
Study of Endogenous Viruses in the Strawberry Plants

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Viral Homologous Sequence Search
2.3. Alignment of Viral Genomes with the Strawberry Genomes
3. Results
3.1. Homologous Sequence Search
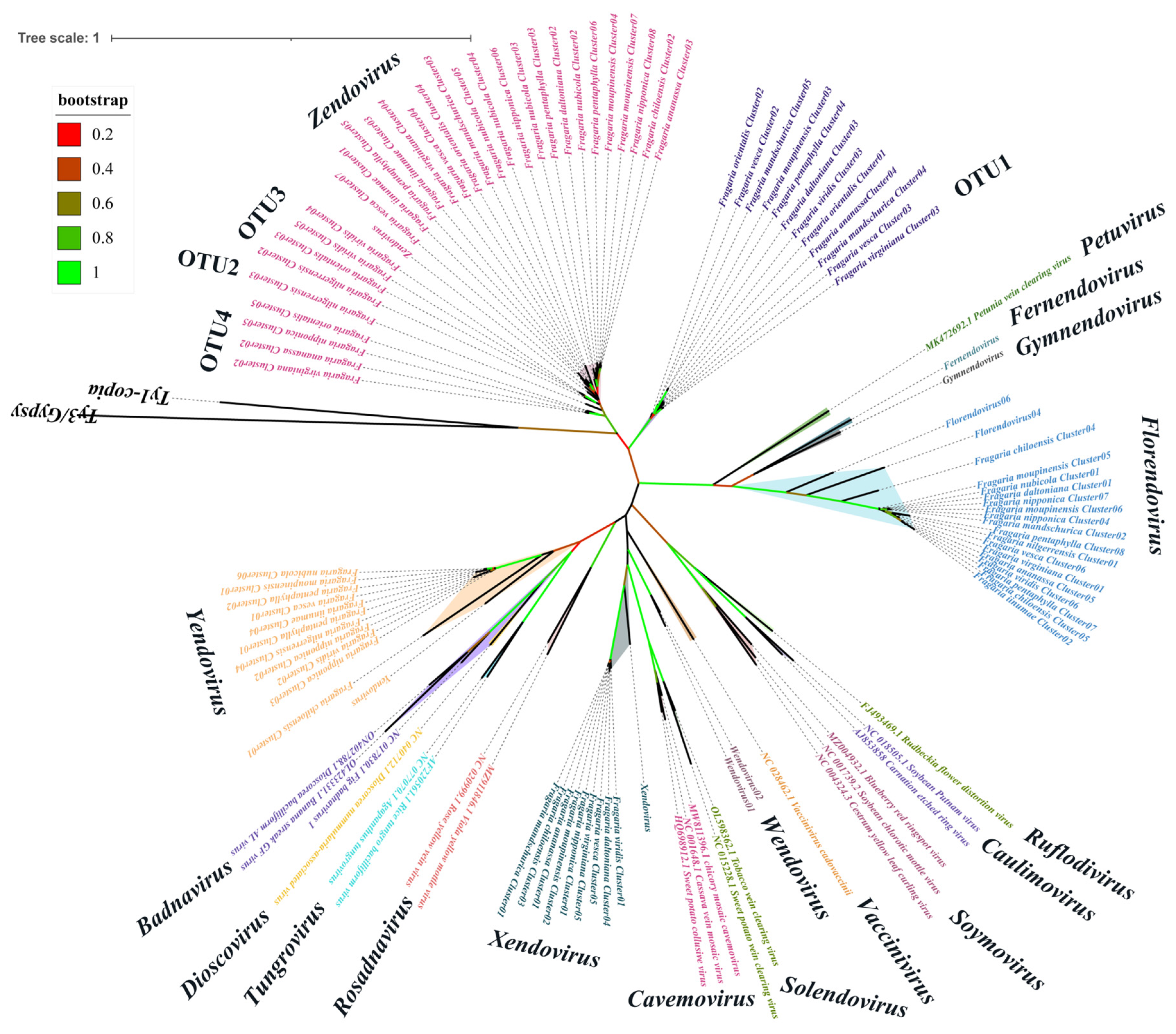
3.1.1. Integration of Virus-like Sequences in the Genomes of Fragaria and Its Related Genera
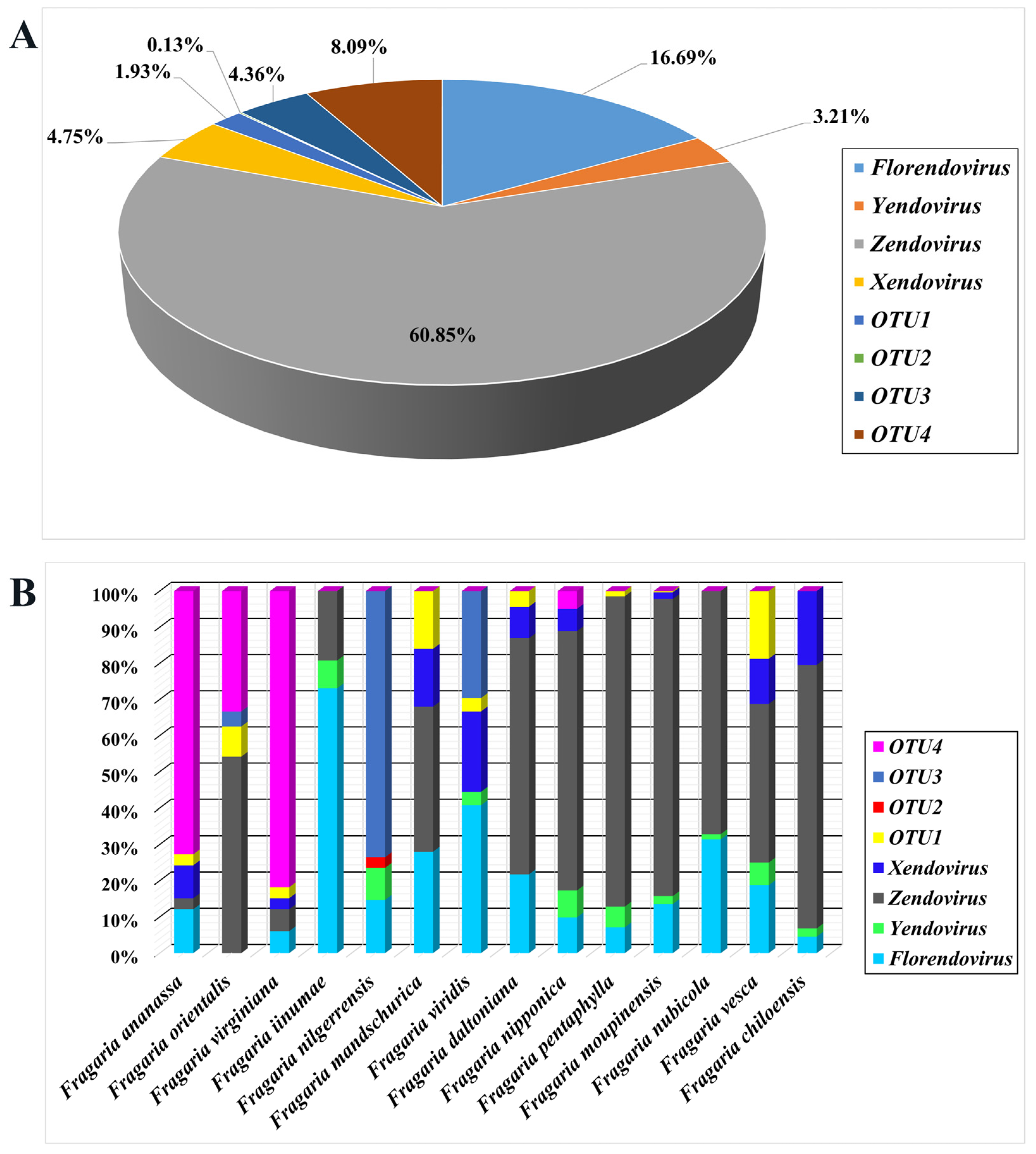
3.1.2. Integration and Classification of EPRVs in the Strawberry Genomes
3.1.3. Diversity and Differences of EPRVs in Strawberry Genomes
3.2. Genome Alignment Results
3.2.1. Virus-like Sequences and Differences in the Genomes of Strawberries
3.2.2. Comparison of Virus-like Sequences in Different Plant Genomes
3.2.3. Integration and Classification of EPRVs in the Strawbery Genomes (Identification of EPRVs in the Strawberry Genomes)
3.3. Comparison of the Two Study Methods
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chu, H.; Jo, Y.; Cho, W.K. Evolution of endogenous non-retroviral genes integrated into plant genomes. Curr. Plant Biol. 2014, 1, 55–59. [Google Scholar] [CrossRef]
- Holmes, E.C. The Evolution of Endogenous Viral Elements. Cell Host Microbe 2011, 10, 368–377. [Google Scholar] [CrossRef]
- Etienne, L. “Paleovirology”: Looking back in time to better understand and control modern viral infections. Virologie 2017, 21, 245–246. [Google Scholar] [PubMed]
- Emerman, M.; Malik, H.S. Paleovirology-Modern Consequences of Ancient Viruses. PLoS Biol. 2010, 8, e1000301. [Google Scholar] [CrossRef]
- Feschotte, C.; Gilbert, C. Endogenous viruses: Insights into viral evolution and impact on host biology. Nat. Rev. Genet. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Umber, M.; Filloux, D.; Muller, E.; Laboureau, N.; Galzi, S.; Roumagnac, P.; Iskra-Caruana, M.-L.; Pavis, C.; Teycheney, P.-Y.; Seal, S.E. The genome of African yam (Dioscorea cayenensis-rotundata complex) hosts endogenous sequences from four distinct badnavirus species. Mol. Plant Pathol. 2014, 15, 790–801. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Fukuhara, T.; Kitazawa, H.; Kormelink, R. Virus Latency and the Impact on Plants. Front. Microbiol. 2019, 10, 2764. [Google Scholar] [CrossRef] [PubMed]
- Boutanaev, A.M.; Nemchinov, L.G. Genome-wide identification of endogenous viral sequences in alfalfa (Medicago sativa L.). Virol. J. 2021, 18, 185. [Google Scholar] [CrossRef]
- Bejarano, E.; Khashoggi, A.; Witty, M.; Lichtenstein, C. Integration of Multiple Repeats of Geminiviral DNA into the Nuclear Genome of Tobacco during Evolution. Proc. Natl. Acad. Sci. USA 1996, 93, 759–764. [Google Scholar] [CrossRef]
- Tomás, C.d.; Vicient, C.M. Genome-wide identification of Reverse Transcriptase domains of recently inserted endogenous plant pararetrovirus (Caulimoviridae). Front. Plant Sci. 2022, 13, 1011565. [Google Scholar] [CrossRef]
- Teycheney, P.-Y.; Geering, A.D.W.; Dasgupta, I.; Hull, R.; Kreuze, J.F.; Lockhart, B.; Muller, E.; Olszewski, N.; Pappu, H.; Pooggin, M.M.; et al. ICTV Virus Taxonomy Profile: Caulimoviridae. J. Gen. Virol. 2020, 101, 1025–1026. [Google Scholar] [CrossRef] [PubMed]
- Geering, A.D.W.; Maumus, F.; Copetti, D.; Choisne, N.; Zwickl, D.J.; Zytnicki, M.; McTaggart, A.R.; Scalabrin, S.; Vezzulli, S.; Wing, R.A.; et al. Endogenous florendoviruses are major components of plant genomes and hallmarks of virus evolution. Nat. Commun. 2014, 5, 5269. [Google Scholar] [CrossRef] [PubMed]
- Sukal, A.C.; Kidanemariam, D.B.; Dale, J.L.; Harding, R.M.; James, A.P. Characterization of a novel member of the family Caulimoviridae infecting Dioscorea nummularia in the Pacific, which may represent a new genus of dsDNA plant viruses. PLoS ONE 2018, 13, e0203038. [Google Scholar] [CrossRef]
- Valli, A.A.; Gonzalo-Magro, I.; Sanchez, D.H. Rearranged Endogenized Plant Pararetroviruses as Evidence of Heritable RNA-based Immunity. Mol. Biol. Evol. 2022, 40, msac240. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, B.E.; Menke, J.; Dahal, G.; Olszewski, N.E. Characterization and genomic analysis of tobacco vein clearing virus, a plant pararetrovirus that is transmitted vertically and related to sequences integrated in the host genome. J. Gen. Virol. 2000, 81, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Ndowora, T.; Dahal, G.; LaFleur, D.; Harper, G.; Hull, R.; Olszewski, N.E.; Lockhart, B. Evidence That Badnavirus Infection in Musa Can Originate from Integrated Pararetroviral Sequences. Virology 1999, 255, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, F.; Carreel, F.; Jenny, C.; Lockhart, B.E.L.; Iskra-Caruana, M.L. Identification of genetic markers linked to banana streak disease expression in inter-specific Musa hybrids. Theor. Appl. Genet. 2003, 106, 594–598. [Google Scholar] [CrossRef]
- Kunii, M.; Kanda, M.; Nagano, H.; Uyeda, I.; Kishima, Y.; Sano, Y. Reconstruction of putative DNA virus from endogenous rice tungro bacilliform virus-like sequences in the rice genome: Implications for integration and evolution. BMC Genom. 2004, 5, 80. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.L.; Liu, R.F.; Koyanagi, K.O.; Kishima, Y. Rice genomes recorded ancient pararetrovirus activities: Virus genealogy and multiple origins of endogenization during rice speciation. Virology 2014, 471, 141–152. [Google Scholar] [CrossRef]
- Chen, S.; Zheng, H.; Kishima, Y. Genomic fossils reveal adaptation of non-autonomous pararetroviruses driven by concerted evolution of noncoding regulatory sequences. PLoS Pathog. 2017, 13, e1006413. [Google Scholar] [CrossRef]
- Richert-Pöggeler, K.R.; Noreen, F.; Schwarzacher, T.; Harper, G.; Hohn, T. Induction of infectious petunia vein clearing (pararetro) virus from endogenous provirus in petunia. Embo J. 2003, 22, 4836–4845. [Google Scholar] [CrossRef] [PubMed]
- Staginnus, C.; Gregor, W.; Mette, M.F.; Teo, C.; Borroto-Fernández, E.; Machado, M.L.; Matzke, M.; Schwarzacher, T. Endogenous pararetroviral sequences in tomato (Solanum lycopersicum) and related species. BMC Plant Biol. 2007, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.N.; Harper, G.; Heslop-Harrison, J.S. Characterisation of pararetrovirus-like sequences in the genome of potato (Solanum tuberosum). Cytogenet. Genome Res. 2005, 110, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Gambley, C.F.; Geering, A.D.W.; Steele, V.; Thomas, J.E. Identification of viral and non-viral reverse transcribing elements in pineapple (Ananas comosus), including members of two new badnavirus species. Arch. Virol. 2008, 153, 1599–1604. [Google Scholar] [CrossRef] [PubMed]
- Pahalawatta, V.; Druffel, K.; Pappu, H. A new and distinct species in the genus Caulimovirus exists as an endogenous plant pararetroviral sequence in its host, Dahlia variabilis. Virology 2008, 376, 253–257. [Google Scholar] [CrossRef]
- Almeyda, C.V.; Raikhy, G.; Pappu, H.R. Characterization and comparative analysis of promoters from three plant pararetroviruses associated with Dahlia (Dahlia variabilis). Virus Genes 2015, 51, 96–104. [Google Scholar] [CrossRef]
- Su, L.; Gao, S.; Huang, Y.W.; Ji, C.Q.; Wang, D.; Ma, Y.; Fang, R.X.; Chen, X.Y. Complete genomic sequence of Dracaena mottle virus, a distinct badnavirus. Virus Genes 2007, 35, 423–429. [Google Scholar] [CrossRef]
- Laney, A.G.; Hassan, M.; Tzanetakis, I.E. An Integrated Badnavirus Is Prevalent in Fig Germplasm. Phytopathology 2012, 102, 1182–1189. [Google Scholar] [CrossRef]
- Marcon, H.S.; Costa-Silva, J.; Lorenzetti, A.P.R.; Marino, C.L.; Domingues, D.S. Genome-wide analysis of EgEVE_1, a transcriptionally active endogenous viral element associated to small RNAs in Eucalyptus genomes. Genet. Mol. Biol. 2017, 40, 217–225. [Google Scholar] [CrossRef]
- Villacreses, J.; Rojas-Herrera, M.; Sánchez, C.; Hewstone, N.; Undurraga, S.; Alzate, J.; Manque, P.; Maracaja-Coutinho, V.; Polanco, V. Deep Sequencing Reveals the Complete Genome and Evidence for Transcriptional Activity of the First Virus-Like Sequences Identified in Aristotelia chilensis (Maqui Berry). Viruses 2015, 7, 1685–1699. [Google Scholar] [CrossRef]
- Yu, H.; Wang, X.; Lu, Z.; Xu, Y.; Deng, X.; Xu, Q. Endogenous pararetrovirus sequences are widely present in Citrinae genomes. Virus Res. 2019, 262, 48–53. [Google Scholar] [CrossRef]
- Diaz-Lara, A.; Mosier, N.J.; Stevens, K.; Keller, K.E.; Martin, R.R. Evidence of Rubus Yellow Net Virus Integration into the Red Raspberry Genome. Cytogenet. Genome Res. 2020, 160, 329–334. [Google Scholar] [CrossRef]
- Bertsch, C.; Beuve, M.; Dolja, V.V.; Wirth, M.; Pelsy, F.; Herrbach, E.; Lemaire, O. Retention of the virus-derived sequences in the nuclear genome of grapevine as a potential pathway to virus resistance. Biol. Direct 2009, 4, 21. [Google Scholar] [CrossRef]
- Serfraz, S.; Sharma, V.; Maumus, F.; Aubriot, X.; Geering, A.D.W.; Teycheney, P.-Y. Insertion of Badnaviral DNA in the Late Blight Resistance Gene (R1a) of Brinjal Eggplant (Solanum melongena). Front. Plant Sci. 2021, 12, 683681. [Google Scholar] [CrossRef] [PubMed]
- Mushegian, A.R.; Elena, S.F. Evolution of plant virus movement proteins from the 30K superfamily and of their homologs integrated in plant genomes. Virology 2015, 476, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Diop, S.I.; Geering, A.D.W.; Alfama-Depauw, F.; Loaec, M.; Teycheney, P.-Y.; Maumus, F. Tracheophyte genomes keep track of the deep evolution of the Caulimoviridae. Sci. Rep. 2018, 8, 572. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Q.; Fu, Y.P.; Xie, J.T.; Cheng, J.S.; Ghabrial, S.A.; Li, G.Q.; Yi, X.H.; Jiang, D.H. Discovery of Novel dsRNA Viral Sequences by In Silico Cloning and Implications for Viral Diversity, Host Range and Evolution. PLoS ONE 2012, 7, e42147. [Google Scholar] [CrossRef] [PubMed]
- Chiba, S.; Kondo, H.; Tani, A.; Saisho, D.; Sakamoto, W.; Kanematsu, S.; Suzuki, N. Widespread Endogenization of Genome Sequences of Non-Retroviral RNA Viruses into Plant Genomes. PLoS Pathog. 2011, 7, e1002146. [Google Scholar] [CrossRef]
- Kondo, H.; Hirano, S.; Chiba, S.; Andika, I.B.; Hirai, M.; Maeda, T.; Tamada, T. Characterization of burdock mottle virus, a novel member of the genus Benyvirus, and the identification of benyvirus-related sequences in the plant and insect genomes. Virus Res. 2013, 177, 75–86. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Erazo-Garcia, M.P.; Aylward, F.O. Endogenous giant viruses contribute to intraspecies genomic variability in the model green alga Chlamydomonas reinhardtii. Virus Evol. 2022, 8, veac102. [Google Scholar] [CrossRef]
- Nibert, M.L.; Vong, M.; Fugate, K.K.; Debat, H.J. Evidence for contemporary plant mitoviruses. Virology 2018, 518, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Maumus, F.; Epert, A.; Nogué, F.; Blanc, G. Plant genomes enclose footprints of past infections by giant virus relatives. Nat. Commun. 2014, 5, 4268. [Google Scholar] [CrossRef] [PubMed]
- Côte, F.X.; Galzi, S.; Folliot, M.; Lamagnère, Y.; Teycheney, P.-Y.; Iskra-Caruana, M.-L. Micropropagation by tissue culture triggers differential expression of infectious endogenous Banana streak virus sequences (eBSV) present in the B genome of natural and synthetic interspecific banana plantains. Mol. Plant Pathol. 2010, 11, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Noreen, F.; Akbergenov, R.; Hohn, T.; Richert-Pöggeler, K.R. Distinct expression of endogenous Petunia vein clearing virus and the DNA transposon dTph1 in two Petunia hybrida lines is correlated with differences in histone modification and siRNA production. Plant J. 2007, 50, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Hull, R.; Harper, G.; Lockhart, B. Viral sequences integrated into plant genomes. Trends Plant Sci. 2000, 5, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Aiewsakun, P.; Katzourakis, A. Endogenous viruses: Connecting recent and ancient viral evolution. Virology 2015, 479–480, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Filloux, D.; Murrell, S.; Koohapitagtam, M.; Golden, M.; Julian, C.; Galzi, S.; Uzest, M.; Rodier-Goud, M.; D’Hont, A.; Vernerey, M.S.; et al. The genomes of many yam species contain transcriptionally active endogenous geminiviral sequences that may be functionally expressed. Virus Evol. 2015, 1, vev002. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Q.; Fu, Y.P.; Jiang, D.H.; Li, G.Q.; Xie, J.T.; Cheng, J.S.; Peng, Y.L.; Ghabrial, S.A.; Yi, X.H. Widespread Horizontal Gene Transfer from Double-Stranded RNA Viruses to Eukaryotic Nuclear Genomes. J. Virol. 2010, 84, 11876–11887. [Google Scholar] [CrossRef] [PubMed]
- Vassilieff, H.; Geering, A.D.W.; Choisne, N.; Teycheney, P.Y.; Maumus, F. Endogenous Caulimovirids: Fossils, Zombies, and Living in Plant Genomes. Biomolecules 2023, 13, 1069. [Google Scholar] [CrossRef]
- Sun, J.; Sun, R.; Liu, H.B.; Chang, L.L.; Li, S.T.; Zhao, M.Z.; Shennan, C.; Lei, J.J.; Dong, J.; Zhong, C.A.F.; et al. Complete chloroplast genome sequencing of ten wild Fragaria species in China provides evidence for phylogenetic evolution of Fragaria. Genomics 2021, 113, 1170–1179. [Google Scholar] [CrossRef]
- Li, C.X.; Cai, C.A.; Tao, Y.T.; Sun, Z.S.; Jiang, M.; Chen, L.X.; Li, J.M. Variation and Evolution of the Whole Chloroplast Genomes of Fragaria spp. (Rosaceae). Front. Plant Sci. 2021, 12, 754209. [Google Scholar] [CrossRef] [PubMed]
- Davalos, N.; Wells, H.; Ali, A. Preliminary Study of Viruses Infecting Strawberry (Fragaria spp.) in the Midsouthern United States. Plant Health Prog. 2022, 23, 7–13. [Google Scholar] [CrossRef]
- Koloniuk, I.; Matyášová, A.; Brázdová, S.; Veselá, J.; Přibylová, J.; Fránová, J.; Elena, S.F. Transmission of Diverse Variants of Strawberry Viruses Is Governed by a Vector Species. Viruses 2022, 14, 1362. [Google Scholar] [CrossRef] [PubMed]
- Torrico, A.K.; Salazar, S.M.; Kirschbaum, D.S.; Conci, V.C. Yield losses of asymptomatic strawberry plants infected with Strawberry mild yellow edge virus. Eur. J. Plant Pathol. 2017, 150, 983–990. [Google Scholar] [CrossRef]
- Thompson, J.R.; Wetzel, S.; Klerks, M.M.; Vašková, D.; Schoen, C.D.; Špak, J.; Jelkmann, W. Multiplex RT-PCR detection of four aphid-borne strawberry viruses in Fragaria spp. in combination with a plant mRNA specific internal control. J. Virol. Methods 2003, 111, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Lee, T.; Cheng, C.H.; Buble, K.; Zheng, P.; Yu, J.; Humann, J.; Ficklin, S.P.; Gasic, K.; Scott, K.; et al. 15 years of GDR: New data and functionality in the Genome Database for Rosaceae. Nucleic Acids Res. 2019, 47, D1137–D1145. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Sipos, B.; Zhao, L.Y. SeqKit2: A Swiss army knife for sequence and alignment processing. iMeta 2024, 3. [Google Scholar] [CrossRef]
- Fu, L.M.; Niu, B.F.; Zhu, Z.W.; Wu, S.T.; Li, W.Z. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.E.; Manivannan, A.; Lee, S.Y.; Han, K.; Yeum, J.G.; Jo, J.; Kim, J.; Rho, I.; Lee, Y.R.; Lee, E.S.; et al. Chromosome Level Assembly of Homozygous Inbred Line ‘Wongyo 3115′ Facilitates the Construction of a High-Density Linkage Map and Identification of QTLs Associated With Fruit Firmness in Octoploid Strawberry (Fragaria x ananassa). Front. Plant Sci. 2021, 12, 696229. [Google Scholar] [CrossRef]
- Sharma, V.; Lefeuvre, P.; Roumagnac, P.; Filloux, D.; Teycheney, P.-Y.; Martin, D.P.; Maumus, F. Large-scale survey reveals pervasiveness and potential function of endogenous geminiviral sequences in plants. Virus Evol. 2020, 6, veaa071. [Google Scholar] [CrossRef]
- Mushegian, A.; Shipunov, A.; Elena, S.F. Changes in the composition of the RNA virome mark evolutionary transitions in green plants. BMC Biol. 2016, 14, 68. [Google Scholar] [CrossRef]
- Maredza, A.T.; Allie, F.; Plata, G.; Rey, M.E.C. Sequences enhancing cassava mosaic disease symptoms occur in the cassava genome and are associated with South African cassava mosaic virus infection. Mol. Genet. Genom. 2015, 291, 1467–1485. [Google Scholar] [CrossRef] [PubMed]
- Staginnus, C.; Richert-Pöggeler, K.R. Endogenous pararetroviruses: Two-faced travelers in the plant genome. Trends Plant Sci. 2006, 11, 485–491. [Google Scholar] [CrossRef]
- Gong, Z.; Han, G.-Z. Euphyllophyte Paleoviruses Illuminate Hidden Diversity and Macroevolutionary Mode of Caulimoviridae. J. Virol. 2018, 92, e02043-17. [Google Scholar] [CrossRef] [PubMed]
- Romeyer Dherbey, J.; Parab, L.; Gallie, J.; Bertels, F. Stepwise Evolution of E. coli C and PhiX174 Reveals Unexpected Lipopolysaccharide (LPS) Diversity. Mol. Biol. Evol. 2023, 40, msad154. [Google Scholar] [CrossRef]
- Labrie, S.J.; Dupuis, M.E.; Tremblay, D.M.; Plante, P.L.; Corbeil, J.; Moineau, S. A New Microviridae Phage Isolated from a Failed Biotechnological Process Driven by Escherichia coli. Appl. Environ. Microbiol. 2014, 80, 6992–7000. [Google Scholar] [CrossRef]
- Hirakawa, H.; Shirasawa, K.; Kosugi, S.; Tashiro, K.; Nakayama, S.; Yamada, M.; Kohara, M.; Watanabe, A.; Kishida, Y.; Fujishiro, T.; et al. Dissection of the Octoploid Strawberry Genome by Deep Sequencing of the Genomes of Fragaria Species. DNA Res. 2014, 21, 169–181. [Google Scholar] [CrossRef]
- Bonneau, P.; Hogue, R.; Tellier, S.; Fournier, V. Evaluation of Various Sources of Viral Infection in Strawberry Fields of Quebec, Canada. J. Econ. Entomol. 2019, 112, 2577–2583. [Google Scholar] [CrossRef]
- Hohn, T. Plant pararetroviruses: Interactions of cauliflower mosaic virus with plants and insects. Curr. Opin. Virol. 2013, 3, 629–638. [Google Scholar] [CrossRef]
- Lutz, L.; Raikhy, G.; Leisner, S.M. Cauliflower mosaic virus major inclusion body protein interacts with the aphid transmission factor, the virion-associated protein, and gene VII product. Virus Res. 2012, 170, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Mette, M.F.; Kanno, T.; Aufsatz, W.; Jakowitsch, J.; van der Winden, J.; Matzke, M.A.; Matzke, A.J.M. Endogenous viral sequences and their potential contribution to heritable virus resistance in plants. Embo J. 2002, 21, 461–469. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position in the Strawberry Genomes | Position in the Virus Genomes | Similarity | Length | Functional Domain | Virus Genome Accession No |
|---|---|---|---|---|---|
| Fragaria mandshurica chr5 18,447,245–18,447,362 | Citrus yellow mosaic virus 5586–5703 | 79% | 119 bp | RT | NC_003382.1 |
| Fragaria nipponica chr1 10,881,625–10,881,742 | Citrus yellow mosaic virus 5586–5703 | 81% | 119 bp | RT | NC_003382.1 |
| Fragaria vesca GG775297.1 536,392–536,509 | Citrus yellow mosaic virus 5586–5703 | 79% | 119 bp | RT | NC_003382.1 |
| Fragaria nipponica chr4 22,846,325–22,846,439 | Banana streak CA virus 5570–5684 | 80% | 119 bp | RT | NC_015506.1 |
| Fragaria nilgerrensis chr3 35,020,921–35,021,035 | Banana streak CA virus 5570–5684 | 81% | 119 bp | RT | NC_015506.1 |
| Fragaria pentaphylla chr2 29,284,759–29,284,886 | Grapevine roditis leaf discoloration-associated virus 5180–5307 | 78% | 133 bp | RT | NC_027131.1 |
| Fragaria pentaphylla chr4 16,387,420–16,387,565 | Birch leaf roll-associated virus 5947–6092 | 77% | 149 bp | RT | NC_040635.1 |
| Fragaria chiloensis Fchil2-B2 20,067,530–20,067,795 | Strawberry vein banding virus 1133–1398 | 95% | 267 bp | Aphid transmission associated protein | NC_001725.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Liu, J.; Qi, X.; Su, D.; Yang, J.; Cui, X. Study of Endogenous Viruses in the Strawberry Plants. Viruses 2024, 16, 1306. https://doi.org/10.3390/v16081306
Wang Z, Liu J, Qi X, Su D, Yang J, Cui X. Study of Endogenous Viruses in the Strawberry Plants. Viruses. 2024; 16(8):1306. https://doi.org/10.3390/v16081306
Chicago/Turabian StyleWang, Zongneng, Jian Liu, Xingyang Qi, Daifa Su, Junyu Yang, and Xiaolong Cui. 2024. "Study of Endogenous Viruses in the Strawberry Plants" Viruses 16, no. 8: 1306. https://doi.org/10.3390/v16081306