
An ISG15-Based High-Throughput Screening Assay for Identification and Characterization of SARS-CoV-2 Inhibitors Targeting Papain-like Protease
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Viruses
2.2. Expression and Purification of SARS-CoV-2 PLpro
2.3. Generation of CFP-3C-ISG15-YFP and CFP-Ubiquitin-YFP Substrate
2.4. FRET Assay for Determination of Enzymatic Activity of PLpro
2.5. Determination of Inhibitory Activity of the Selected Compounds on PLpro and USP25
2.6. Inhibition Mechanism of SARS-CoV-2 PLpro Inhibitors
2.7. Cellular Cytotoxicity
2.8. Antiviral Plaque Reduction Assay (PRA)
2.9. Antiviral Immunofluorescence Assay (IFA)
2.10. Inhibition of Cleavage of Protein Substrate by Inhibitors
2.11. Surface Plasmon Resonance
2.12. Molecular Docking
3. Results
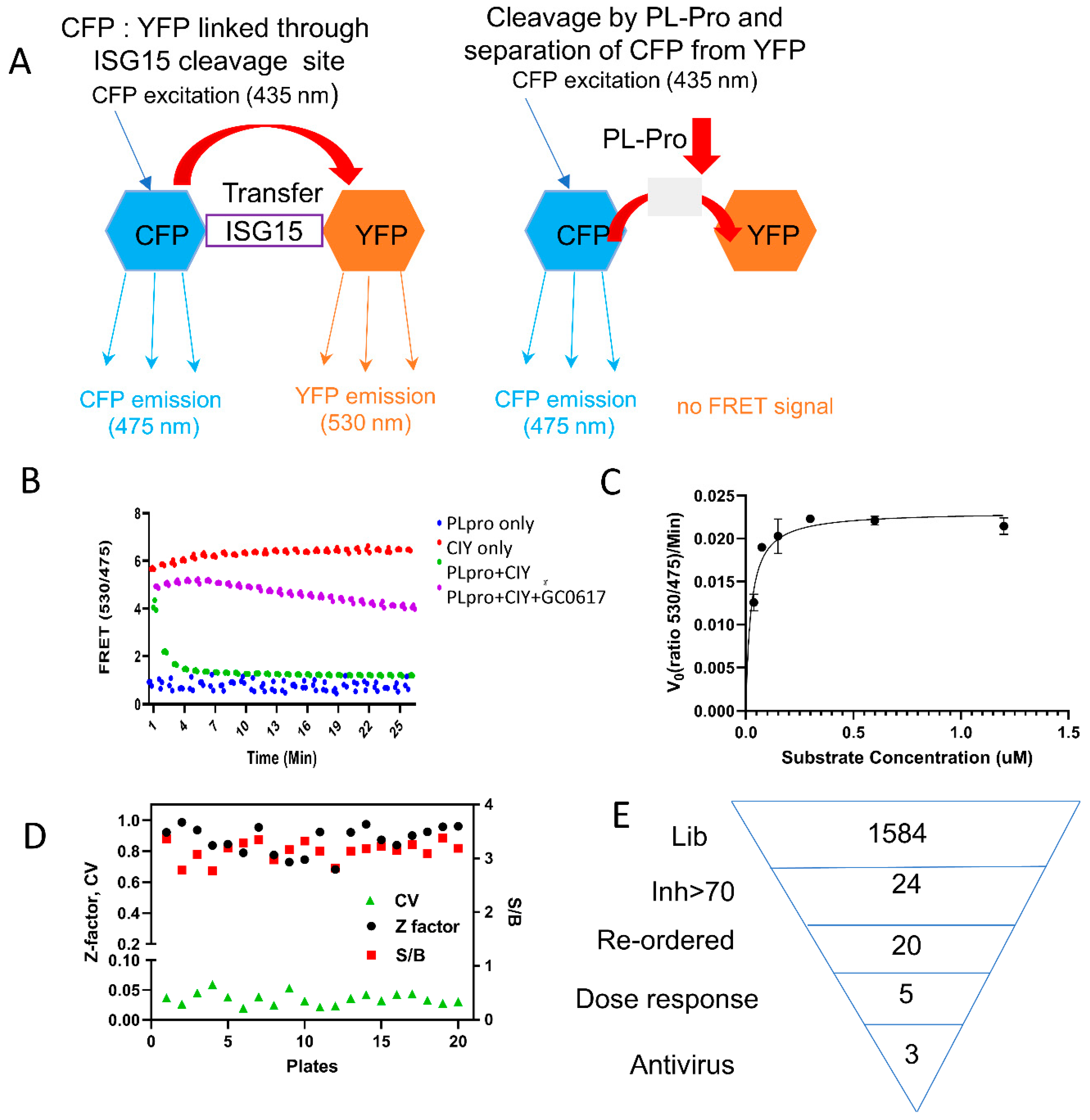
3.1. ISG15-Based PLpro Assay
3.2. High-Throughput Screening
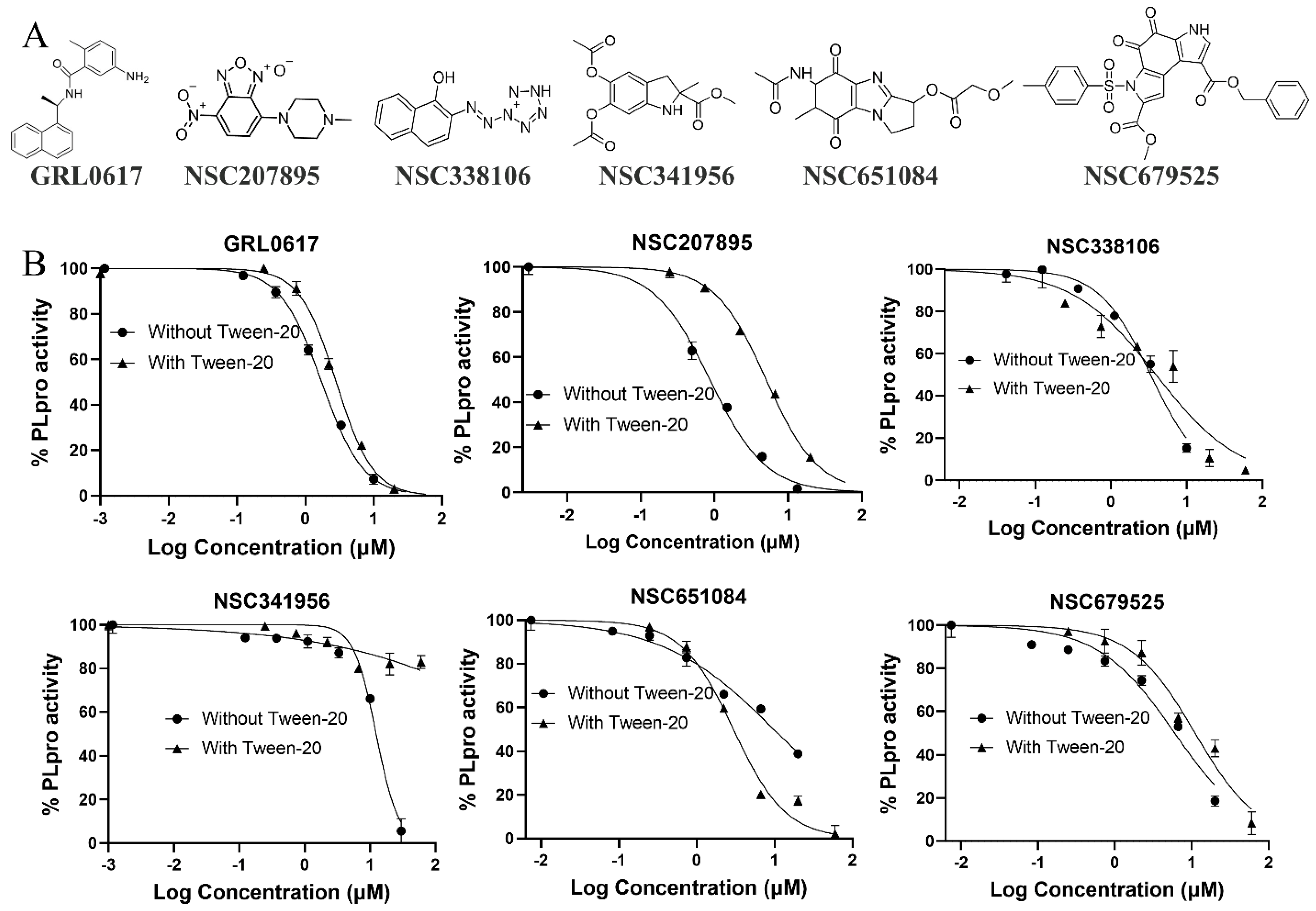
3.3. Specificity
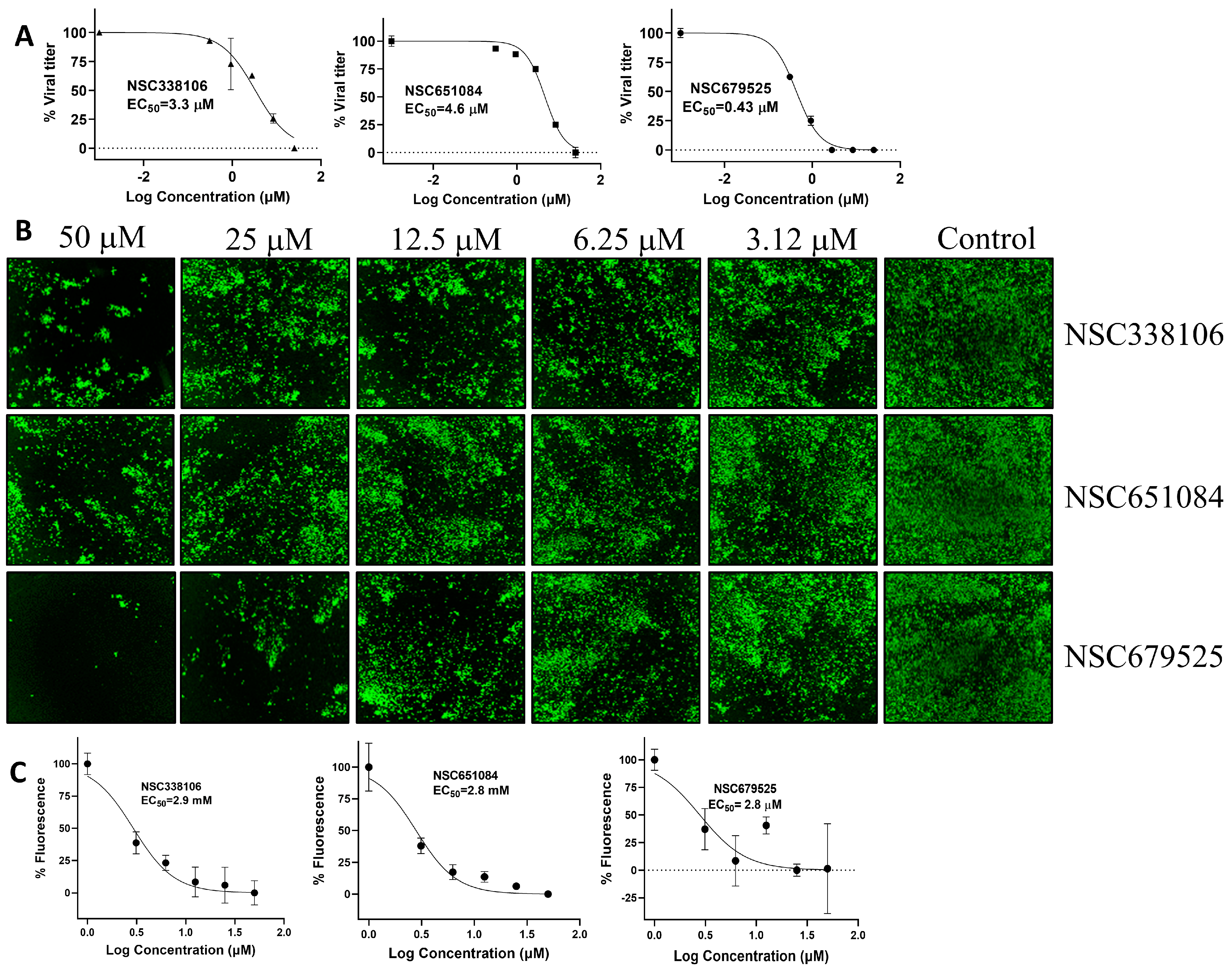
3.4. Inhibition of SARS-CoV-2 Replication by PLpro Inhibitors
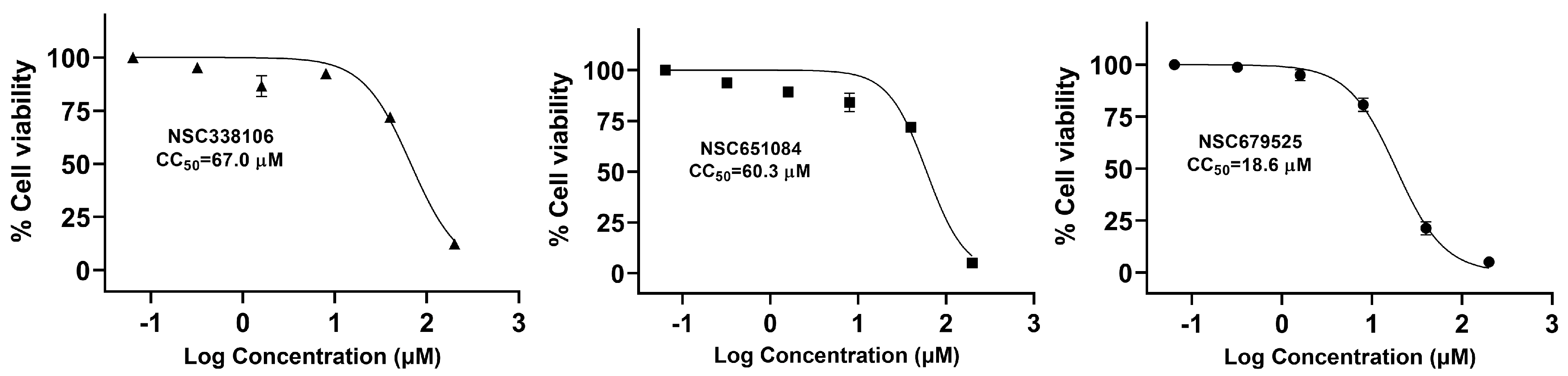
3.5. Cytotoxicity of Candidate PLpro Inhibitors
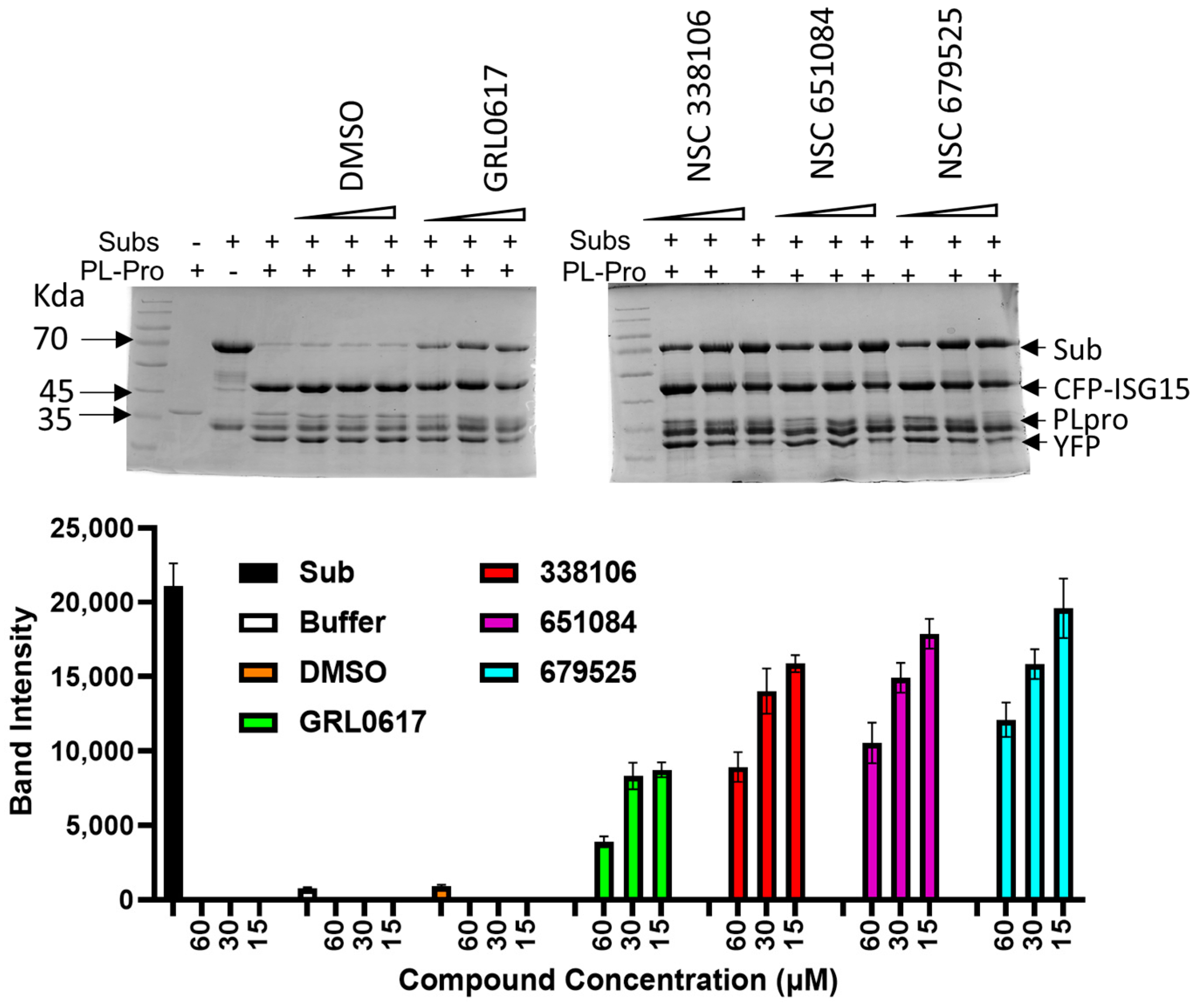
3.6. PAGE Analysis for Cleavage of the CFP-3C-ISG15-YFP Substrate by PLpro
3.7. Analysis of Bimolecular Interactions between PLpro and Inhibitors
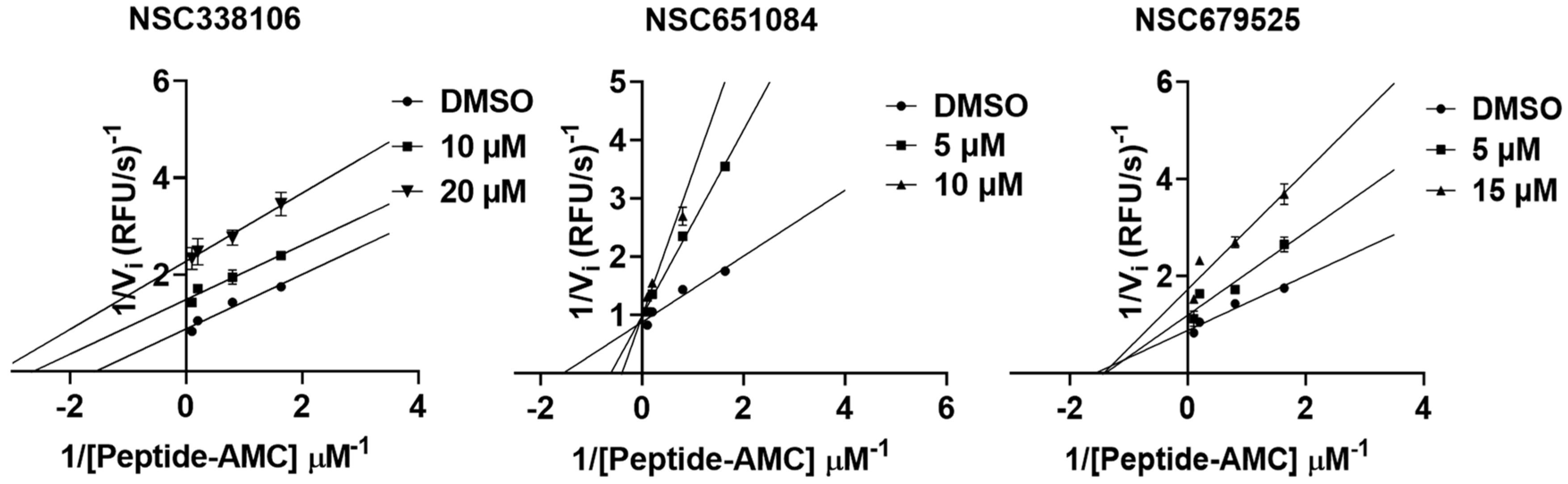
3.8. Mechanism of Inhibition
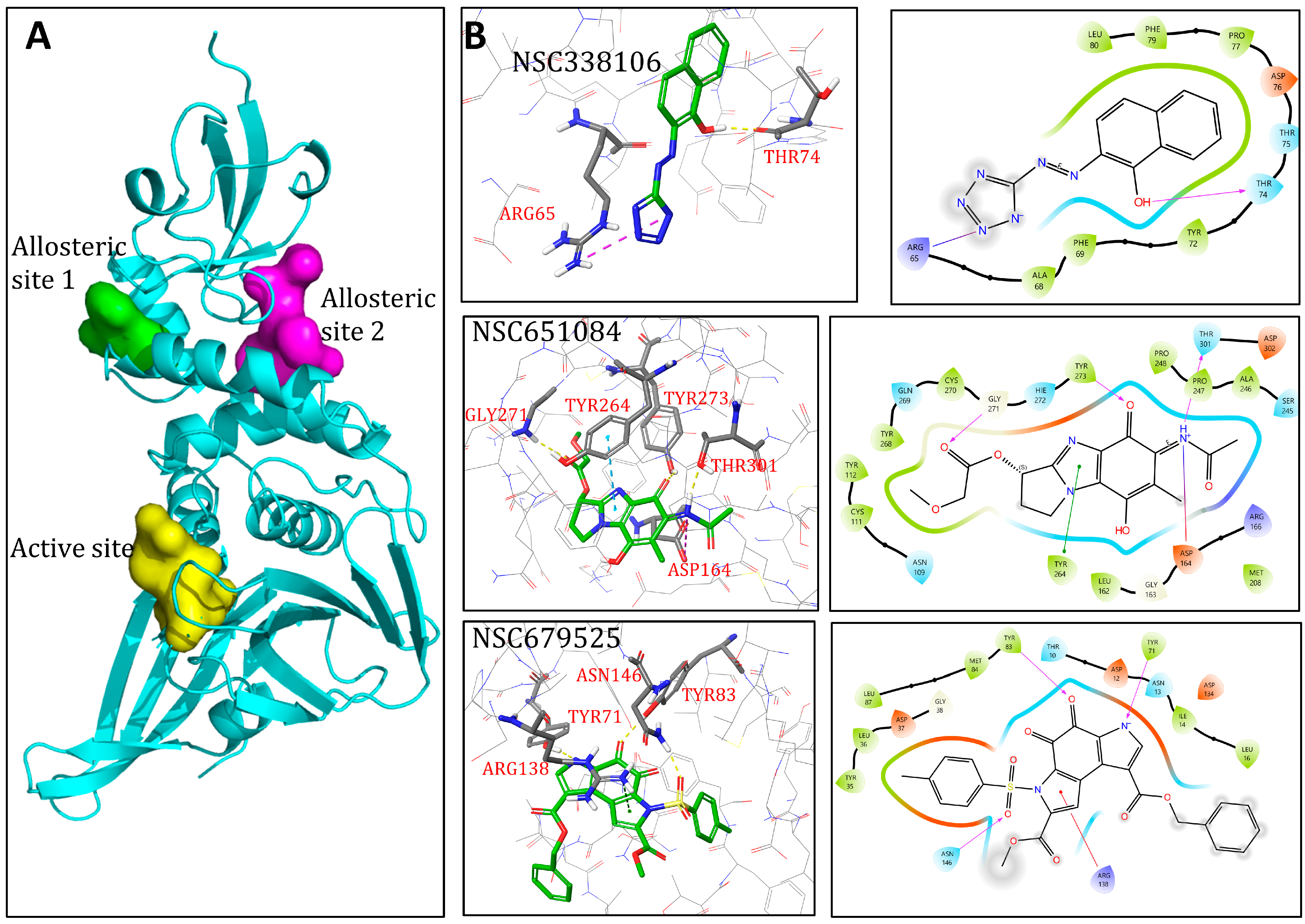
3.9. Molecular Modelling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lipsitch, M.; Krammer, F.; Regev-Yochay, G.; Lustig, Y.; Balicer, R.D. SARS-CoV-2 breakthrough infections in vaccinated individuals: Measurement, causes and impact. Nat. Rev. Immunol. 2022, 22, 57–65. [Google Scholar] [CrossRef]
- Puhach, O.; Adea, K.; Hulo, N.; Sattonnet, P.; Genecand, C.; Iten, A.; Jacquérioz, F.; Kaiser, L.; Vetter, P.; Eckerle, I.; et al. Infectious viral load in unvaccinated and vaccinated individuals infected with ancestral, Delta or Omicron SARS-CoV-2. Nat. Med. 2022, 28, 1491–1500. [Google Scholar] [CrossRef]
- Flacco, M.E.; Acuti Martellucci, C.; Baccolini, V.; De Vito, C.; Renzi, E.; Villari, P.; Manzoli, L. COVID-19 vaccines reduce the risk of SARS-CoV-2 reinfection and hospitalization: Meta-analysis. Front. Med. 2022, 9, 1023507. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.T.; Kwan, A.T.; Rodríguez-Barraquer, I.; Singer, B.J.; Park, H.J.; Lewnard, J.A.; Sears, D.; Lo, N.C. Infectiousness of SARS-CoV-2 breakthrough infections and reinfections during the Omicron wave. Nat. Med. 2023, 29, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.W.X.; Chiew, C.J.; Ang, L.W.; Mak, T.M.; Cui, L.; Toh, M.; Lim, Y.D.; Lee, P.H.; Lee, T.H.; Chia, P.Y.; et al. Clinical and Virological Features of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Variants of Concern: A Retrospective Cohort Study Comparing B.1.1.7 (Alpha), B.1.351 (Beta), and B.1.617.2 (Delta). Clin. Infect. Dis. 2022, 75, e1128–e1136. [Google Scholar] [CrossRef] [PubMed]
- Bolze, A.; Luo, S.; White, S.; Cirulli, E.T.; Wyman, D.; Dei Rossi, A.; Machado, H.; Cassens, T.; Jacobs, S.; Schiabor Barrett, K.M.; et al. SARS-CoV-2 variant Delta rapidly displaced variant Alpha in the United States and led to higher viral loads. Cell Rep. Med. 2022, 3, 100564. [Google Scholar] [CrossRef] [PubMed]
- Samieefar, N.; Rashedi, R.; Akhlaghdoust, M.; Mashhadi, M.; Darzi, P.; Rezaei, N. Delta Variant: The New Challenge of COVID-19 Pandemic, an Overview of Epidemiological, Clinical, and Immune Characteristics. Acta Biomed. 2022, 93, e2022179. [Google Scholar] [CrossRef]
- Ganesh, B.; Rajakumar, T.; Malathi, M.; Manikandan, N.; Nagaraj, J.; Santhakumar, A.; Elangovan, A.; Malik, Y.S. Epidemiology and pathobiology of SARS-CoV-2 (COVID-19) in comparison with SARS, MERS: An updated overview of current knowledge and future perspectives. Clin. Epidemiol. Glob. Health 2021, 10, 100694. [Google Scholar] [CrossRef] [PubMed]
- Malik, Y.S.; Kumar, N.; Sircar, S.; Kaushik, R.; Bhat, S.; Dhama, K.; Gupta, P.; Goyal, K.; Singh, M.P.; Ghoshal, U.; et al. Coronavirus Disease Pandemic (COVID-19): Challenges and a Global Perspective. Pathogens 2020, 9, 519. [Google Scholar] [CrossRef]
- Samrat, S.K.; Tharappel, A.M.; Li, Z.; Li, H. Prospect of SARS-CoV-2 spike protein: Potential role in vaccine and therapeutic development. Virus Res. 2020, 288, 198141. [Google Scholar] [CrossRef]
- Tharappel, A.M.; Samrat, S.K.; Li, Z.; Li, H. Targeting Crucial Host Factors of SARS-CoV-2. ACS Infect. Dis. 2020, 6, 2844–2865. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.Y.; Yang, J.S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Finkel, Y.; Mizrahi, O.; Nachshon, A.; Weingarten-Gabbay, S.; Morgenstern, D.; Yahalom-Ronen, Y.; Tamir, H.; Achdout, H.; Stein, D.; Israeli, O.; et al. The coding capacity of SARS-CoV-2. Nature 2021, 589, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Anderson-Daniels, J.; Gribble, J.; Denison, M. Proteolytic Processing of the Coronavirus Replicase Nonstructural Protein 14 Exonuclease Is Not Required for Virus Replication but Alters RNA Synthesis and Viral Fitness. J. Virol. 2022, 96, e0084122. [Google Scholar] [CrossRef] [PubMed]
- Weizhu, Y.; Yanhui, Z.; Xiaotao, Z.; Bin, H.; Wei, C. Structural biology of SARS-CoV-2: Open the door for novel therapies. Signal Transduct. Target Ther. 2022, 7, 26. [Google Scholar] [CrossRef]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Wu, G. Potential 3-chymotrypsin-like cysteine protease cleavage sites in the coronavirus polyproteins pp1a and pp1ab and their possible relevance to COVID-19 vaccine and drug development. FASEB J. 2021, 35, e21573. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.; Chiaravalli, J.; Gellenoncourt, S.; Brownridge, P.; Bryne, D.P.; Daly, L.A.; Grauslys, A.; Walter, M.; Agou, F.; Chakrabarti, L.A.; et al. Characterising proteolysis during SARS-CoV-2 infection identifies viral cleavage sites and cellular targets with therapeutic potential. Nat. Commun. 2021, 12, 5553. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Ackerley, D.F.; Calcott, M.J. High-Throughput Screening for Inhibitors of the SARS-CoV-2 Protease Using a FRET-Biosensor. Molecules 2020, 25, 4666. [Google Scholar] [CrossRef]
- Osipiuk, J.; Azizi, S.A.; Dvorkin, S.; Endres, M.; Jedrzejczak, R.; Jones, K.A.; Kang, S.; Kathayat, R.S.; Kim, Y.; Lisnyak, V.G.; et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021, 12, 743. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Ullrich, S.; Nitsche, C. SARS-CoV-2 Papain-Like Protease: Structure, Function and Inhibition. Chembiochem 2022, 23, e202200327. [Google Scholar] [CrossRef]
- Stasiulewicz, A.; Maksymiuk, A.W.; Nguyen, M.L.; Bełza, B.; Sulkowska, J.I. SARS-CoV-2 Papain-Like Protease Potential Inhibitors-In Silico Quantitative Assessment. Int. J. Mol. Sci. 2021, 22, 3957. [Google Scholar] [CrossRef]
- Dong, Y.; Dai, T.; Liu, J.; Zhang, L.; Zhou, F. Coronavirus in Continuous Flux: From SARS-CoV to SARS-CoV-2. Adv. Sci. 2020, 7, 2001474. [Google Scholar] [CrossRef]
- Moustaqil, M.; Ollivier, E.; Chiu, H.P.; Van Tol, S.; Rudolffi-Soto, P.; Stevens, C.; Bhumkar, A.; Hunter, D.J.B.; Freiberg, A.N.; Jacques, D.; et al. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): Implications for disease presentation across species. Emerg. Microbes Infect. 2021, 10, 178–195. [Google Scholar] [CrossRef]
- Ratia, K.; Kilianski, A.; Baez-Santos, Y.M.; Baker, S.C.; Mesecar, A. Structural Basis for the Ubiquitin-Linkage Specificity and deISGylating activity of SARS-CoV papain-like protease. PLoS Pathog. 2014, 10, e1004113. [Google Scholar] [CrossRef]
- Ratia, K.; Pegan, S.; Takayama, J.; Sleeman, K.; Coughlin, M.; Baliji, S.; Chaudhuri, R.; Fu, W.; Prabhakar, B.S.; Johnson, M.E.; et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [Google Scholar] [CrossRef]
- Zhao, Y.; Du, X.; Duan, Y.; Pan, X.; Sun, Y.; You, T.; Han, L.; Jin, Z.; Shang, W.; Yu, J.; et al. High-throughput screening identifies established drugs as SARS-CoV-2 PLpro inhibitors. Protein Cell 2021, 12, 877–888. [Google Scholar] [CrossRef]
- Shen, Z.; Ratia, K.; Cooper, L.; Kong, D.; Lee, H.; Kwon, Y.; Li, Y.; Alqarni, S.; Huang, F.; Dubrovskyi, O.; et al. Design of SARS-CoV-2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity. J. Med. Chem. 2022, 65, 2940–2955. [Google Scholar] [CrossRef]
- Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Zhu, W.; Cao, D.; et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021, 12, 488. [Google Scholar] [CrossRef]
- Freitas, B.T.; Durie, I.A.; Murray, J.; Longo, J.E.; Miller, H.C.; Crich, D.; Hogan, R.J.; Tripp, R.A.; Pegan, S.D. Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect. Dis. 2020, 6, 2099–2109. [Google Scholar] [CrossRef]
- Tan, B.; Zhang, X.; Ansari, A.; Jadhav, P.; Tan, H.; Li, K.; Chopra, A.; Ford, A.; Chi, X.; Ruiz, F.X.; et al. Design of a SARS-CoV-2 papain-like protease inhibitor with antiviral efficacy in a mouse model. Science 2024, 383, 1434–1440. [Google Scholar] [CrossRef]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, e106275. [Google Scholar] [CrossRef]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; El Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti-COVID-19 drug design. Sci. Adv. 2020, 6, abd4596. [Google Scholar] [CrossRef]
- Smith, E.; Davis-Gardner, M.E.; Garcia-Ordonez, R.D.; Nguyen, T.T.; Hull, M.; Chen, E.; Baillargeon, P.; Scampavia, L.; Strutzenberg, T.; Griffin, P.R.; et al. High-Throughput Screening for Drugs That Inhibit Papain-Like Protease in SARS-CoV-2. SLAS Discov. 2020, 25, 1152–1161. [Google Scholar] [CrossRef]
- Zang, Y.; Su, M.; Wang, Q.; Cheng, X.; Zhang, W.; Zhao, Y.; Chen, T.; Jiang, Y.; Shen, Q.; Du, J.; et al. High-throughput screening of SARS-CoV-2 main and papain-like protease inhibitors. Protein Cell 2023, 14, 17–27. [Google Scholar] [CrossRef]
- Ma, C.; Sacco, M.D.; Xia, Z.; Lambrinidis, G.; Townsend, J.A.; Hu, Y.; Meng, X.; Szeto, T.; Ba, M.; Zhang, X.; et al. Discovery of SARS-CoV-2 Papain-like Protease Inhibitors through a Combination of High-Throughput Screening and a FlipGFP-Based Reporter Assay. ACS Cent. Sci. 2021, 7, 1245–1260. [Google Scholar] [CrossRef]
- Samrat, S.K.; Bashir, Q.; Zhang, R.; Huang, Y.; Liu, Y.; Wu, X.; Brown, T.; Wang, W.; Zheng, Y.G.; Zhang, Q.Y.; et al. A universal fluorescence polarization high throughput screening assay to target the SAM-binding sites of SARS-CoV-2 and other viral methyltransferases. Emerg. Microbes Infect. 2023, 12, 2204164. [Google Scholar] [CrossRef] [PubMed]
- Samrat, S.K.; Bashir, Q.; Huang, Y.; Trieshmann, C.W.; Tharappel, A.M.; Zhang, R.; Chen, K.; Zheng, Y.G.; Li, Z.; Li, H. Broad-Spectrum Small-Molecule Inhibitors Targeting the SAM-Binding Site of Flavivirus NS5 Methyltransferase. ACS Infect. Dis. 2023, 9, 1319–1333. [Google Scholar] [CrossRef]
- Samrat, S.K.; Xu, J.; Xie, X.; Gianti, E.; Chen, H.; Zou, J.; Pattis, J.G.; Elokely, K.; Lee, H.; Li, Z.; et al. Allosteric inhibitors of the main protease of SARS-CoV-2. Antivir. Res. 2022, 205, 105381. [Google Scholar] [CrossRef]
- Li, Z.; Brecher, M.; Deng, Y.Q.; Zhang, J.; Sakamuru, S.; Liu, B.; Huang, R.; Koetzner, C.A.; Allen, C.A.; Jones, S.A.; et al. Existing drugs as broad-spectrum and potent inhibitors for Zika virus by targeting NS2B-NS3 interaction. Cell Res. 2017, 27, 1046–1064. [Google Scholar] [CrossRef]
- Brecher, M.; Li, Z.; Liu, B.; Zhang, J.; Koetzner, C.A.; Alifarag, A.; Jones, S.A.; Lin, Q.; Kramer, L.D.; Li, H. A conformational switch high-throughput screening assay and allosteric inhibition of the flavivirus NS2B-NS3 protease. PLoS Pathog. 2017, 13, e1006411. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Sakamuru, S.; Huang, R.; Brecher, M.; Koetzner, C.A.; Zhang, J.; Chen, H.; Qin, C.F.; Zhang, Q.Y.; Zhou, J.; et al. Erythrosin B is a potent and broad-spectrum orthosteric inhibitor of the flavivirus NS2B-NS3 protease. Antivir. Res. 2018, 150, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Madahar, V.; Liao, J. Development of FRET assay into quantitative and high-throughput screening technology platforms for protein-protein interactions. Ann. Biomed. Eng. 2011, 39, 1224–1234. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, B.H.; Jukneliene, D.; Kanjanahaluethai, A.; Bechill, J.; Severson, K.M.; Smith, C.M.; Rota, P.A.; Baker, S.C. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 2004, 78, 13600–13612. [Google Scholar] [CrossRef]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef] [PubMed]
- Wydorski, P.M.; Osipiuk, J.; Lanham, B.T.; Tesar, C.; Endres, M.; Engle, E.; Jedrzejczak, R.; Mullapudi, V.; Michalska, K.; Fidelis, K.; et al. Dual domain recognition determines SARS-CoV-2 PLpro selectivity for human ISG15 and K48-linked di-ubiquitin. Nat. Commun. 2023, 14, 2366. [Google Scholar] [CrossRef]
- Perng, Y.C.; Lenschow, D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018, 16, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.Y.; Song, Y.; Liu, Y. A new trend to determine biochemical parameters by quantitative FRET assays. Acta Pharmacol. Sin. 2015, 36, 1408–1415. [Google Scholar] [CrossRef]
- Felber, L.M.; Cloutier, S.M.; Kündig, C.; Kishi, T.; Brossard, V.; Jichlinski, P.; Leisinger, H.J.; Deperthes, D. Evaluation of the CFP-substrate-YFP system for protease studies: Advantages and limitations. Biotechniques 2004, 36, 878–885. [Google Scholar] [CrossRef]
- Liu, Y.; Kieslich, C.A.; Morikis, D.; Liao, J. Engineering pre-SUMO4 as efficient substrate of SENP2. Protein Eng. Des. Sel. 2014, 27, 117–126. [Google Scholar] [CrossRef]
- Iversen, P.W.; Beck, B.; Chen, Y.F.; Dere, W.; Devanarayan, V.; Eastwood, B.J.; Farmen, M.W.; Iturria, S.J.; Montrose, C.; Moore, R.A.; et al. HTS Assay Validation. In Assay Guidance Manual; Markossian, S., Grossman, A., Arkin, M., Auld, D., Austin, C., Baell, J., Brimacombe, K., Chung, T.D.Y., Coussens, N.P., Dahlin, J.L., et al., Eds.; Eli Lilly & Company: Sydney, Australia; National Center for Advancing Translational Sciences: Bethesda, MA, USA, 2004. [Google Scholar]
- Swaim, C.D.; Canadeo, L.A.; Monte, K.J.; Khanna, S.; Lenschow, D.J.; Huibregtse, J.M. Modulation of Extracellular ISG15 Signaling by Pathogens and Viral Effector Proteins. Cell Rep. 2020, 31, 107772. [Google Scholar] [CrossRef]
- Lindner, H.A.; Lytvyn, V.; Qi, H.; Lachance, P.; Ziomek, E.; Menard, R. Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain-like protease. Arch. Biochem. Biophys. 2007, 466, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, B.; Leach, C.A.; Goldenberg, S.J.; Francis, D.M.; Kodrasov, M.P.; Tian, X.; Shanks, J.; Sterner, D.E.; Bernal, A.; Mattern, M.R.; et al. Characterization of ubiquitin and ubiquitin-like-protein isopeptidase activities. Protein Sci. 2008, 17, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Van Dycke, J.; Dai, W.; Stylianidou, Z.; Li, J.; Cuvry, A.; Roux, E.; Li, B.; Rymenants, J.; Bervoets, L.; de Witte, P.; et al. A Novel Class of Norovirus Inhibitors Targeting the Viral Protease with Potent Antiviral Activity In Vitro and In Vivo. Viruses 2021, 13, 1852. [Google Scholar] [CrossRef]
- Yuan, S.; Gao, X.; Tang, K.; Cai, J.P.; Hu, M.; Luo, P.; Wen, L.; Ye, Z.W.; Luo, C.; Tsang, J.O.; et al. Targeting papain-like protease for broad-spectrum coronavirus inhibition. Protein Cell 2022, 13, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Magiorkinis, G. On the evolution of SARS-CoV-2 and the emergence of variants of concern. Trends Microbiol. 2023, 31, 5–8. [Google Scholar] [CrossRef]
- Singh, H.; Dahiya, N.; Yadav, M.; Sehrawat, N. Emergence of SARS-CoV-2 New Variants and Their Clinical Significance. Can. J. Infect. Dis. Med. Microbiol. 2022, 2022, 7336309. [Google Scholar] [CrossRef]
- Perlinska, A.P.; Stasiulewicz, A.; Nguyen, M.L.; Swiderska, K.; Zmudzinski, M.; Maksymiuk, A.W.; Drag, M.; Sulkowska, J.I. Amino acid variants of SARS-CoV-2 papain-like protease have impact on drug binding. PLoS Comput. Biol. 2022, 18, e1010667. [Google Scholar] [CrossRef]
- Tian, D.; Sun, Y.; Zhou, J.; Ye, Q. The global epidemic of SARS-CoV-2 variants and their mutational immune escape. J. Med. Virol. 2022, 94, 847–857. [Google Scholar] [CrossRef]
- Serafim, M.S.M.; Pantaleão, S.Q.; da Silva, E.B.; McKerrow, J.H.; O’Donoghue, A.J.; Mota, B.E.F.; Honorio, K.M.; Maltarollo, V.G. The importance of good practices and false hits for QSAR-driven virtual screening real application: A SARS-CoV-2 main protease (Mpro) case study. Front. Drug Discov. 2023, 3, 1237655. [Google Scholar] [CrossRef]
- Chen, X.; Varghese, S.; Zhang, Z.; Du, J.; Ruan, B.; Baell, J.B.; Liu, X. Drug discovery and optimization based on the co-crystal structure of natural product with target. Eur. J. Med. Chem. 2024, 266, 116126. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50-PLpro (µM) | IC50-PLpro-tw (µM) | IC50- USP25 (µM) | SI | EC50-IFA (µM) | EC50-PRA (µM) | EC50-Omi (µM) | CC50 (µM) | KD (µM) |
|---|---|---|---|---|---|---|---|---|---|
| Tween-20 | − | + | − | − | − | − | + | ||
| GRL0617 | 1.7 | 2.8 | |||||||
| 207895 | 0.9 | 5.0 | |||||||
| 338106 | 3.3 | 3.9 | 37 | 11.2 | 2.9 | 3.3 | 6.9 | 67.0 | 6.6 (±0.1) |
| 341956 | 12.4 | >200 | |||||||
| 651084 | 4.9 | 2.9 | 23 | 4.7 | 2.8 | 4.6 | 5.9 | 60.3 | 13.9 (±0.9) |
| 679525 | 6.0 | 11 | 41 | 6.8 | 2.8 | 0.4 | 5.4 | 18.6 | 4.1 (±2.9) |
| Compounds | Docking Scores | Putative Mode of Action | ||
|---|---|---|---|---|
| Active Site | Allosteric Site 1 | Allosteric Site 2 | ||
| NSC338106 | −6.16 | −7.53 | −7.25 | Uncompetitive |
| NSC651084 | −8.44 | −6.8 | −7.10 | Competitive |
| NSC679525 | −3.79 | −6.62 | −7.16 | Non-competitive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samrat, S.K.; Kumar, P.; Liu, Y.; Chen, K.; Lee, H.; Li, Z.; Chen, Y.; Li, H. An ISG15-Based High-Throughput Screening Assay for Identification and Characterization of SARS-CoV-2 Inhibitors Targeting Papain-like Protease. Viruses 2024, 16, 1239. https://doi.org/10.3390/v16081239
Samrat SK, Kumar P, Liu Y, Chen K, Lee H, Li Z, Chen Y, Li H. An ISG15-Based High-Throughput Screening Assay for Identification and Characterization of SARS-CoV-2 Inhibitors Targeting Papain-like Protease. Viruses. 2024; 16(8):1239. https://doi.org/10.3390/v16081239
Chicago/Turabian StyleSamrat, Subodh Kumar, Prashant Kumar, Yuchen Liu, Ke Chen, Hyun Lee, Zhong Li, Yin Chen, and Hongmin Li. 2024. "An ISG15-Based High-Throughput Screening Assay for Identification and Characterization of SARS-CoV-2 Inhibitors Targeting Papain-like Protease" Viruses 16, no. 8: 1239. https://doi.org/10.3390/v16081239