
Longitudinal Monitoring of the Effects of Anti-Adenoviral Treatment Regimens in a Permissive In Vivo Model
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Compounds
2.3. Animals
2.4. Measuring Radiance
2.5. Statistical Analysis
3. Results
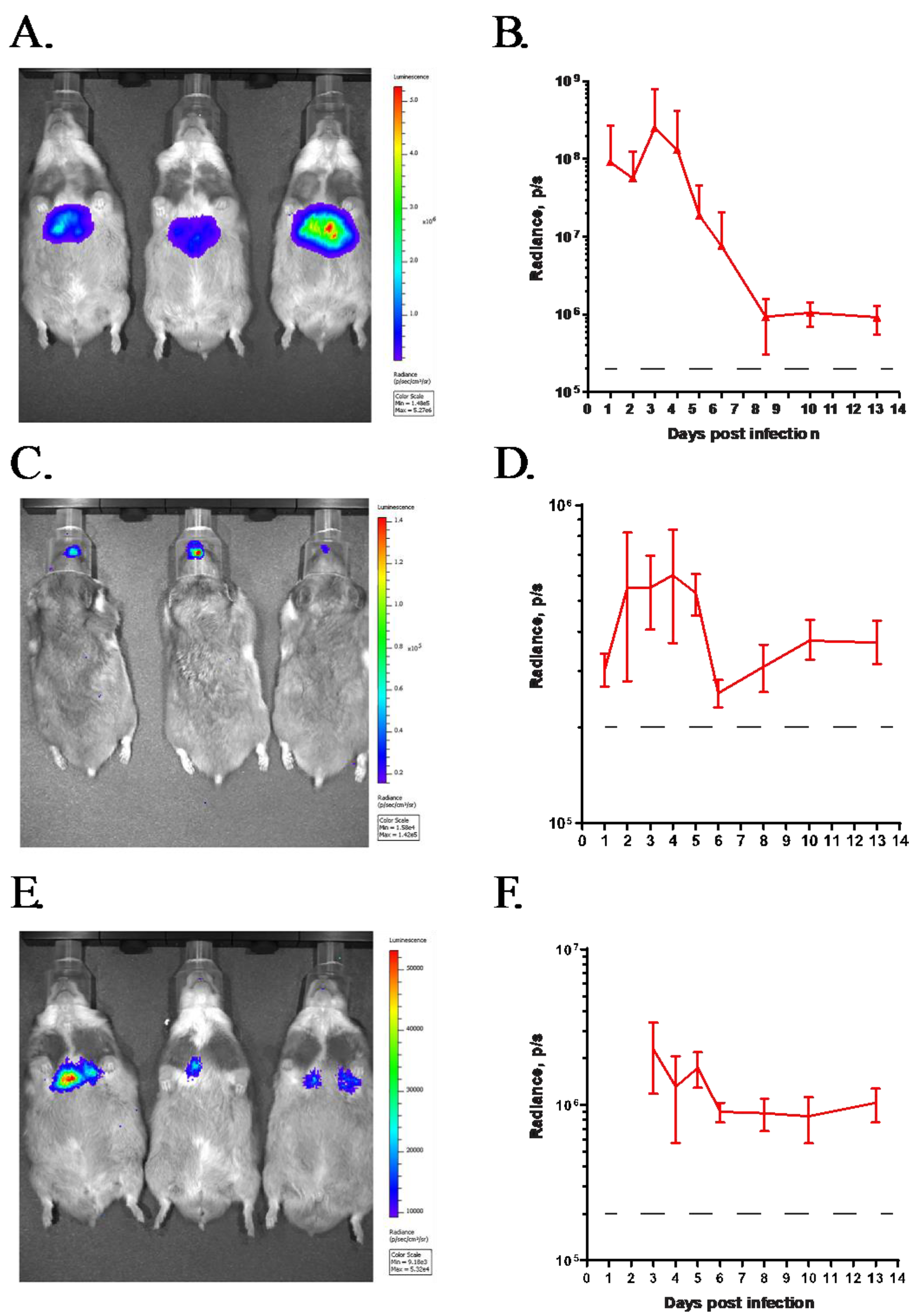
3.1. GZ3Luc, a Replication-Competent HAdV Vector, Can Be Used to Track Virus Replication In Vivo after Intravenous or Intranasal Challenge
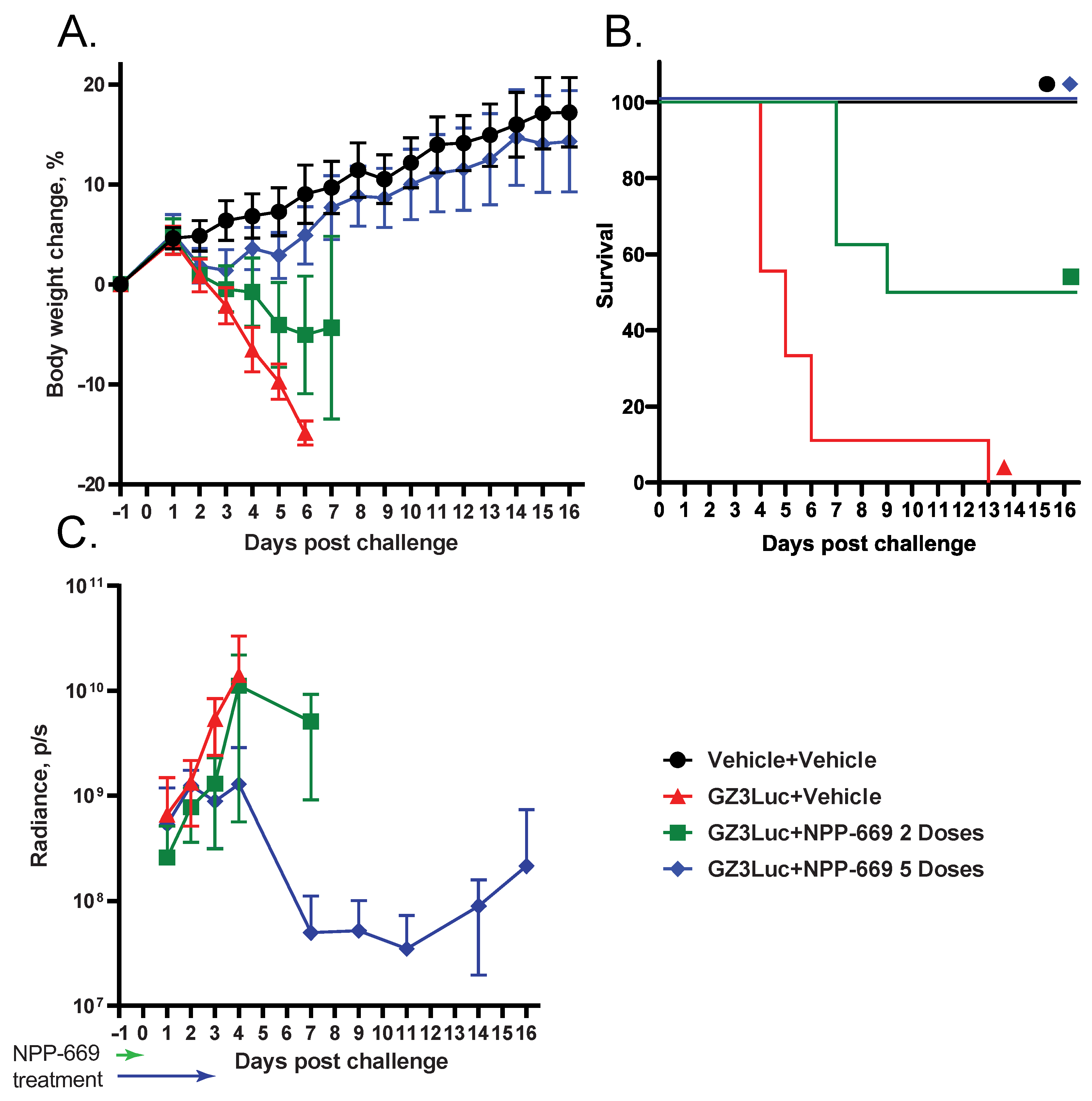
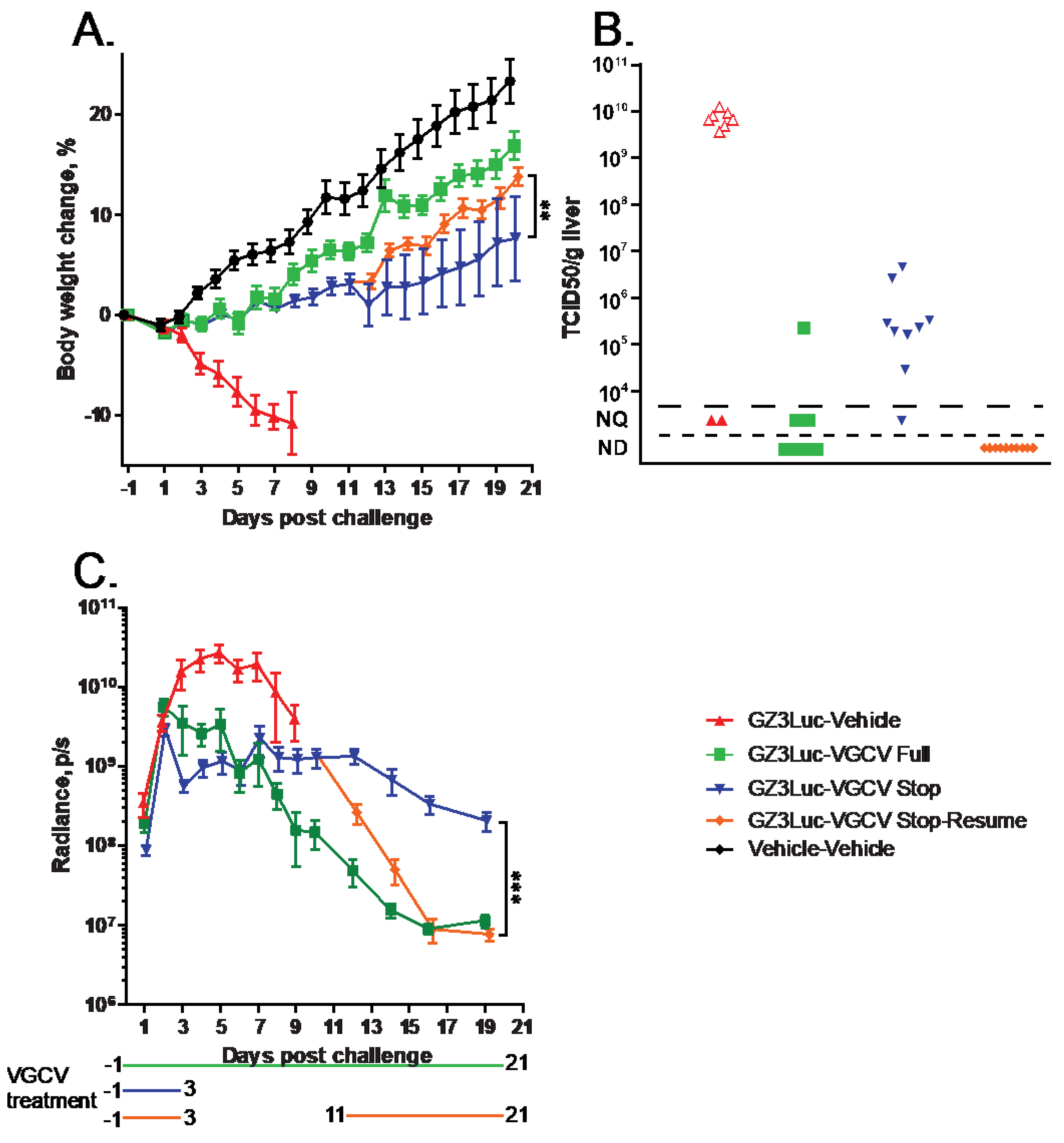
3.2. HAdV-Induced Pathology Can Be Mitigated with Short-Course Treatment with an Antiviral Compound; However, Low-Grade Virus Replication Persists
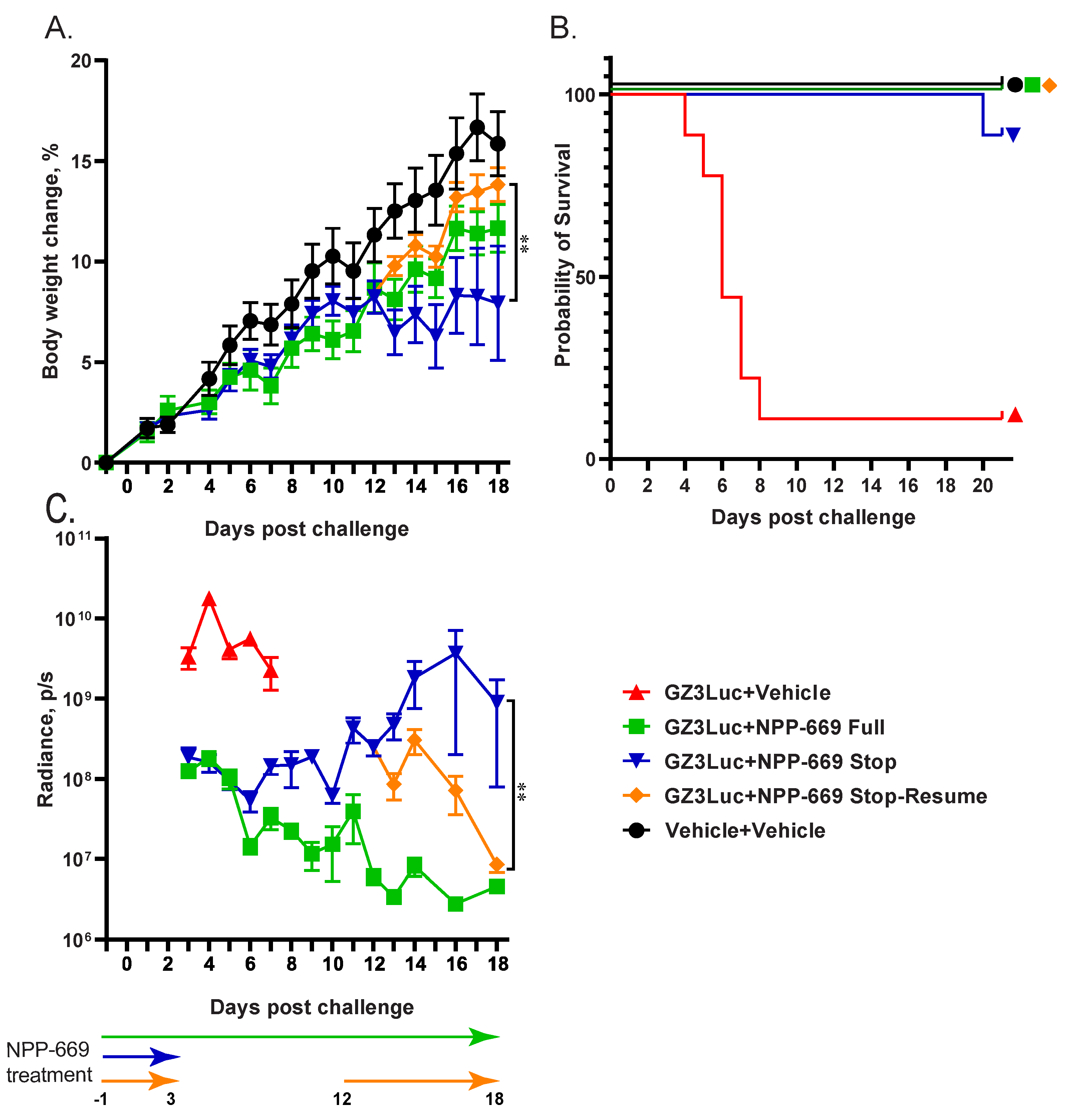
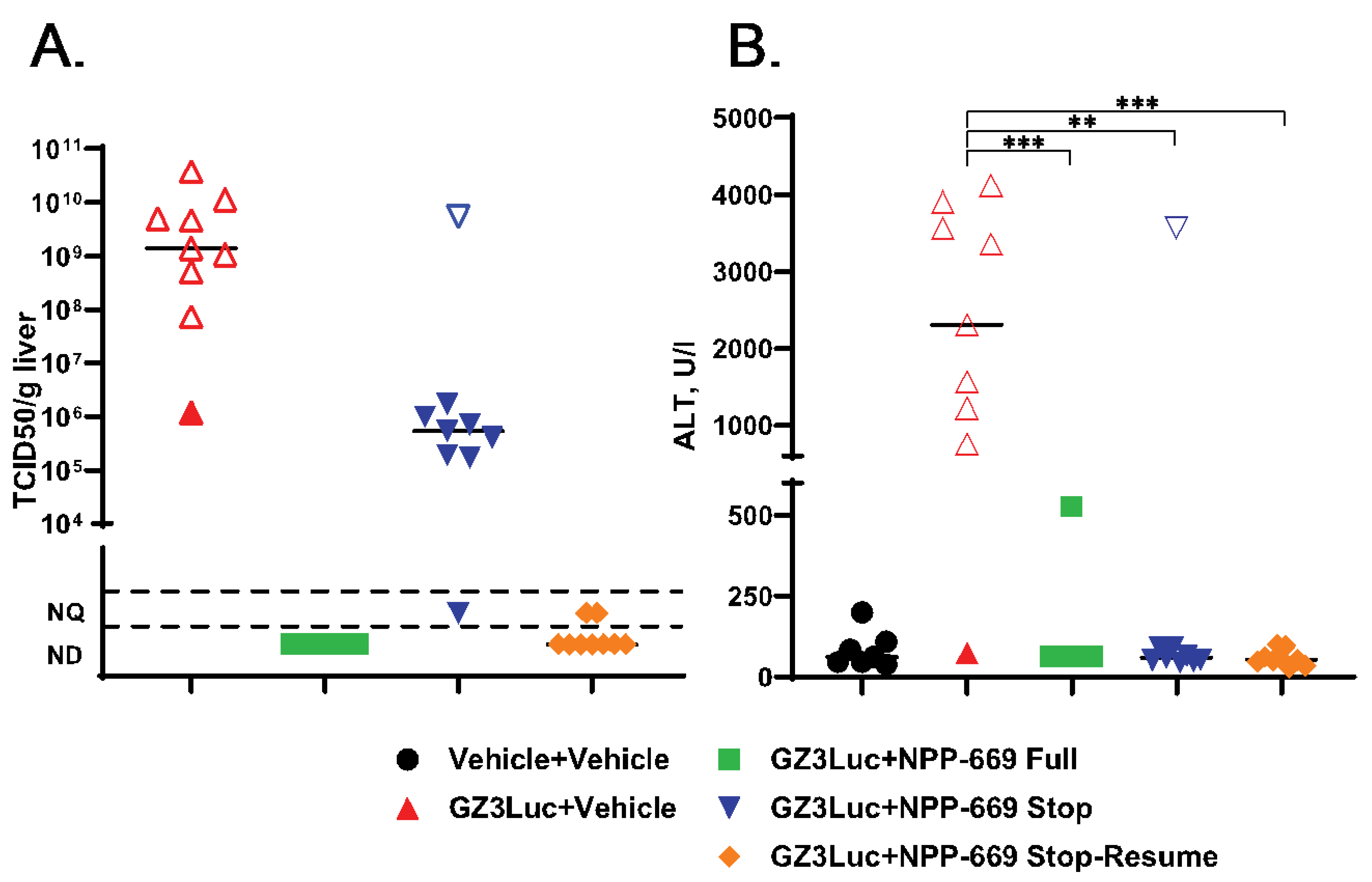
3.3. Persistent HAdV Infection and the Resulting Pathology Can Be Managed with Intermittent Treatment with Antiviral Compounds
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wold, W.S.M.; Ison, M.G. Adenoviruses. In Fields Virology, 7th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2022; Volume DNA Viruses, pp. 267–366. [Google Scholar]
- Group, H.A.W. Available online: http://hadvwg.gmu.edu/ (accessed on 31 May 2024).
- Lion, T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin. Microbiol. Rev. 2014, 27, 441–462. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.P., 3rd; Kajon, A.E. Adenovirus: Epidemiology, Global Spread of Novel Types, and Approach to Treatment. Semin. Respir. Crit. Care Med. 2021, 42, 800–821. [Google Scholar] [CrossRef] [PubMed]
- Rozwadowski, F.; Caulcrick-Grimes, M.; McHugh, L.; Haldeman, A.; Fulton, T.; Killerby, M.; Schneider, E.; Lu, X.; Sakthivel, S.K.; Bhatnagar, J.; et al. Notes from the Field: Fatalities Associated with Human Adenovirus Type 7 at a Substance Abuse Rehabilitation Facility—New Jersey, 2017. MMWR. Morb. Mortal. Wkly. Rep. 2018, 67, 371–372. [Google Scholar] [CrossRef] [PubMed]
- Romaní Vidal, A.; Vaughan, A.; Innocenti, F.; Colombe, S.; Nerlander, L.; Rachwal, N.; Ciancio, B.C.; Mougkou, A.; Carvalho, C.; Delgado, E.; et al. Hepatitis of unknown aetiology in children—Epidemiological overview of cases reported in Europe, 1 January to 16 June 2022. Eurosurveillance 2022, 27, 2200483. [Google Scholar] [CrossRef] [PubMed]
- Grand, R.J. Pathogenicity and virulence of human adenovirus F41: Possible links to severe hepatitis in children. Virulence 2023, 14, 2242544. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Williams, D.J.; Arnold, S.R.; Ampofo, K.; Bramley, A.M.; Reed, C.; Stockmann, C.; Anderson, E.J.; Grijalva, C.G.; Self, W.H.; et al. Community-acquired pneumonia requiring hospitalization among U.S. children. N. Engl. J. Med. 2015, 372, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.A.; Mayer, B.T.; Xie, H.; Leisenring, W.M.; Huang, M.L.; Stevens-Ayers, T.; Milano, F.; Delaney, C.; Sorror, M.L.; Sandmaier, B.M.; et al. The cumulative burden of double-stranded DNA virus detection after allogeneic HCT is associated with increased mortality. Blood 2017, 129, 2316–2325. [Google Scholar] [CrossRef] [PubMed]
- Sandkovsky, U.; Vargas, L.; Florescu, D.F. Adenovirus: Current epidemiology and emerging approaches to prevention and treatment. Curr. Infect. Dis. Rep. 2014, 16, 416. [Google Scholar] [CrossRef] [PubMed]
- Lion, T. Adenovirus persistence, reactivation, and clinical management. FEBS Lett. 2019, 593, 3571–3582. [Google Scholar] [CrossRef] [PubMed]
- King, C.R.; Zhang, A.; Mymryk, J.S. The Persistent Mystery of Adenovirus Persistence. Trends Microbiol. 2016, 24, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Houldcroft, C.J.; Roy, S.; Morfopoulou, S.; Margetts, B.K.; Depledge, D.P.; Cudini, J.; Shah, D.; Brown, J.R.; Romero, E.Y.; Williams, R.; et al. Use of Whole-Genome Sequencing of Adenovirus in Immunocompromised Pediatric Patients to Identify Nosocomial Transmission and Mixed-Genotype Infection. J. Infect. Dis. 2018, 218, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Grimley, M.S.; Chemaly, R.F.; Englund, J.A.; Kurtzberg, J.; Chittick, G.; Brundage, T.M.; Bae, A.; Morrison, M.E.; Prasad, V.K. Brincidofovir for asymptomatic adenovirus viremia in pediatric and adult allogeneic hematopoietic cell transplant recipients: A randomized placebo-controlled Phase II trial. Biol. Blood Marrow Transplant. 2017, 23, 512–521. [Google Scholar] [CrossRef] [PubMed]
- El Helou, G.; Razonable, R.R. Safety considerations with current and emerging antiviral therapies for cytomegalovirus infection in transplantation. Expert Opin. Drug Saf. 2019, 18, 1017–1030. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Holy, A. Acyclic nucleoside phosphonates: A key class of antiviral drugs. Nat. Rev. Drug Discov. 2005, 4, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, J.; Sortino, K.; Sethna, P.; Bae, A.; Lanier, R.; Bambara, R.A.; Dewhurst, S. Cidofovir diphosphate inhibits adenovirus 5 DNA polymerase via both non-obligate chain termination and direct inhibition, and polymerase mutations confer cidofovir resistance on intact virus. Antimicrob. Agents Chemother. 2018, 63, e01925-18. [Google Scholar] [PubMed]
- Cihlar, T.; Lin, D.C.; Pritchard, J.B.; Fuller, M.D.; Mendel, D.B.; Sweet, D.H. The antiviral nucleotide analogs cidofovir and adefovir are novel substrates for human and rat renal organic anion transporter 1. Mol. Pharmacol. 1999, 56, 570–580. [Google Scholar] [CrossRef] [PubMed]
- Vora, S.B.; Brothers, A.W.; Englund, J.A. Renal Toxicity in Pediatric Patients Receiving Cidofovir for the Treatment of Adenovirus Infection. J. Pediatric. Infect. Dis. Soc. 2017, 6, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Florescu, D.F.; Keck, M.A. Development of CMX001 (Brincidofovir) for the treatment of serious diseases or conditions caused by dsDNA viruses. Expert Rev. Anti-Infect. Ther. 2014, 12, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Florescu, D.F.; Pergam, S.A.; Neely, M.N.; Qiu, F.; Johnston, C.; Way, S.; Sande, J.; Lewinsohn, D.A.; Guzman-Cottrill, J.A.; Graham, M.L.; et al. Safety and efficacy of CMX001 as salvage therapy for severe adenovirus infections in immunocompromised patients. Biol. Blood Marrow Transplant. 2012, 18, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Toth, K.; Spencer, J.F.; Dhar, D.; Sagartz, J.E.; Buller, R.M.; Painter, G.R.; Wold, W.S.M. Hexadecyloxypropyl-cidofovir, CMX001, prevents adenovirus-induced mortality in a permissive, immunosuppressed animal model. Proc. Natl. Acad. Sci. USA 2008, 105, 7293–7297. [Google Scholar] [CrossRef]
- Marty, F.M.; Winston, D.J.; Chemaly, R.F.; Mullane, K.M.; Shore, T.B.; Papanicolaou, G.A.; Chittick, G.; Brundage, T.M.; Wilson, C.; Morrison, M.E.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase 3 Trial of Oral Brincidofovir for Cytomegalovirus Prophylaxis in Allogeneic Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2019, 25, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Detweiler, C.J.; Mueller, S.B.; Sung, A.D.; Saullo, J.L.; Prasad, V.K.; Cardona, D.M. Brincidofovir (CMX001) Toxicity Associated With Epithelial Apoptosis and Crypt Drop Out in a Hematopoietic Cell Transplant Patient: Challenges in Distinguishing Drug Toxicity From GVHD. J. Pediatr. Hematol. Oncol. 2018, 40, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Painter, W.; Robertson, A.; Trost, L.C.; Godkin, S.; Lampert, B.; Painter, G. First pharmacokinetic and safety study in humans of the novel lipid antiviral conjugate CMX001, a broad-spectrum oral drug active against double-stranded DNA viruses. Antimicrob. Agents Chemother. 2012, 56, 2726–2734. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Cardona, J.J.; Whited, L.K.; Chemaly, R.F. Brincidofovir: Understanding its unique profile and potential role against adenovirus and other viral infections. Future Microbiol. 2020, 15, 389–400. [Google Scholar] [CrossRef]
- FDA TEMBEXA (Brincidofovir) Tablets, for Oral Use. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214460s001,214461s001lbl.pdf (accessed on 27 June 2024).
- Wold, W.S.; Toth, K. Syrian hamster as an animal model to study oncolytic adenoviruses and to evaluate the efficacy of antiviral compounds. Adv. Cancer Res. 2012, 115, 69–92. [Google Scholar] [PubMed]
- Tollefson, A.E.; Ying, B.; Spencer, J.F.; Sagartz, J.E.; Wold, W.S.M.; Toth, K. Pathology in permissive Syrian hamsters after infection with species C human adenovirus (HAdV-C) is the result of virus replication: HAdV-C6 replicates more and causes more pathology than HAdV-C5. J. Virol. 2017, 91, e00284-17. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ying, B.; Liu, Y.; Spencer, J.F.; Miao, J.; Tollefson, A.E.; Brien, J.D.; Wang, Y.; Wold, W.S.M.; Wang, Z.; et al. Generation and characterization of an IL2RG knockout Syrian hamster model for XSCID and HAdV-C6 infection in immunocompromised patients. Dis. Models Mech. 2020, 13, dmm044602. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Ying, B.; Li, R.; Tollefson, A.E.; Spencer, J.F.; Wold, W.S.M.; Song, S.H.; Kong, I.K.; Toth, K.; Wang, Y.; et al. Characterization of an N-terminal non-core domain of RAG1 gene disrupted Syrian hamster model generated by CRISPR Cas9. Viruses 2018, 10, 243. [Google Scholar] [CrossRef] [PubMed]
- Toth, K.; Lee, S.R.; Ying, B.; Spencer, J.F.; Tollefson, A.E.; Sagartz, J.E.; Kong, I.K.; Wang, Z.; Wold, W.S. STAT2 knockout Syrian hamsters support enhanced replication and pathogenicity of human adenovirus, revealing an important role of type I interferon response in viral control. PLoS Pathog. 2015, 11, e1005084. [Google Scholar] [CrossRef] [PubMed]
- Ying, B.; Spencer, J.F.; Tollefson, A.E.; Wold, W.S.M.; Toth, K. Male Syrian hamsters are more susceptible to intravenous infection with species C human adenoviruses than are females. Virology 2018, 514, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Ying, B.; Toth, K.; Spencer, J.F.; Aurora, R.; Wold, W.S.M. Transcriptome sequencing and development of an expression microarray platform for liver infection in adenovirus type 5-infected Syrian golden hamsters. Virology 2015, 485, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Lipka, E.; Chadderdon, A.M.; Harteg, C.C.; Doherty, M.K.; Simon, E.S.; Domagala, J.M.; Reyna, D.M.; Hutchings, K.M.; Gan, X.; White, A.D.; et al. NPP-669, a Novel Broad-Spectrum Antiviral Therapeutic with Excellent Cellular Uptake, Antiviral Potency, Oral Bioavailability, Preclinical Efficacy, and a Promising Safety Margin. Mol. Pharm. 2023, 20, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Tollefson, A.E.; Riemann, S.B.; Ying, B.; Spencer, J.F.; Overhulse, J.M.; Kashemirov, B.A.; Wold, W.S.M.; McKenna, C.E.; Toth, K. Oral USC-093, a novel homoserinamide analogue of the tyrosinamide (S)-HPMPA prodrug USC-087 has decreased nephrotoxicity while maintaining antiviral efficacy against human adenovirus infection of Syrian hamsters. Antiviral Res. 2024, 222, 105799. [Google Scholar] [CrossRef] [PubMed]
- Tollefson, A.E.; Spencer, J.F.; Ying, B.; Buller, R.M.; Wold, W.S.; Toth, K. Cidofovir and brincidofovir reduce the pathology caused by systemic infection with human type 5 adenovirus in immunosuppressed Syrian hamsters, while ribavirin is largely ineffective in this model. Antiviral Res. 2014, 112, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Toth, K.; Hussein, I.T.M.; Tollefson, A.E.; Ying, B.; Spencer, J.F.; Eagar, J.; James, S.H.; Prichard, M.N.; Wold, W.S.M.; Bowlin, T.L. Filociclovir Is a Potent In Vitro and In Vivo Inhibitor of Human Adenoviruses. Antimicrob. Agents Chemother. 2020, 64, e01299-20. [Google Scholar] [CrossRef] [PubMed]
- Toth, K.; Spencer, J.F.; Ying, B.; Tollefson, A.E.; Hartline, C.B.; Richard, E.T.; Fan, J.; Lyu, J.; Kashemirov, B.A.; Harteg, C.; et al. USC-087 protects Syrian hamsters against lethal challenge with human species C adenoviruses. Antiviral Res. 2018, 153, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Toth, K.; Spencer, J.F.; Ying, B.; Tollefson, A.E.; Wold, W.S.M. HAdV-C6 is a more relevant challenge virus than HAdV-C5 for testing antiviral drugs with the immunosuppressed Syrian hamster model. Viruses 2017, 9, 147. [Google Scholar] [CrossRef] [PubMed]
- Toth, K.; Tollefson, A.E.; Spencer, J.F.; Ying, B.; Wold, W.S.M. Combination therapy with brincidofovir and valganciclovir against species C adenovirus infection in the immunosuppressed Syrian hamster model allows for substantial reduction of dose for both compounds. Antiviral Res. 2017, 146, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Toth, K.; Ying, B.; Tollefson, A.E.; Spencer, J.F.; Balakrishnan, L.; Sagartz, J.E.; Buller, R.M.; Wold, W.S. Valganciclovir inhibits human adenovirus replication and pathology in permissive immunosuppressed female and male Syrian hamsters. Viruses 2015, 7, 1409–1428. [Google Scholar] [CrossRef] [PubMed]
- Ying, B.; Tollefson, A.E.; Spencer, J.F.; Balakrishnan, L.; Dewhurst, S.; Capella, C.; Buller, R.M.; Toth, K.; Wold, W.S. Ganciclovir inhibits human adenovirus replication and pathogenicity in permissive immunosuppressed Syrian hamsters. Antimicrob. Agents Chemother. 2014, 58, 7171–7181. [Google Scholar] [CrossRef] [PubMed]
- Schaar, K.; Geisler, A.; Kraus, M.; Pinkert, S.; Pryshliak, M.; Spencer, J.F.; Tollefson, A.E.; Ying, B.; Kurreck, J.; Wold, W.S.; et al. Anti-adenoviral artificial microRNAs expressed from AAV9 vectors inhibit human adenovirus infection in immunosuppressed Syrian hamsters. Mol. Ther. Nucleic Acids 2017, 8, 300–316. [Google Scholar] [CrossRef] [PubMed]
- Geisler, A.; Dieringer, B.; Elsner, L.; Klingel, K.; Klopfleisch, R.; Vornlocher, H.P.; Kurreck, J.; Fechner, H. Lipid nanoparticle-encapsulated, chemically modified anti-adenoviral siRNAs inhibit hepatic adenovirus infection in immunosuppressed Syrian hamsters. Mol. Ther. Nucleic Acids 2023, 32, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Berastegui-Cabrera, J.; Ye, N.; Carretero-Ledesma, M.; Pachón-Díaz, J.; Chen, H.; Pachón-Ibáñez, M.E.; Sánchez-Céspedes, J.; Zhou, J. Discovery of Novel Substituted N-(4-Amino-2-chlorophenyl)-5-chloro-2-hydroxybenzamide Analogues as Potent Human Adenovirus Inhibitors. J. Med. Chem. 2020, 63, 12830–12852. [Google Scholar] [CrossRef] [PubMed]
- Tollefson, A.E.; Cline-Smith, A.B.; Spencer, J.F.; Reyna, D.M.; Lipka, E.; Toth, K. NPP-669, a prodrug of cidofovir, is highly efficacious against human adenovirus infection in the permissive Syrian hamster model. Antimicrob. Agents Chemother. 2024, 68, e0048924. [Google Scholar] [CrossRef] [PubMed]
- Young, B.A.; Spencer, J.F.; Ying, B.; Tollefson, A.E.; Toth, K.; Wold, W.S. The role of cyclophosphamide in enhancing antitumor efficacy of an adenovirus oncolytic vector in subcutaneous Syrian hamster tumors. Cancer Gene Ther. 2013, 20, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Young, B.A.; Spencer, J.F.; Ying, B.; Toth, K.; Wold, W.S. The effects of radiation on antitumor efficacy of an oncolytic adenovirus vector in the Syrian hamster model. Cancer Gene Ther. 2013, 20, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, D.L.; Toth, K.; Doronin, K.; Tollefson, A.E.; Wold, W.S.M. Functions and mechanisms of action of the adenovirus E3 proteins. Int. Rev. Immunol. 2004, 23, 75–111. [Google Scholar] [CrossRef] [PubMed]
- Doronin, K.; Toth, K.; Kuppuswamy, M.; Krajcsi, P.; Tollefson, A.E.; Wold, W.S.M. Overexpression of the ADP (E3-11.6K) protein increases cell lysis and spread of adenovirus. Virology 2003, 305, 378–387. [Google Scholar] [CrossRef]
- Tollefson, A.E.; Scaria, A.; Hermiston, T.W.; Ryerse, J.S.; Wold, L.J.; Wold, W.S. The adenovirus death protein (E3-11.6K) is required at very late stages of infection for efficient cell lysis and release of adenovirus from infected cells. J. Virol. 1996, 70, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Moghadamnia, M.; Eshaghi, H.; Alimadadi, H.; Dashti-Khavidaki, S. A quick algorithmic review on management of viral infectious diseases in pediatric solid organ transplant recipients. Front Pediatr 2023, 11, 1252495. [Google Scholar] [CrossRef] [PubMed]
- Humar, A.; Gregson, D.; Caliendo, A.M.; McGeer, A.; Malkan, G.; Krajden, M.; Corey, P.; Greig, P.; Walmsley, S.; Levy, G.; et al. Clinical utility of quantitative cytomegalovirus viral load determination for predicting cytomegalovirus disease in liver transplant recipients. Transplantation 1999, 68, 1305–1311. [Google Scholar] [CrossRef] [PubMed]
- Razonable, R.R.; Humar, A. Cytomegalovirus in solid organ transplant recipients-Guidelines of the American Society of Transplantation Infectious Diseases Community of Practice. Clin. Transplant. 2019, 33, e13512. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Winston, D.J.; Razonable, R.R.; Lyon, G.M.; Silveira, F.P.; Wagener, M.M.; Stevens-Ayers, T.; Edmison, B.; Boeckh, M.; Limaye, A.P. Effect of Preemptive Therapy vs Antiviral Prophylaxis on Cytomegalovirus Disease in Seronegative Liver Transplant Recipients With Seropositive Donors: A Randomized Clinical Trial. JAMA 2020, 323, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Florescu, D.F.; Schaenman, J.M. Adenovirus in solid organ transplant recipients: Guidelines from the American Society of Transplantation Infectious Diseases Community of Practice. Clin. Transplant. 2019, 33, e13527. [Google Scholar] [CrossRef] [PubMed]
- Cesaro, S. Adenovirus infection in allogeneic hematopoietic cell transplantation. Transpl. Infect. Dis. 2023, 25 (Suppl. S1), e14173. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, M.G.; Yee, M.B.; Flot, J.S.; Liu, D.; Geiler, B.W.; Kinchington, P.R.; Moffat, J.F. Development of Robust Varicella Zoster Virus Luciferase Reporter Viruses for In Vivo Monitoring of Virus Growth and Its Antiviral Inhibition in Culture, Skin, and Humanized Mice. Viruses 2022, 14, 826. [Google Scholar] [CrossRef] [PubMed]
- Al-Heeti, O.M.; Cathro, H.P.; Ison, M.G. Adenovirus Infection and Transplantation. Transplantation 2022, 106, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Doronin, K.; Shashkova, E.V.; May, S.M.; Hofherr, S.E.; Barry, M.A. Chemical Modification with High Molecular Weight Polyethylene Glycol Reduces Transduction of Hepatocytes and Increases Efficacy of Intravenously Delivered Oncolytic Adenovirus. Hum. Gene Ther. 2009, 20, 975–988. [Google Scholar] [CrossRef] [PubMed]
- Shashkova, E.V.; Doronin, K.; Senac, J.S.; Barry, M.A. Macrophage depletion combined with anticoagulant therapy increases therapeutic window of systemic treatment with oncolytic adenovirus. Cancer Res. 2008, 68, 5896–5904. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tollefson, A.E.; Cline-Smith, A.; Spencer, J.F.; Ying, B.; Reyna, D.M.; Lipka, E.; James, S.H.; Toth, K. Longitudinal Monitoring of the Effects of Anti-Adenoviral Treatment Regimens in a Permissive In Vivo Model. Viruses 2024, 16, 1200. https://doi.org/10.3390/v16081200
Tollefson AE, Cline-Smith A, Spencer JF, Ying B, Reyna DM, Lipka E, James SH, Toth K. Longitudinal Monitoring of the Effects of Anti-Adenoviral Treatment Regimens in a Permissive In Vivo Model. Viruses. 2024; 16(8):1200. https://doi.org/10.3390/v16081200
Chicago/Turabian StyleTollefson, Ann E., Anna Cline-Smith, Jacqueline F. Spencer, Baoling Ying, Dawn M. Reyna, Elke Lipka, Scott H. James, and Karoly Toth. 2024. "Longitudinal Monitoring of the Effects of Anti-Adenoviral Treatment Regimens in a Permissive In Vivo Model" Viruses 16, no. 8: 1200. https://doi.org/10.3390/v16081200