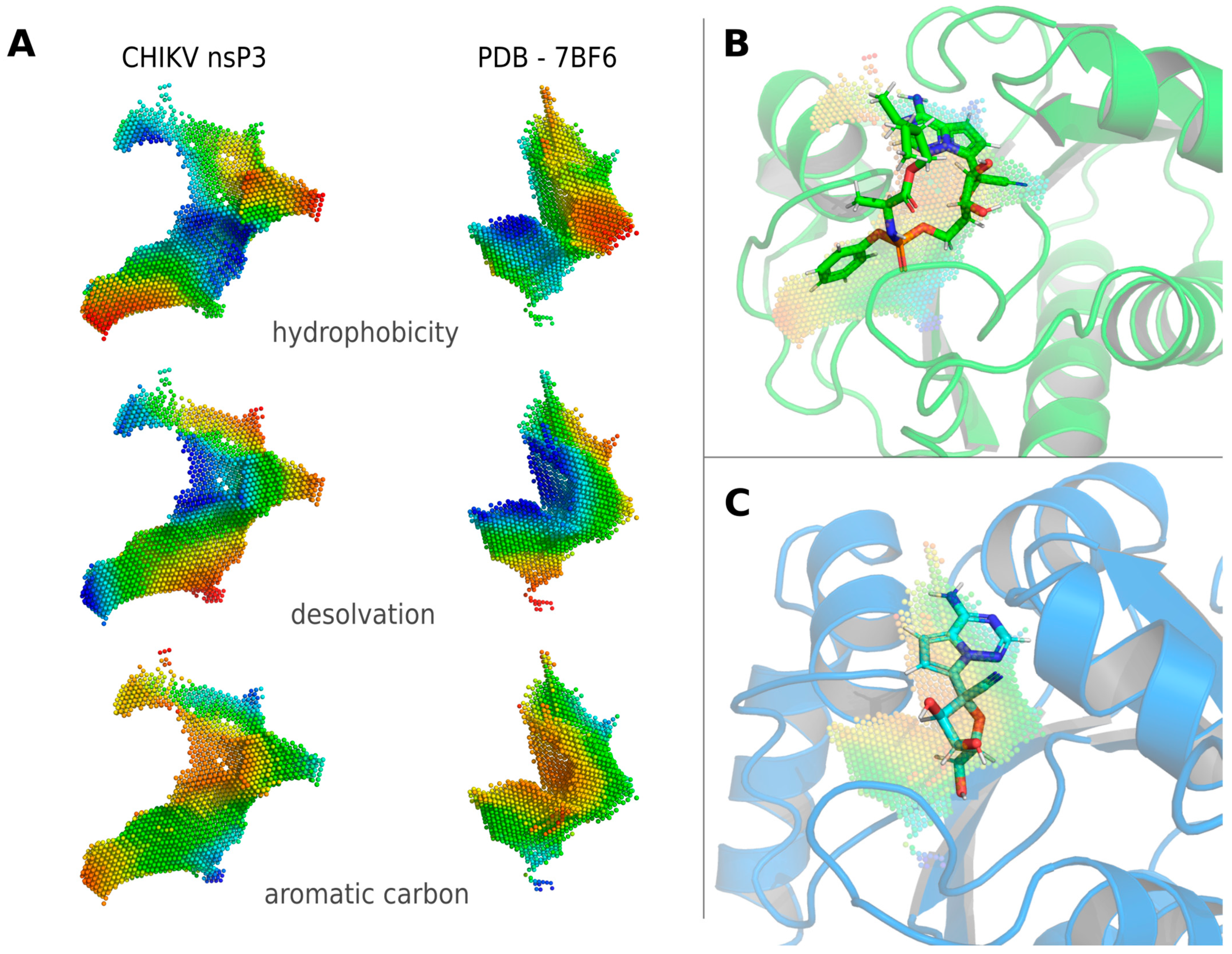
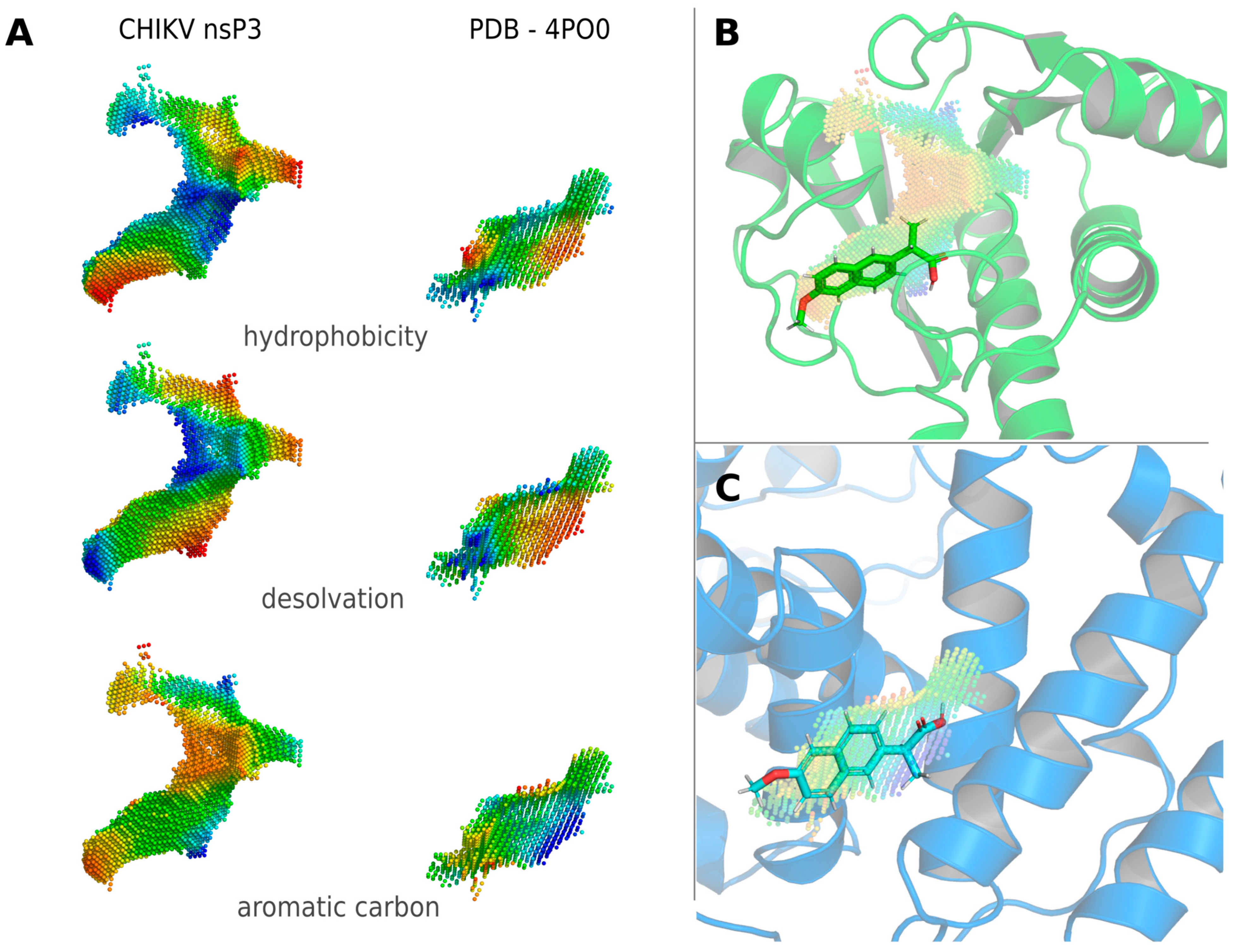
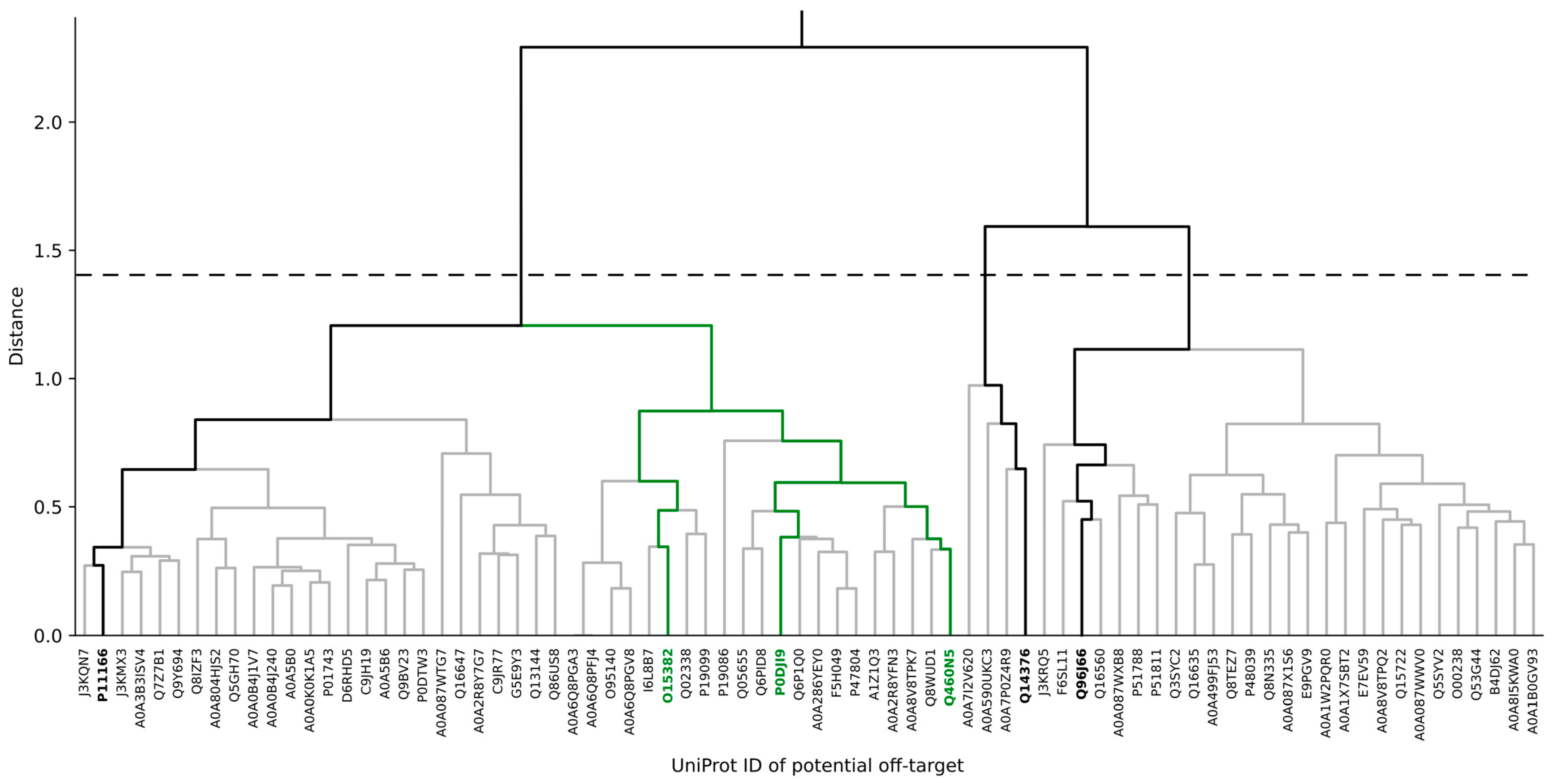
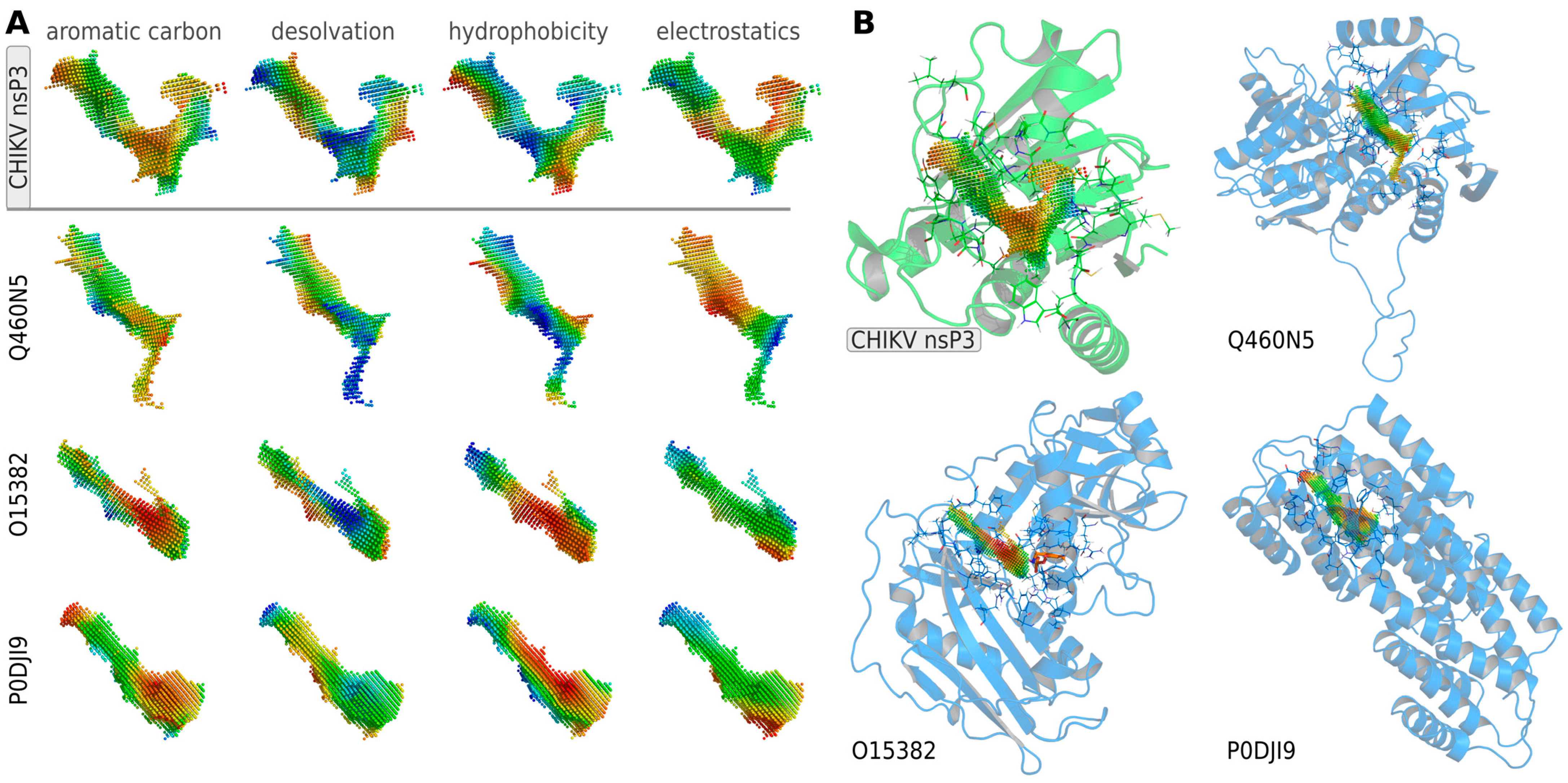
3.2. Identification of Potential Human Off-Target Sites for nsP3 Inhibitors
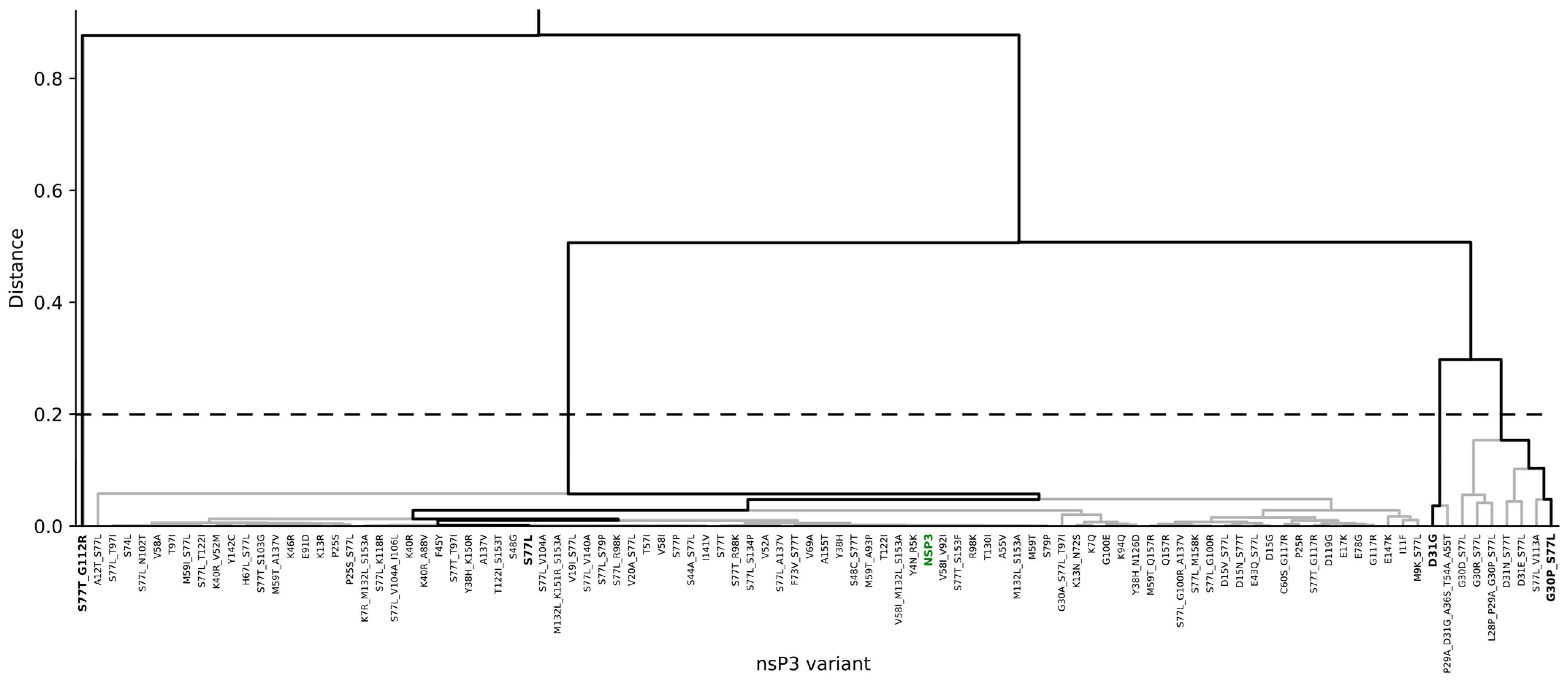
3.3. Identification of Concerning Viral Variants
3.4. Refinement of Small Molecules towards Safe and Effective Antivirals
3.4.1. Inhibitor Refinement for Binding the Related Viral Species EEEV and SINV
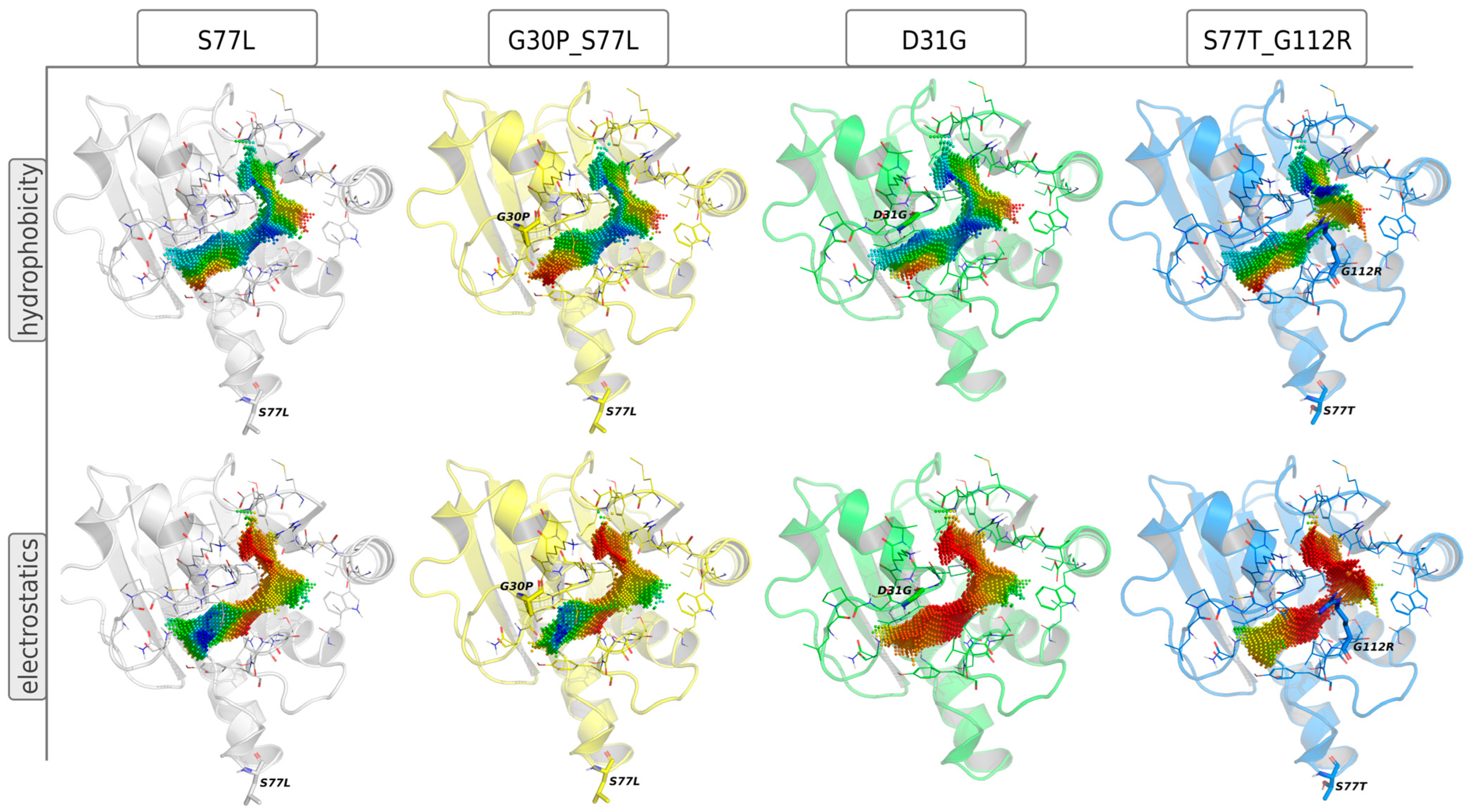
3.4.2. Inhibitor Refinement for Binding to Several Viral Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, B.M.; Mulkey, S.B.; Campos, J.M.; DeBiasi, R.L. Laboratory diagnosis of CNS infections in children due to emerging and re-emerging neurotropic viruses. Pediatr. Res. 2024, 95, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Filippone, C.; Legros, V.; Jeannin, P.; Choumet, V.; Butler-Browne, G.; Zoladek, J.; Mouly, V.; Gessain, A.; Ceccaldi, P.E. Arboviruses and Muscle Disorders: From Disease to Cell Biology. Viruses 2020, 12, 616. [Google Scholar] [CrossRef] [PubMed]
- Socha, W.; Kwasnik, M.; Larska, M.; Rola, J.; Rozek, W. Vector-Borne Viral Diseases as a Current Threat for Human and Animal Health-One Health Perspective. J. Clin. Med. 2022, 11, 3026. [Google Scholar] [CrossRef] [PubMed]
- Tajudeen, Y.A.; Oladunjoye, I.O.; Mustapha, M.O.; Mustapha, S.T.; Ajide-Bamigboye, N.T. Tackling the global health threat of arboviruses: An appraisal of the three holistic approaches to health. Health Promot. Perspect. 2021, 11, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Diseases, T.L.I. Twin threats: Climate change and zoonoses. Lancet Infect. Dis. 2022, 23, 1. [Google Scholar] [CrossRef] [PubMed]
- Kharwadkar, S.; Herath, N. Clinical manifestations of dengue, Zika and chikungunya in the Pacific Islands: A systematic review and meta-analysis. Rev. Med. Virol. 2024, 34, e2521. [Google Scholar] [CrossRef] [PubMed]
- Laporta, G.Z.; Potter, A.M.; Oliveira, J.F.A.; Bourke, B.P.; Pecor, D.B.; Linton, Y.M. Global Distribution of Aedes aegypti and Aedes albopictus in a Climate Change Scenario of Regional Rivalry. Insects 2023, 14, 49. [Google Scholar] [CrossRef] [PubMed]
- Pujhari, S. Recent Advances in Arboviral Vaccines: Emerging Platforms and Promising Innovations. Biologics 2023, 4, 1–16. [Google Scholar] [CrossRef]
- Troppens, D. Challenges in combating arboviral infections. Nat. Commun. 2024, 15, 3350. [Google Scholar] [CrossRef]
- Feracci, M.; Eydoux, C.; Fattorini, V.; Bello, L.L.; Gauffre, P.; Selisko, B.; Sutto-Ortiz, P.; Shannon, A.; Xia, H.; Shi, P.-Y.; et al. AT-752 targets multiple sites and activities on the Dengue virus replication enzyme NS5. Antivir. Res. 2023, 212, 105574. [Google Scholar] [CrossRef]
- Goethals, O.; Kaptein, S.J.F.; Kesteleyn, B.; Bonfanti, J.-F.; Van Wesenbeeck, L.; Bardiot, D.; Verschoor, E.J.; Verstrepen, B.E.; Fagrouch, Z.; Putnak, J.R.; et al. Blocking NS3–NS4B interaction inhibits dengue virus in non-human primates. Nature 2023, 615, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef] [PubMed]
- Dowden, H.; Munro, J. Trends in clinical success rates and therapeutic focus. Nat. Rev. Drug Discov. 2019, 18, 495–496. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.K. Phase II and phase III failures: 2013–2015. Nat. Rev. Drug Discov. 2016, 15, 817–818. [Google Scholar] [CrossRef]
- Hetmann, M.; Parigger, L.; Sirelkhatim, H.; Stern, A.; Krassnigg, A.; Gruber, K.; Steinkellner, G.; Ruau, D.; Gruber, C.C. Folding the human proteome using BioNeMo: A fused dataset of structural models for machine learning purposes. Sci. Data 2024, 11, 591. [Google Scholar] [CrossRef] [PubMed]
- Struelens, M.J.; Ludden, C.; Werner, G.; Sintchenko, V.; Jokelainen, P.; Ip, M. Real-time genomic surveillance for enhanced control of infectious diseases and antimicrobial resistance. Front. Sci. 2024, 2, 1298248. [Google Scholar] [CrossRef]
- Parigger, L.; Krassnigg, A.; Schopper, T.; Singh, A.; Tappler, K.; Köchl, K.; Hetmann, M.; Gruber, K.; Steinkellner, G.; Gruber, C.C. Recent changes in the mutational dynamics of the SARS-CoV-2 main protease substantiate the danger of emerging resistance to antiviral drugs. Front. Med. 2022, 9, 1061142. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Schwartz, O.; Albert, M.L. Biology and pathogenesis of chikungunya virus. Nat. Rev. Microbiol. 2010, 8, 491–500. [Google Scholar] [CrossRef]
- Gao, Y.; Goonawardane, N.; Ward, J.; Tuplin, A.; Harris, M. Multiple roles of the non-structural protein 3 (nsP3) alphavirus unique domain (AUD) during Chikungunya virus genome replication and transcription. PLoS Pathog. 2019, 15, e1007239. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, J.F.; Martins, D.O.S.; McPhillie, M.J.; Roberts, G.C.; Zothner, C.; Merits, A.; Harris, M.; Jardim, A.C.G. Is the ADP ribose site of the Chikungunya virus NSP3 Macro domain a target for antiviral approaches? Acta Tropica 2020, 207, 105490. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information (NCBI). Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; [1988]. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 14 June 2024).
- Cock, P.J.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Gruber, K.; Steinkellner, G.; Gruber, C. Determining Novel Enzymatic Functionalities Using Three-Dimensional Point Clouds Representing Physico Chemical Properties of Protein Cavities. 2014. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014080005 (accessed on 14 June 2024).
- Steinkellner, G.; Gruber, C.C.; Pavkov-Keller, T.; Binter, A.; Steiner, K.; Winkler, C.; Łyskowski, A.; Schwamberger, O.; Oberer, M.; Schwab, H.; et al. Identification of promiscuous ene-reductase activity by mining structural databases using active site constellations. Nat. Commun. 2014, 5, 4150. [Google Scholar] [CrossRef] [PubMed]
- Hendlich, M.; Rippmann, F.; Barnickel, G. LIGSITE: Automatic and efficient detection of potential small molecule-binding sites in proteins. J. Mol. Graph. Model. 1997, 15, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Durmaz, V.; Köchl, K.; Krassnigg, A.; Parigger, L.; Hetmann, M.; Singh, A.; Nutz, D.; Korsunsky, A.; Kahler, U.; König, C.; et al. Structural-bioinformatics analysis of SARS-CoV-2 variants reveals higher hACE2 receptor binding affinity for Omicron B.1.1.529 spike RBD compared to wild-type reference. Sci. Rep. 2021, 12, 14534. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Zdrazil, B.; Felix, E.; Hunter, F.; Manners, E.J.; Blackshaw, J.; Corbett, S.; de Veij, M.; Ioannidis, H.; Lopez, D.M.; Mosquera, J.F.; et al. The ChEMBL Database in 2023: A drug discovery platform spanning multiple bioactivity data types and time periods. Nucleic Acids Res. 2024, 52, D1180–D1192. [Google Scholar] [CrossRef] [PubMed]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nature Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Helfrich, S.; Herzel, A.; Ruzika, S.; Thielen, C. Using scalarizations for the approximation of multiobjective optimization problems: Towards a general theory. Math. Meth. Oper. Res. 2023. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Waskom, M. seaborn: Statistical data visualization. JOSS 2021, 6, 3021. [Google Scholar] [CrossRef]
- Hetmann, M.; Langner, C.; Durmaz, V.; Cespugli, M.; Köchl, K.; Krassnigg, A.; Blaschitz, K.; Groiss, S.; Loibner, M.; Ruau, D.; et al. Identification and validation of fusidic acid and flufenamic acid as inhibitors of SARS-CoV-2 replication using DrugSolver CavitomiX. Sci. Rep. 2023, 13, 11783. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.; Schröder, M.; Olieric, V.; Sharpe, M.E.; Hernandez-Olmos, V.; Proschak, E.; Merk, D.; Knapp, S.; Chaikuad, A. Structural Insights into Plasticity and Discovery of Remdesivir Metabolite GS-441524 Binding in SARS-CoV-2 Macrodomain. ACS Med. Chem. Lett. 2021, 12, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins Struct. Funct. Bioinform. 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 2010, 26, 889–895. [Google Scholar] [CrossRef]
- Bujacz, A.; Zielinski, K.; Sekula, B. Structural studies of bovine, equine, and leporine serum albumin complexes with naproxen: Structures of BSA, ESA, and LSA with Naproxen. Proteins 2014, 82, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- McTigue, M.; Murray, B.W.; Chen, J.H.; Deng, J.L.; Solowiej, J.; Kania, R.S. Molecular conformations, interactions, and properties associated with drug efficiency and clinical performance among VEGFR TK inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 18281–18289. [Google Scholar] [CrossRef] [PubMed]
- Koeberle, S.C.; Romir, J.; Fischer, S.; Koeberle, A.; Schattel, V.; Albrecht, W.; Grütter, C.; Werz, O.; Rauh, D.; Stehle, T.; et al. Skepinone-L is a selective p38 mitogen-activated protein kinase inhibitor. Nat. Chem. Biol. 2012, 8, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zheng, X.; Zhang, H.; Zhai, J.; Zhang, L.; Li, C.; Zeng, K.; Chen, Y.; Li, Q.; Hu, X. In vitro inhibition of AKR1Cs by sulphonylureas and the structural basis. Chem.-Biol. Interact. 2015, 240, 310–315. [Google Scholar] [CrossRef]
- Wishart, D.S. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Final Rank | #Gen | Structure | BE to Viral Species [kcal/mol] CHIKV|EEEV|SINV | BE to Off-Targets [kcal/mol] Q460N5|O15382|P0DJI9 | BE to 3ZYA [kcal/mol] |
|---|---|---|---|---|---|
| Original Molecule—2-Amino-Phenylamino-Dibenzosuberone | |||||
| - | 0 |  | −10.69|−9.88|−9.70 | −8.72|−8.78|−11.70 | (−10.67) |
| Batch 1|Considering off-target effects | |||||
| 1 | 1 | 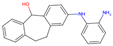 | −10.07|−9.59|−9.31 +0.62|+0.29|+0.39 | −8.78|−8.27|−7.12 −0.06|+0.51|+4.58 | (−9.78) (+0.89) |
| 2 | 19 | 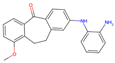 | −9.72|−9.62|−9.63 +0.97|+0.28|+0.07 | −7.43|−8.37|−9.15 +1.29|+0.41|+2.55 | (−7.77) (+2.90) |
| 3 | 15 | 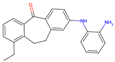 | −9.51|−9.88|−9.74 +1.18|0|−0.04 | −8.74|−8.07|−8.66 −0.02|+0.71|+3.04 | (−8.16) (+2.51) |
| Batch 2|Without considering off-target effects | |||||
| 1 | 8 |  | −11.37|−10.91|−10.38 −0.68|−1.03|−0.68 | (−9.71|−10.40|−11.59) (−0.99|−1.62|+0.11) | (−11.55) (−0.88) |
| 2 | 2 | 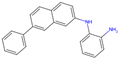 | −10.87|−10.85|−10.10 −0.18|−0.97|−0.40 | (−9.03|−10.80|−12.15) (−0.31|−2.02|−0.45) | (−11.42) (−0.75) |
| 3 | 17 | 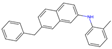 | −11.02|−10.38|−10.06 −0.33|−0.50|−0.36 | (−9.57|−10.39|−12.03) (−0.85|−1.61|−0.33) | (−10.20) (+0.47) |
| Final Rank | #Gen | Structure | BE to Viral Variants S77L|S77T_G112R|D31G|G30P_S77L [kcal/mol] | BE to Off-Target Sites Q460N5|P11166|Q96J66|Q14376 [kcal/mol] | BE to 4ZFC [kcal/mol] |
|---|---|---|---|---|---|
| Original Molecule—Gliclazide | |||||
| - | 0 | 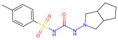 | −10.97|−11.71|−10.71|−10.03 | −8.16|−10.56|−8.52|−9.70 | (−9.14) |
| Batch 1 | |||||
| 1 | 15 | 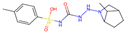 | −12.18|−12.55|−12.02|−10.74 −1.21|−0.84|−1.31|−0.71 | −8.19|−9.94|−8.49|−8.79 −0.03|+0.62|+0.03|+0.91 | (−8.93) (+0.21) |
| 2 | 6 | 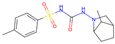 | −11.38|−12.10|−11.16|−10.59 −0.41|−0.39|−0.45|−0.56 | −7.68|−9.80|−8.07|−8.46 +0.48|+0.76|+0.45|+1.24 | (−9.17) (−0.03) |
| 3 | 8 | 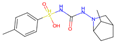 | −11.37|−12.13|−11.17|−10.61 −0.40|−0.42|−0.46|−0.58 | −7.82|−9.73|−8.18|−8.85 +0.34|+0.83|+0.34|+0.85 | (−8.81) (+0.33) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parigger, L.; Krassnigg, A.; Hetmann, M.; Hofmann, A.; Gruber, K.; Steinkellner, G.; Gruber, C.C. CavitOmiX Drug Discovery: Engineering Antivirals with Enhanced Spectrum and Reduced Side Effects for Arboviral Diseases. Viruses 2024, 16, 1186. https://doi.org/10.3390/v16081186
Parigger L, Krassnigg A, Hetmann M, Hofmann A, Gruber K, Steinkellner G, Gruber CC. CavitOmiX Drug Discovery: Engineering Antivirals with Enhanced Spectrum and Reduced Side Effects for Arboviral Diseases. Viruses. 2024; 16(8):1186. https://doi.org/10.3390/v16081186
Chicago/Turabian StyleParigger, Lena, Andreas Krassnigg, Michael Hetmann, Anna Hofmann, Karl Gruber, Georg Steinkellner, and Christian C. Gruber. 2024. "CavitOmiX Drug Discovery: Engineering Antivirals with Enhanced Spectrum and Reduced Side Effects for Arboviral Diseases" Viruses 16, no. 8: 1186. https://doi.org/10.3390/v16081186
APA StyleParigger, L., Krassnigg, A., Hetmann, M., Hofmann, A., Gruber, K., Steinkellner, G., & Gruber, C. C. (2024). CavitOmiX Drug Discovery: Engineering Antivirals with Enhanced Spectrum and Reduced Side Effects for Arboviral Diseases. Viruses, 16(8), 1186. https://doi.org/10.3390/v16081186