
Phylodynamic and Epistatic Analysis of Coxsackievirus A24 and Its Variant

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Specimen Collection and Ethics Statement
2.2. Viral RNA Extraction, RT-PCR, and Viral Genome Sequencing
2.3. Sequence Variation and Phylodynamic Analysis
2.4. Detection of Sequence Variation
2.5. GenBank Accession Numbers
3. Results
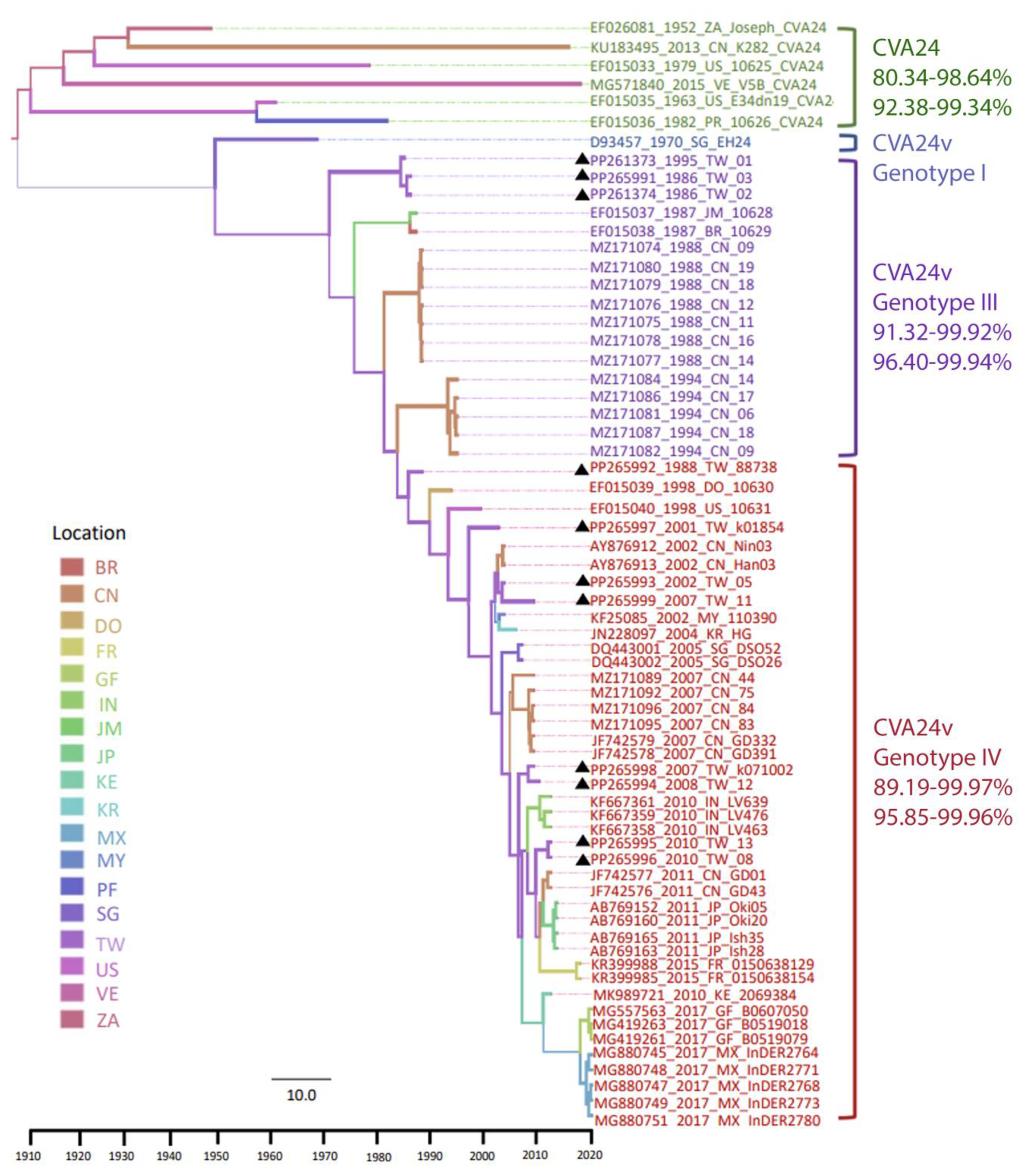
3.1. Phylodynamic Trees Constructed from the Genome
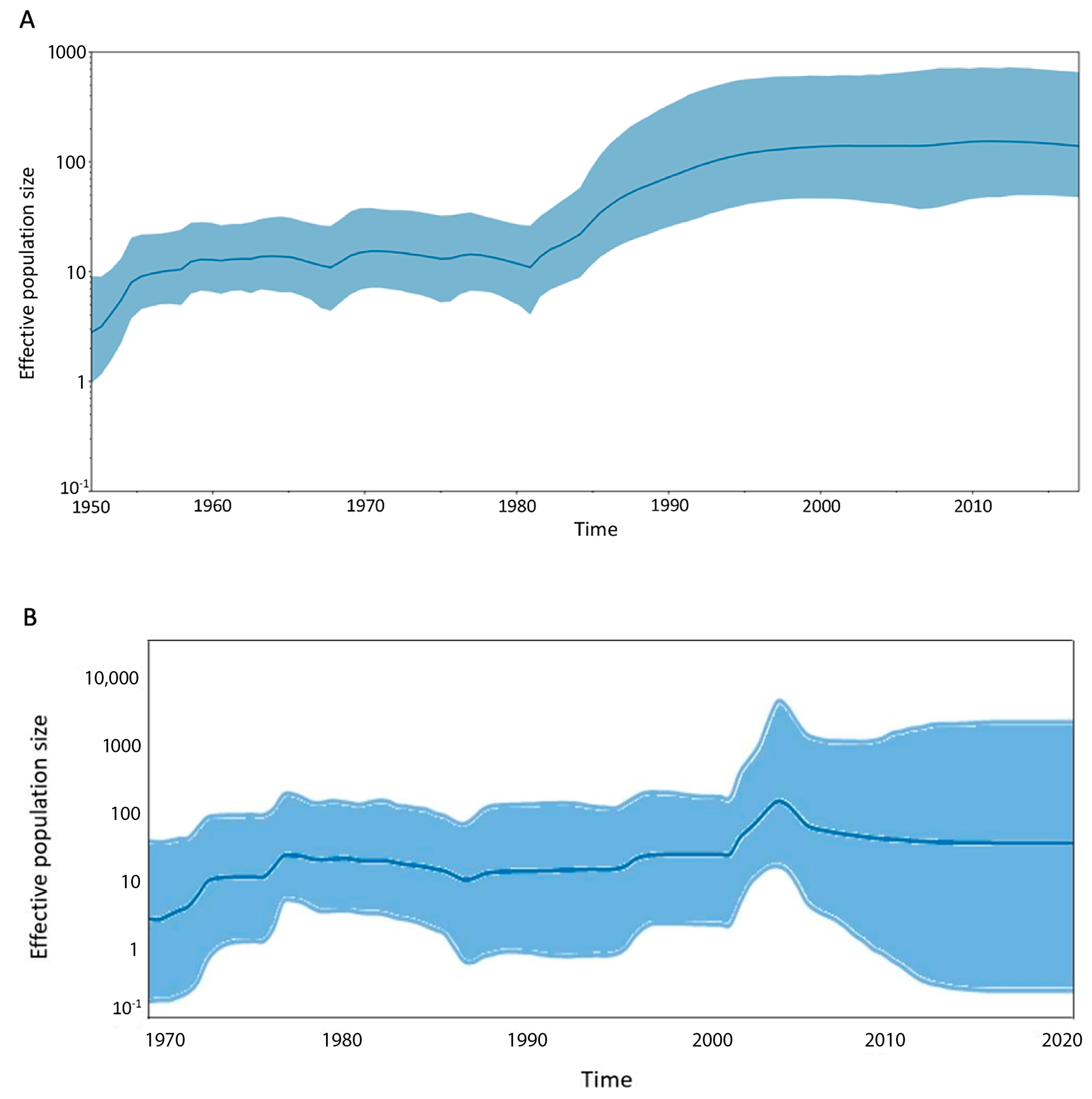
3.2. Viral Demographic History and Geographic Transmission
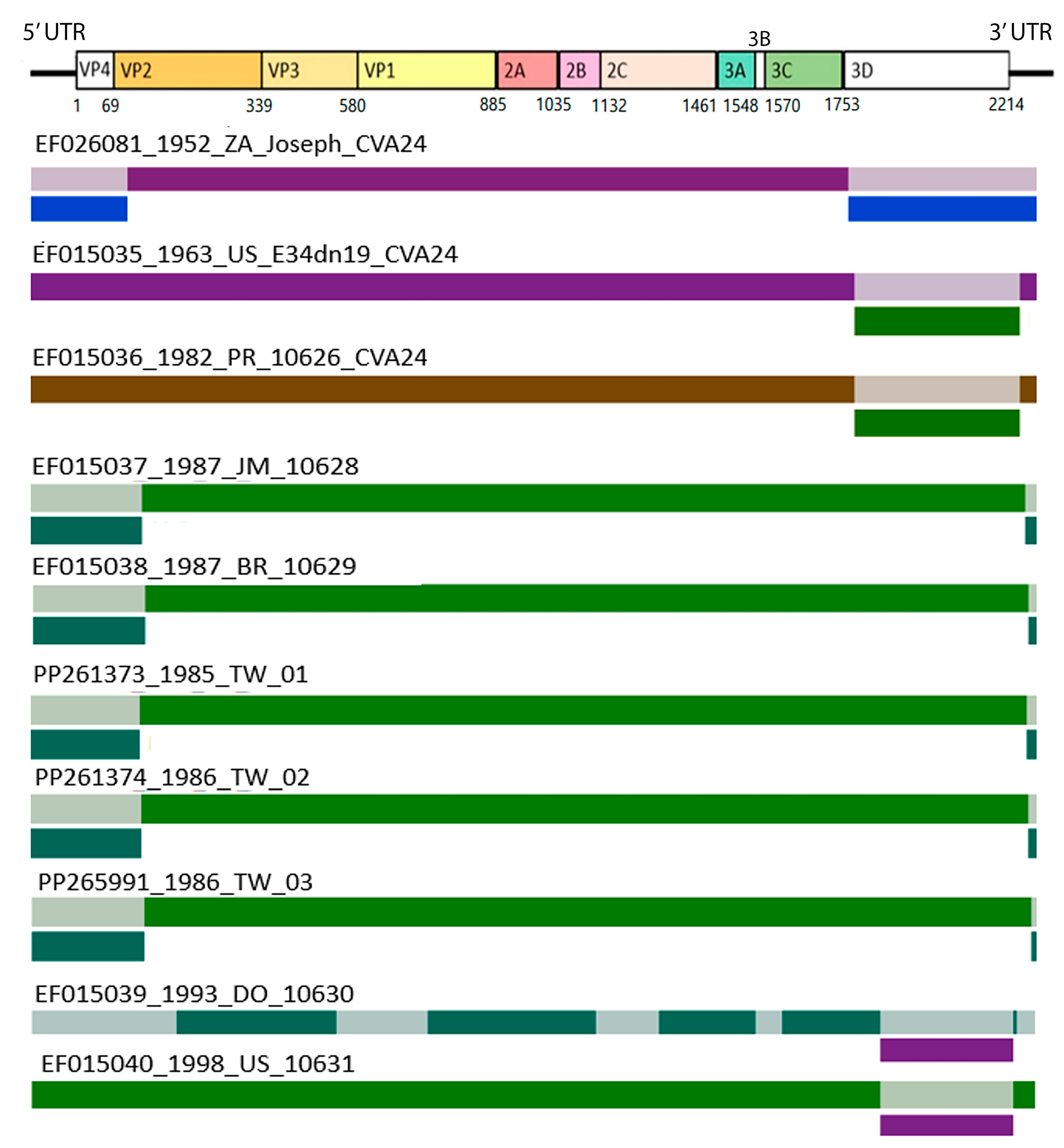
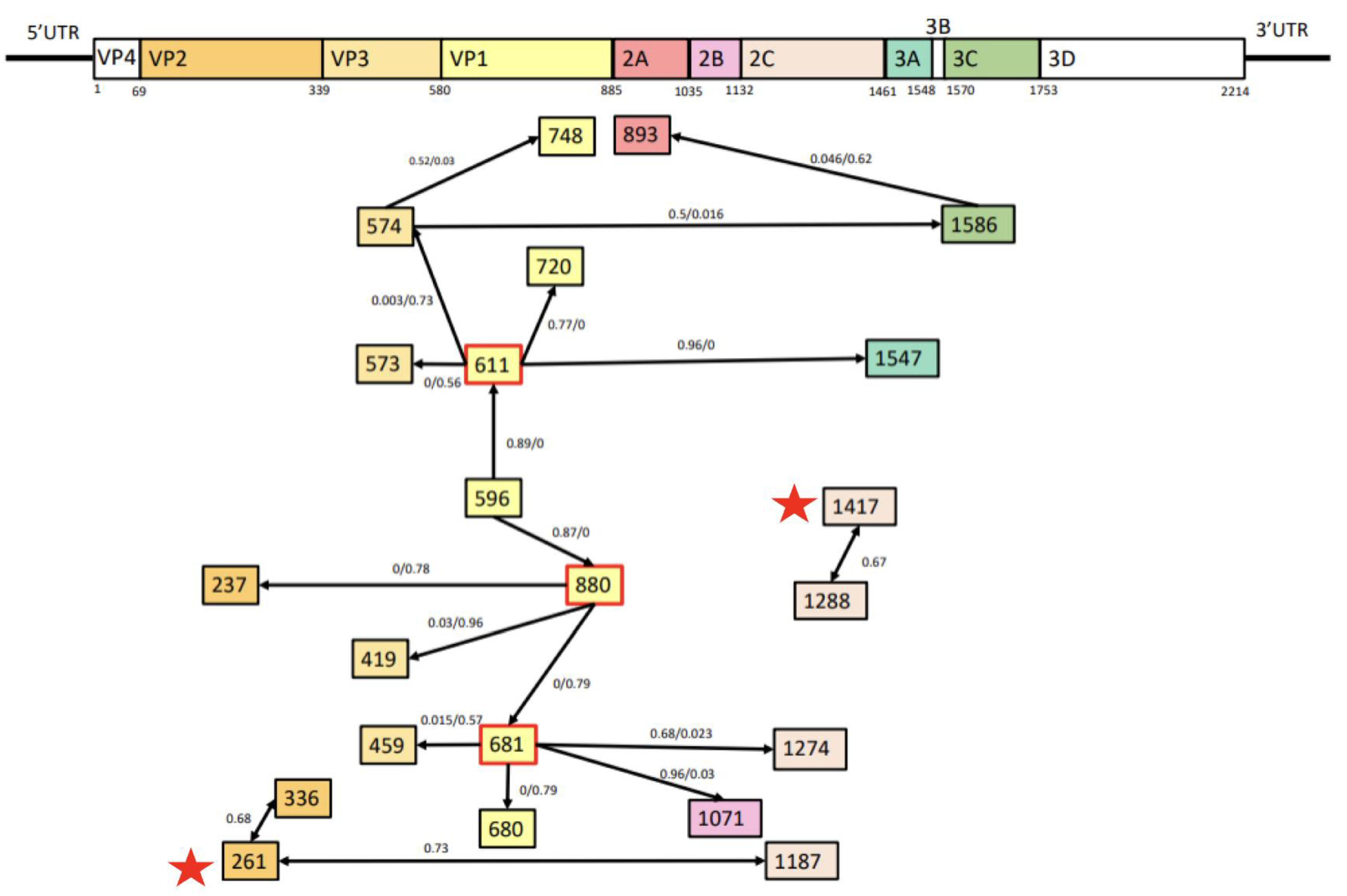
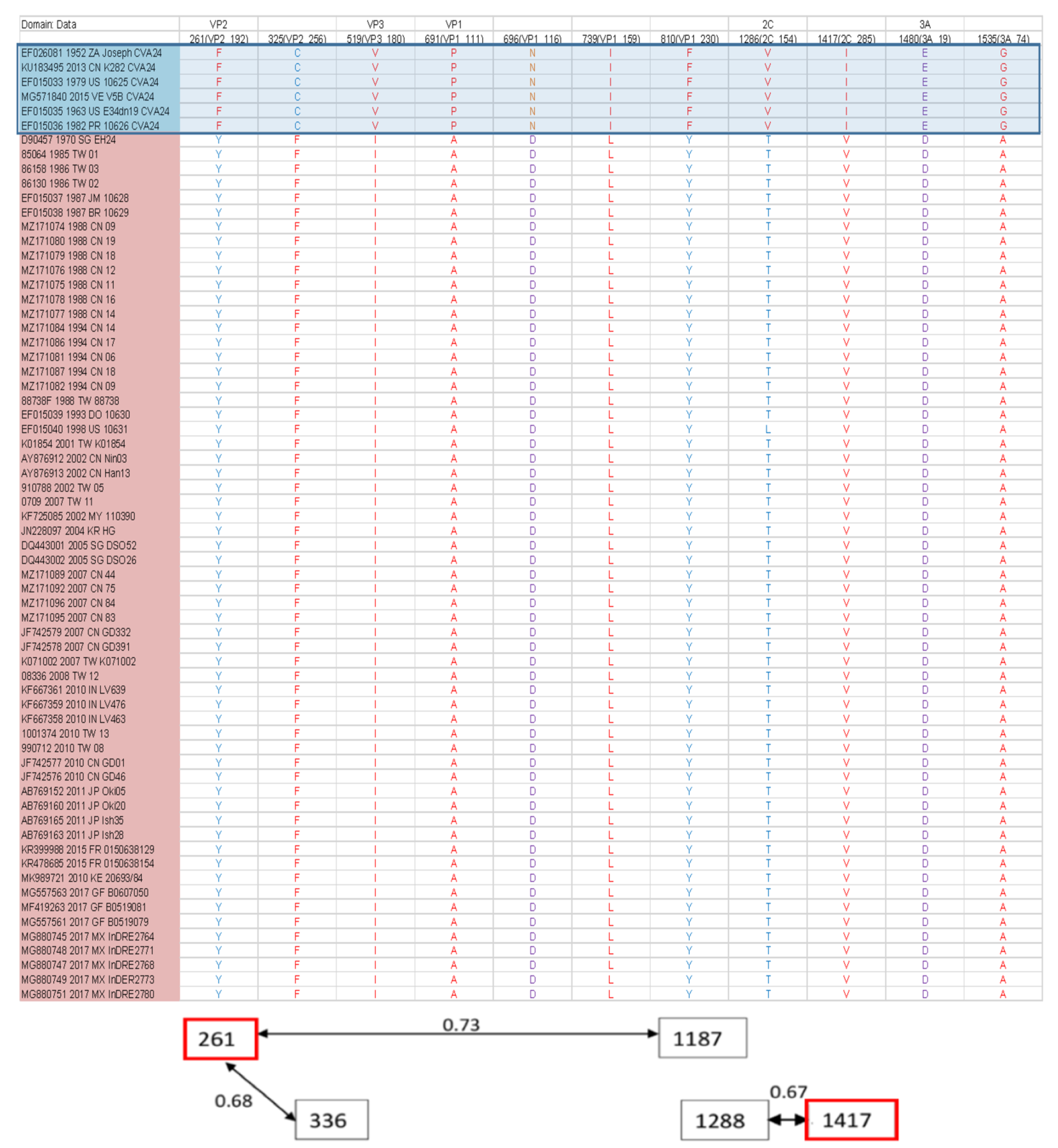
3.3. Recombination and Epistatic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, B. Universal PCR Primers Are Critical for Direct Sequencing-Based Enterovirus Genotyping. J. Clin. Microbiol. 2017, 55, 339–340. [Google Scholar] [CrossRef] [PubMed]
- Laxmivandana, R.; Yergolkar, P.; Rajeshwari, M.; Chitambar, S.D. Genomic characterization of coxsackievirus type A24 strains associated with acute flaccid paralysis and rarely identified Hopkins syndrome. Arch. Virol. 2014, 159, 3125–3129. [Google Scholar] [CrossRef] [PubMed]
- Murphy, O.C.; Messacar, K.; Benson, L.; Bove, R.; Carpenter, J.L.; Crawford, T.; Dean, J.; DeBiasi, R.; Desai, J.; Elrick, M.J.; et al. Acute flaccid myelitis: Cause, diagnosis, and management. Lancet 2021, 397, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, J.; Zhang, H.; Guo, C.; Xia, L.; Yang, F.; Yang, H.; Yang, Q.; Yang, Z.; Ma, S. Complete genome analysis of coxsackievirus A24 isolated in Yunnan, China, in 2013. Arch. Virol. 2016, 161, 1705–1709. [Google Scholar] [CrossRef] [PubMed]
- Yen, Y.C.; Chu, P.H.; Lu, P.L.; Lin, Y.C.; Shi, Y.Y.; Chou, L.C.; Wang, C.F.; Lin, Y.Y.; Su, H.J.; Lin, C.C.; et al. Phylodynamic Characterization of an Ocular-Tropism Coxsackievirus A24 Variant. PLoS ONE 2016, 11, e0160672. [Google Scholar] [CrossRef] [PubMed]
- Ghazali, O.; Chua, K.B.; Ng, K.P.; Hooi, P.S.; Pallansch, M.A.; Oberste, M.S.; Chua, K.H.; Mak, J.W. An outbreak of acute haemorrhagic conjunctivitis in Melaka, Malaysia. Singap. Med. J. 2003, 44, 511–516. [Google Scholar]
- Fonseca, M.C.; Pupo-Merino, M.; Garcia-Gonzalez, L.A.; Mune, M.; Resik, S.; Norder, H.; Sarmiento, L. Molecular Characterization of Coxsackievirus A24v from Feces and Conjunctiva Reveals Epidemiological Links. Microorganisms 2021, 9, 531. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Lin, X.J.; Ji, F.; Li, Y.; Wang, S.T.; Liu, Y.; Tao, Z.X.; Xu, A.Q. Evolutionary phylogeography reveals novel genotypes of coxsackievirus A24 variant and updates the spatiotemporal dynamics in the population with acute hemorrhagic conjunctivitis. Int. J. Infect. Dis. 2022, 124, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Baggen, J.; Hurdiss, D.L.; Zocher, G.; Mistry, N.; Roberts, R.W.; Slager, J.J.; Guo, H.; van Vliet, A.L.W.; Wahedi, M.; Benschop, K.; et al. Role of enhanced receptor engagement in the evolution of a pandemic acute hemorrhagic conjunctivitis virus. Proc. Natl. Acad. Sci. USA 2018, 115, 397–402. [Google Scholar] [CrossRef]
- Huang, H.W.; Chu, P.H.; Pan, C.H.; Wang, C.F.; Lin, C.C.; Lu, P.L.; Chen, Y.S.; Shi, Y.Y.; Su, H.J.; Chou, L.C.; et al. Evolutionary histories of coxsackievirus B5 and swine vesicular disease virus reconstructed by phylodynamic and sequence variation analyses. Sci. Rep. 2018, 8, 8821. [Google Scholar] [CrossRef]
- Huang, H.W.; Chen, Y.S.; Chen, J.Y.; Lu, P.L.; Lin, Y.C.; Chen, B.C.; Chou, L.C.; Wang, C.F.; Su, H.J.; Huang, Y.C.; et al. Phylodynamic reconstruction of the spatiotemporal transmission and demographic history of coxsackievirus B2. BMC Bioinform. 2015, 16, 302. [Google Scholar] [CrossRef] [PubMed]
- Dultz, G.; Srikakulam, S.K.; Konetschnik, M.; Shimakami, T.; Doncheva, N.T.; Dietz, J.; Sarrazin, C.; Biondi, R.M.; Zeuzem, S.; Tampé, R.; et al. Epistatic interactions promote persistence of NS3-Q80K in HCV infection by compensating for protein folding instability. J. Biol. Chem. 2021, 297, 101031. [Google Scholar] [CrossRef]
- Duan, S.; Govorkova, E.A.; Bahl, J.; Zaraket, H.; Baranovich, T.; Seiler, P.; Prevost, K.; Webster, R.G.; Webby, R.J. Epistatic interactions between neuraminidase mutations facilitated the emergence of the oseltamivir-resistant H1N1 influenza viruses. Nat. Commun. 2014, 5, 5029. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.P.; Chen, Y.; Takata, Y.; Tomida, M.W.; Lin, K.; Kirk, J.S.; Simper, M.S.; Mikulec, C.D.; Rundhaug, J.E.; Fischer, S.M.; et al. Systematic evaluation of RNA-Seq preparation protocol performance. BMC Genom. 2019, 20, 571. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, H.; Moreno, K.; Lima, I.A.B.; Santos, C.S.; Costa, B.G.G.; de Almeida, B.L.; Dos Santos, R.A.; Francisco, M.; Sampaio, M.P.S.; de Lima, M.M.; et al. Phylogenetic Reconstructions Reveal the Circulation of a Novel Dengue Virus-1V Clade and the Persistence of a Dengue Virus-2 III Genotype in Northeast Brazil. Viruses 2023, 15, 1073. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, L.; Feng, Z.; Xu, H.; Li, F.; Shen, Y.; Zhang, D.; Liu, W.J.; Gao, G.F.; Wang, Q. Characterisation of SARS-CoV-2 variants in Beijing during 2022: An epidemiological and phylogenetic analysis. Lancet 2023, 401, 664–672. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.Y.; Ke, G.M.; Chang, C.H.; Lin, J.C.; Sun, C.Y.; Huang, W.L.; Tsai, Y.C.; Ke, L.Y.; Lin, K.H. Molecular epidemiology of coxsackie A type 24 variant in Taiwan, 2000–2007. J. Clin. Virol. 2009, 45, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.H.; Chern, C.L.; Chu, P.Y.; Chang, C.H.; Wang, H.L.; Sheu, M.M.; Huang, W.L.; Pongsuwanna, Y.; Yamamoto, S.; Yoshino, S.; et al. Genetic analysis of recent Taiwanese isolates of a variant of coxsackievirus A24. J. Med. Virol. 2001, 64, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Mirkovic, R.R.; Schmidt, N.J.; Yin-Murphy, M.; Melnick, J.L. Enterovirus etiology of the 1970 Singapore epidemic of acute conjunctivitis. Intervirology 1974, 4, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Qi, X.; Xu, K.; Ji, H.; Zhu, Y.; Tang, F.; Zhou, M. Genetic characteristics of the coxsackievirus A24 variant causing outbreaks of acute hemorrhagic conjunctivitis in Jiangsu, China, 2010. PLoS ONE 2014, 9, e86883. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.H.; Wang, H.L.; Sheu, M.M.; Huang, W.L.; Chen, C.W.; Yang, C.S.; Takeda, N.; Kato, N.; Miyamura, K.; Yamazaki, S. Molecular epidemiology of a variant of coxsackievirus A24 in Taiwan: Two epidemics caused by phylogenetically distinct viruses from 1985 to 1989. J. Clin. Microbiol. 1993, 31, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.M.; Koelle, K.; Bedford, T. Viral phylodynamics. PLoS Comput. Biol. 2013, 9, e1002947. [Google Scholar] [CrossRef]
- Fragoso-Fonseca, D.E.; Escobar-Escamilla, N.; Rodríguez-Maldonado, A.P.; Barrera-Badillo, G.; Garcés-Ayala, F.; Mendieta-Condado, E.; González-Durán, E.; Puerto, F.I.; Hernández-Rivas, L.; López-Martínez, I.; et al. Complete genome sequence of a coxsackievirus type A24 variant causing an outbreak of acute haemorrhagic conjunctivitis in southeastern Mexico in 2017. Arch. Virol. 2020, 165, 1015–1018. [Google Scholar] [CrossRef]
- Li, J.; Huang, F.; Zhang, Y.; Ji, T.; Zhu, S.; Wang, D.; Han, Z.; Xiao, J.; Si, F.; Xu, W.; et al. Molecular analysis of Coxsackievirus A24 variant isolates from three outbreaks of acute hemorrhagic conjunctivitis in 1988, 1994 and 2007 in Beijing, China. Virol. Sin. 2022, 37, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.M.; Mulkey, S.B.; Campos, J.M.; DeBiasi, R.L. Laboratory diagnosis of CNS infections in children due to emerging and re-emerging neurotropic viruses. Pediatr. Res. 2024, 95, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Miyamura, K.; Yamashita, K.; Takeda, N.; Ogino, T.; Utagawa, E.; Yamazaki, S.; Fukumura, K.; Uehara, T.; Shinjo, N. The first epidemic of acute hemorrhagic conjunctivitis due to a coxsackievirus A24 variant in Okinawa, Japan, in 1985–1986. Jpn. J. Med. Sci. Biol. 1988, 41, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Santos Ede, O.; Macedo, O.; Gomes Mde, L.; Nakauth, C.M. Acute hemorrhagic conjunctivitis caused by a variant of coxsackievirus A24, in Belém, Pará, Brazil, 1987. Rev. Inst. Med. Trop. Sao Paulo 1989, 31, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, M.C.; Pupo-Meriño, M.; García-González, L.A.; Resik, S.; Hung, L.H.; Muné, M.; Rodríguez, H.; Morier, L.; Norder, H.; Sarmiento, L. Molecular evolution of coxsackievirus A24v in Cuba over 23-years, 1986–2009. Sci. Rep. 2020, 10, 13761. [Google Scholar] [CrossRef] [PubMed]
- Brandful, J.A.; Yoshii, T.; Addy, E.T.; Adiku, T.K.; Osei-Kwasi, M.; Mingle, J.A. Epidemic acute haemorrhagic conjunctivitis due to Coxsackie virus A24 variant in Ghana. East Afr. Med. J. 1990, 67, 878–886. [Google Scholar] [PubMed]
- Smura, T.; Blomqvist, S.; Vuorinen, T.; Ivanova, O.; Samoilovich, E.; Al-Hello, H.; Savolainen-Kopra, C.; Hovi, T.; Roivainen, M. Recombination in the evolution of enterovirus C species sub-group that contains types CVA-21, CVA-24, EV-C95, EV-C96 and EV-C99. PLoS ONE 2014, 9, e94579. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Welch, J. Frequency and dynamics of recombination within different species of human enteroviruses. J. Virol. 2006, 80, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, L.; Benschop, K.S.M.; Nguyen, D.; Kamau, E.; Pajkrt, D.; Simmonds, P.; Wolthers, K.C. Recombination Analysis of Non-Poliovirus Members of the Enterovirus C Species; Restriction of Recombination Events to Members of the Same 3DPol Cluster. Viruses 2020, 12, 706. [Google Scholar] [CrossRef]
- Zhou, J.; Shi, Y.; Miao, L.; Zhang, C.; Liu, Y. Molecular epidemiology and recombination of Enterovirus A71 in mainland China from 1987 to 2017. Int. Microbiol. 2021, 24, 291–299. [Google Scholar] [CrossRef]
- Noisumdaeng, P.; Sangsiriwut, K.; Prasertsopon, J.; Klinmalai, C.; Payungporn, S.; Mungaomklang, A.; Chokephaibulkit, K.; Buathong, R.; Thitithanyanont, A.; Puthavathana, P. Complete genome analysis demonstrates multiple introductions of enterovirus 71 and coxsackievirus A16 recombinant strains into Thailand during the past decade. Emerg. Microbes Infect. 2018, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Runckel, C.; Westesson, O.; Andino, R.; DeRisi, J.L. Identification and manipulation of the molecular determinants influencing poliovirus recombination. PLoS Pathog. 2013, 9, e1003164. [Google Scholar] [CrossRef] [PubMed]
- Chrisman, B.S.; Paskov, K.; Stockham, N.; Tabatabaei, K.; Jung, J.Y.; Washington, P.; Varma, M.; Sun, M.W.; Maleki, S.; Wall, D.P. Indels in SARS-CoV-2 occur at template-switching hotspots. BioData Min. 2021, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Souii, A.; Ben M’hadheb-Gharbi, M.; Gharbi, J. Role of RNA structure motifs in IRES-dependent translation initiation of the coxsackievirus B3: New insights for developing live-attenuated strains for vaccines and gene therapy. Mol. Biotechnol. 2013, 55, 179–202. [Google Scholar] [CrossRef]
- Muslin, C.; Mac Kain, A.; Bessaud, M.; Blondel, B.; Delpeyroux, F. Recombination in Enteroviruses, a Multi-Step Modular Evolutionary Process. Viruses 2019, 11, 859. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Liu, X.; Wu, Y.; He, Y.; Zheng, H.; Liu, H.; Liu, Q. Identification of a neutralizing linear epitope within the VP1 protein of coxsackievirus A10. Virol. J. 2022, 19, 203. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.H.; Chong, W.L.; Lee, V.S.; Abdullah, S.; Jasni, K.; Suarni, S.Q.; Perera, D.; Sam, I.C.; Chan, Y.F. Substitution of Coxsackievirus A16 VP1 BC and EF Loop Altered the Protective Immune Responses in Chimera Enterovirus A71. Vaccines 2023, 11, 1363. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Peng, W.; Ren, J.; Hu, Z.; Xu, J.; Lou, Z.; Li, X.; Yin, W.; Shen, X.; Porta, C.; et al. A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71. Nat. Struct. Mol. Biol. 2012, 19, 424–429. [Google Scholar] [CrossRef]
- Chen, D.; Duggan, C.; Texada, D.E.; Reden, T.B.; Kooragayala, L.M.; Langford, M.P. Immunogenicity of enterovirus 70 capsid protein VP1 and its non-overlapping N- and C-terminal fragments. Antivir. Res. 2005, 66, 111–117. [Google Scholar] [CrossRef]
- Füzik, T.; Moravcová, J.; Kalynych, S.; Plevka, P. Structure of Human Enterovirus 70 and Its Inhibition by Capsid-Binding Compounds. J. Virol. 2022, 96, e0060422. [Google Scholar] [CrossRef]
- Liu, Y.; Fu, C.; Wu, S.; Chen, X.; Shi, Y.; Zhou, B.; Zhang, L.; Zhang, F.; Wang, Z.; Zhang, Y.; et al. A novel finding for enterovirus virulence from the capsid protein VP1 of EV71 circulating in mainland China. Virus Genes 2014, 48, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.X.; Jin, W.P.; Wei, Z.N.; Lv, S.Y.; Wang, M.J.; Meng, S.L.; Guo, J.; Wang, Z.J.; Shen, S. Identification of specific and shared epitopes at the extreme N-terminal VP1 of Coxsackievirus A4, A2 and A5 by monoclonal antibodies. Virus Res. 2023, 328, 199074. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, C.-C.; Chu, P.-H.; Huang, H.-W.; Ke, G.-M.; Ke, L.-Y.; Chu, P.-Y. Phylodynamic and Epistatic Analysis of Coxsackievirus A24 and Its Variant. Viruses 2024, 16, 1267. https://doi.org/10.3390/v16081267
Cheng C-C, Chu P-H, Huang H-W, Ke G-M, Ke L-Y, Chu P-Y. Phylodynamic and Epistatic Analysis of Coxsackievirus A24 and Its Variant. Viruses. 2024; 16(8):1267. https://doi.org/10.3390/v16081267
Chicago/Turabian StyleCheng, Chia-Chi, Pei-Huan Chu, Hui-Wen Huang, Guan-Ming Ke, Liang-Yin Ke, and Pei-Yu Chu. 2024. "Phylodynamic and Epistatic Analysis of Coxsackievirus A24 and Its Variant" Viruses 16, no. 8: 1267. https://doi.org/10.3390/v16081267
APA StyleCheng, C.-C., Chu, P.-H., Huang, H.-W., Ke, G.-M., Ke, L.-Y., & Chu, P.-Y. (2024). Phylodynamic and Epistatic Analysis of Coxsackievirus A24 and Its Variant. Viruses, 16(8), 1267. https://doi.org/10.3390/v16081267