
Enhanced Anti-Tumor Response Elicited by a Novel Oncolytic Pseudorabies Virus Engineered with a PD-L1 Inhibitor

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses and Cells
2.2. Virus Titration
2.3. Cell Viability Assay
2.4. Western Blotting
2.5. Animal Experiment
2.6. Ethics Statement
2.7. Construct the Recombinant Viruses rPRV-iPD-L1
2.8. Histological Examination
2.9. Immunofluorescence (IF) Staining
2.10. Statistical Analysis
3. Results
3.1. PRV Can Infect and Kill Tumor Cells In Vitro
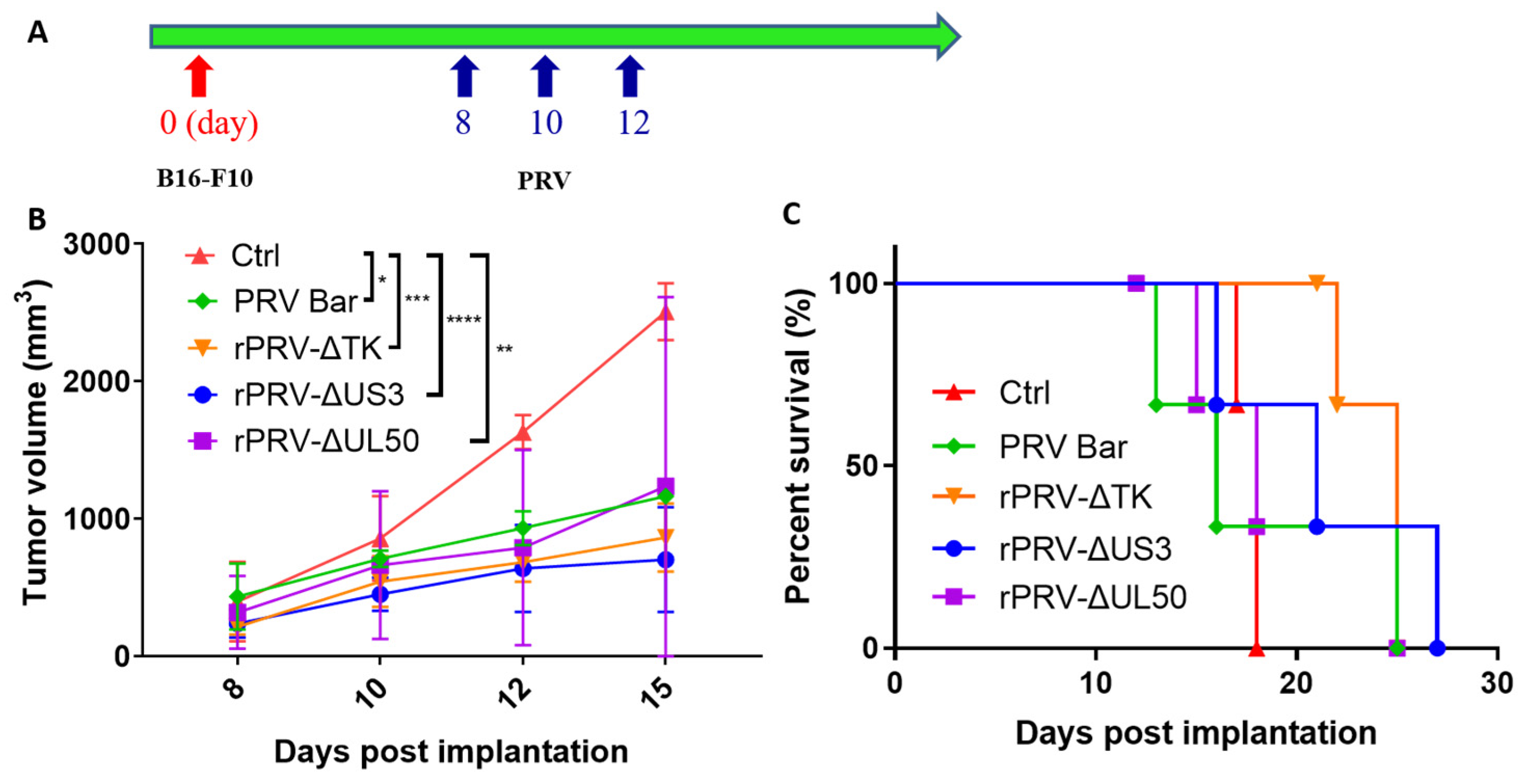
3.2. PRV Reduces Tumor Growth Rates and Enhances Survival in an Immunocompetent Model
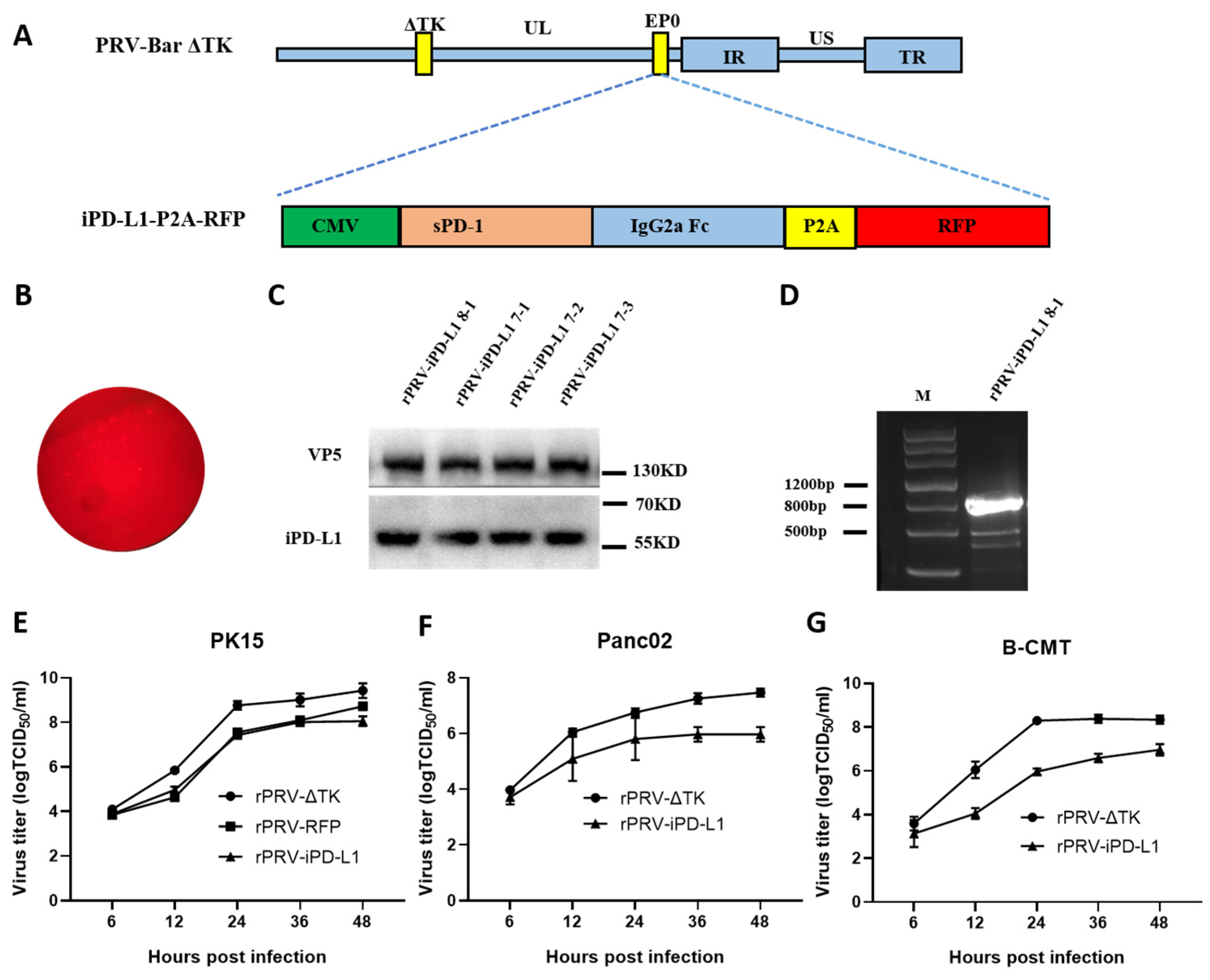
3.3. Construction of the Recombinant PRV Expressing iPD-L1 by CRISPR/Cas9
3.4. Anti-tumor Activity of the Recombinant Strain rPRV-iPD-L1 In Vivo
3.5. PRV Treatment Increases the Infiltration of Lymphocytes within the Tumor Microenvironment
3.6. PRV Treatment Increased the PD-L1 Expression in the Tumor Microenvironment
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.; Zhang, M.; Yang, J.; Zhu, Z.; Cao, W.; Dong, C. Therapeutic cancer vaccines: Advancements, challenges, and prospects. Signal Transduct. Target. Ther. 2023, 8, 450. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.D.; Lai, J.; Slaney, C.Y.; Kallies, A.; Beavis, P.A.; Darcy, P.K. Cellular networks controlling T cell persistence in adoptive cell therapy. Nat. Rev. Immunol. 2021, 21, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Hsueh, P.-C.; Li, Z.; Ho, P.-C. Microenvironment-driven metabolic adaptations guiding CD8+ T cell anti-tumor immunity. Immunity 2023, 56, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Korman, A.J.; Garrett-Thomson, S.C.; Lonberg, N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat. Rev. Drug Discov. 2022, 21, 509–528. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 324–328. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, T.; Zheng, L.; Liu, H.; Song, W.; Liu, D.; Li, Z.; Pan, C.X. Combination strategies to maximize the benefits of cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 156. [Google Scholar] [CrossRef]
- Butterfield, L.H.; Najjar, Y.G. Immunotherapy combination approaches: Mechanisms, biomarkers and clinical observations. Nat. Rev. Immunol. 2024, 24, 399–416. [Google Scholar] [CrossRef]
- Zhu, Z.; McGray, A.J.R.; Jiang, W.; Lu, B.; Kalinski, P.; Guo, Z.S. Improving cancer immunotherapy by rationally combining oncolytic virus with modulators targeting key signaling pathways. Mol. Cancer 2022, 21, 196. [Google Scholar] [CrossRef]
- Shalhout, S.Z.; Miller, D.M.; Emerick, K.S.; Kaufman, H.L. Therapy with oncolytic viruses: Progress and challenges. Nat. Rev. Clin. Oncol. 2023, 20, 160–177. [Google Scholar] [CrossRef] [PubMed]
- Ramelyte, E.; Tastanova, A.; Balázs, Z.; Ignatova, D.; Turko, P.; Menzel, U.; Guenova, E.; Beisel, C.; Krauthammer, M.; Levesque, M.P.; et al. Oncolytic virotherapy-mediated anti-tumor response: A single-cell perspective. Cancer Cell 2021, 39, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chard Dunmall, L.S.; Cheng, Z.; Wang, Y. Remodeling the tumor microenvironment by oncolytic viruses: Beyond oncolysis of tumor cells for cancer treatment. J. Immunother. Cancer 2022, 10, e004167. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Shen, Y.; Liang, T. Oncolytic virotherapy: Basic principles, recent advances and future directions. Signal Transduct. Target. Ther. 2023, 8, 156. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ramachandran, M.; Jin, C.; Quijano-Rubio, C.; Martikainen, M.; Yu, D.; Essand, M. Characterization of virus-mediated immunogenic cancer cell death and the consequences for oncolytic virus-based immunotherapy of cancer. Cell Death Dis. 2020, 11, 48. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2019, 20, 95–112. [Google Scholar] [CrossRef]
- Tian, Y.; Xie, D.; Yang, L. Engineering strategies to enhance oncolytic viruses in cancer immunotherapy. Signal Transduct. Target. Ther. 2022, 7, 117. [Google Scholar] [CrossRef]
- Uche, I.K.; Fowlkes, N.; Vu, L.; Watanabe, T.; Carossino, M.; Nabi, R.; Del Piero, F.; Rudd, J.S.; Kousoulas, K.G.; Rider, P.J.F.; et al. The Novel Oncolytic Herpes Simplex Virus Type-1 (HSV-1) VC2 Promotes Long-lasting, Systemic Anti-melanoma Tumor Immune Responses and Increased Survival in an Immunocompetent B16F10-derived Mouse Melanoma Model. J. Virol. 2020, 95, e01359-20. [Google Scholar] [CrossRef]
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 2005, 69, 462–500. [Google Scholar] [CrossRef]
- Zheng, H.-H.; Fu, P.-F.; Chen, H.-Y.; Wang, Z.-Y. Pseudorabies Virus: From Pathogenesis to Prevention Strategies. Viruses 2022, 14, 1638. [Google Scholar] [CrossRef]
- Laval, K.; Vernejoul, J.B.; Van Cleemput, J.; Koyuncu, O.O.; Enquist, L.W. Virulent Pseudorabies Virus Infection Induces a Specific and Lethal Systemic Inflammatory Response in Mice. J. Virol. 2018, 92, e01614-18. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zha, Z.; Huang, P.; Sun, H.; Huang, Y.; He, M.; Chen, T.; Lin, L.; Chen, Z.; Kong, Z.; et al. Structures of pseudorabies virus capsids. Nat. Commun. 2022, 13, 1533. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Abid, M.; Cao, S.; Zhu, S. Recombinant Pseudorabies Virus Usage in Vaccine Development against Swine Infectious Disease. Viruses 2023, 15, 370. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Cao, J.; Gui, M.; Huang, P.; Zhang, L.; Qi, R.; Chen, R.; Lin, L.; Han, Q.; Lin, Y.; et al. The potential of swine pseudorabies virus attenuated vaccine for oncolytic therapy against malignant tumors. J. Exp. Clin. Cancer Res. 2023, 42, 284. [Google Scholar] [CrossRef] [PubMed]
- Xu, A.; Qin, C.; Lang, Y.; Wang, M.; Lin, M.; Li, C.; Zhang, R.; Tang, J. A simple and rapid approach to manipulate pseudorabies virus genome by CRISPR/Cas9 system. Biotechnol. Lett. 2015, 37, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Chen, S.; Zhang, Y.; Wang, M.; Qin, C.; Yu, C.; Zhang, Y.; Li, Y.; Chen, L.; Zhang, X.; et al. Pseudorabies Virus DNA Polymerase Processivity Factor UL42 Inhibits Type I IFN Response by Preventing ISGF3-ISRE Interaction. J. Immunol. 2021, 207, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wu, H.; Sun, Y.; Zhu, J.; Tang, J.; Kuang, Y.; Li, G. A Novel Canine Mammary Cancer Cell Line: Preliminary Identification and Utilization for Drug Screening Studies. Front. Vet. Sci. 2021, 8, 665906. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Li, L.; Lou, H.; Sun, H.; Ngai, S.M.; Shao, G.; Tang, J. The ribosomal protein S26 regulates p53 activity in response to DNA damage. Oncogene 2014, 33, 2225–2235. [Google Scholar] [CrossRef]
- Bo, Z.; Li, X. A Review of Pseudorabies Virus Variants: Genomics, Vaccination, Transmission, and Zoonotic Potential. Viruses 2022, 14, 1003. [Google Scholar] [CrossRef]
- Zhang, R.; Xu, A.; Qin, C.; Zhang, Q.; Chen, S.; Lang, Y.; Wang, M.; Li, C.; Feng, W.; Zhang, R.; et al. Pseudorabies Virus dUTPase UL50 Induces Lysosomal Degradation of Type I Interferon Receptor 1 and Antagonizes the Alpha Interferon Response. J. Virol. 2017, 91, e01148-17. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Zhang, R.; Lang, Y.; Shao, A.; Xu, A.; Feng, W.; Han, J.; Wang, M.; He, W.; Yu, C.; et al. Bclaf1 critically regulates the type I interferon response and is degraded by alphaherpesvirus US3. PLoS Pathog. 2019, 15, e1007559. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Kang, X.; Chen, K.S.; Jehng, T.; Jones, L.; Chen, J.; Huang, X.F.; Chen, S.-Y. An engineered oncolytic virus expressing PD-L1 inhibitors activates tumor neoantigen-specific T cell responses. Nat. Commun. 2020, 11, 1395. [Google Scholar] [CrossRef]
- Chai, C.; Zhang, J.; Zhou, Y.; Yin, H.; Zhang, F.; Diao, Y.; Zan, X.; Ma, Y.; Wang, Y.; Wu, Y.; et al. The Effects of Oncolytic Pseudorabies Virus Vaccine Strain Inhibited the Growth of Colorectal Cancer HCT-8 Cells In Vitro and In Vivo. Animals. 2022, 12, 2416. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Sun, J.; Lv, X.; Tang, X.; Zheng, Y.; Ma, J.; Sun, Y. A Recombinant Oncolytic Pseudorabies Virus Expressing Interleukin-18, Interferon-Gamma and PH20 Genes Promotes Systemic Antitumor Immunity. Microorganisms 2023, 11, 1850. [Google Scholar] [CrossRef]
- Wollmann, G.; Tattersall, P.; van den Pol, A.N. Targeting human glioblastoma cells: Comparison of nine viruses with oncolytic potential. J. Virol. 2005, 79, 6005–6022. [Google Scholar] [CrossRef]
- Boldogkoi, Z.; Bratincsak, A.; Fodor, I. Evaluation of pseudorabies virus as a gene transfer vector and an oncolytic agent for human tumor cells. Anticancer. Res. 2002, 22, 2153–2159. [Google Scholar]
- Brittle, E.E.; Reynolds, A.E.; Enquist, L.W. Two modes of pseudorabies virus neuroinvasion and lethality in mice. J. Virol. 2004, 78, 12951–12963. [Google Scholar] [CrossRef]
- Cong, X.; Lei, J.L.; Xia, S.L.; Wang, Y.M.; Li, Y.; Li, S.; Luo, Y.; Sun, Y.; Qiu, H.J. Pathogenicity and immunogenicity of a gE/gI/TK gene-deleted pseudorabies virus variant in susceptible animals. Vet. Microbiol. 2016, 182, 170–177. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, L.-Q.; Zheng, H.-H.; Yang, Y.-R.; Liu, F.; Zheng, L.-L.; Jin, Y.; Chen, H.-Y. Construction and immunogenicity of a gE/gI/TK-deleted PRV based on porcine pseudorabies virus variant. Mol. Cell. Probes 2020, 53, 101605. [Google Scholar] [CrossRef]
- Huang, A.C.; Zappasodi, R. A decade of checkpoint blockade immunotherapy in melanoma: Understanding the molecular basis for immune sensitivity and resistance. Nat. Immunol. 2022, 23, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-Term Outcomes with Nivolumab Plus Ipilimumab or Nivolumab Alone Versus Ipilimumab in Patients with Advanced Melanoma. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Loo, K.; Smithy, J.W.; Postow, M.A.; Betof Warner, A. Factors Determining Long-Term Antitumor Responses to Immune Checkpoint Blockade Therapy in Melanoma. Front. Immunol. 2021, 12, 810388. [Google Scholar] [CrossRef] [PubMed]
- Ravi, A.; Hellmann, M.D.; Arniella, M.B.; Holton, M.; Freeman, S.S.; Naranbhai, V.; Stewart, C.; Leshchiner, I.; Kim, J.; Akiyama, Y.; et al. Genomic and transcriptomic analysis of checkpoint blockade response in advanced non-small cell lung cancer. Nat. Genet. 2023, 55, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Melcher, A.; Harrington, K.; Vile, R. Oncolytic virotherapy as immunotherapy. Science 2021, 374, 1325–1326. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.R.; Espitia, C.M.; Zhao, W.; Wu, K.; Visconte, V.; Anwer, F.; Calton, C.M.; Carew, J.S.; Nawrocki, S.T. Oncolytic reovirus sensitizes multiple myeloma cells to anti-PD-L1 therapy. Leukemia 2017, 32, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Liu, G.; Zhong, J.; Zheng, K.; Xiao, H.; Li, C.; Song, X.; Li, Y.; Xu, C.; Wu, H.; et al. Immune Checkpoints in Viral Infections. Viruses 2020, 12, 1051. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, W.; Wang, R.; Zhang, N.; Shang, H.; Bi, Y.; Chen, D.; Zhang, C.; Li, L.; Yin, J.; et al. Reshaping the Immune Microenvironment by Oncolytic Herpes Simplex Virus in Murine Pancreatic Ductal Adenocarcinoma. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 29, 744–761. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.-C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef]
- Ju, F.; Luo, Y.; Lin, C.; Jia, X.; Xu, Z.; Tian, R.; Lin, Y.; Zhao, M.; Chang, Y.; Huang, X.; et al. Oncolytic virus expressing PD-1 inhibitors activates a collaborative intratumoral immune response to control tumor and synergizes with CTLA-4 or TIM-3 blockade. J. Immunother. Cancer 2022, 10, e004762. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, G.; Wang, M.; Wang, P.; Li, R.; Gao, C.; Li, Y.; Liang, X.; Liu, Y.; Xu, A.; Tang, J. Enhanced Anti-Tumor Response Elicited by a Novel Oncolytic Pseudorabies Virus Engineered with a PD-L1 Inhibitor. Viruses 2024, 16, 1228. https://doi.org/10.3390/v16081228
Xiang G, Wang M, Wang P, Li R, Gao C, Li Y, Liang X, Liu Y, Xu A, Tang J. Enhanced Anti-Tumor Response Elicited by a Novel Oncolytic Pseudorabies Virus Engineered with a PD-L1 Inhibitor. Viruses. 2024; 16(8):1228. https://doi.org/10.3390/v16081228
Chicago/Turabian StyleXiang, Guangtao, Mengdong Wang, Pu Wang, Rifei Li, Chao Gao, Yue Li, Xinxin Liang, Yun Liu, Aotian Xu, and Jun Tang. 2024. "Enhanced Anti-Tumor Response Elicited by a Novel Oncolytic Pseudorabies Virus Engineered with a PD-L1 Inhibitor" Viruses 16, no. 8: 1228. https://doi.org/10.3390/v16081228