
Modeling the 2014–2015 Vesicular Stomatitis Outbreak in the United States Using an SEIR-SEI Approach

 , , , , , , , , , , and
, , , , , , , , , , and
Abstract
:1. Introduction
2. Data and Methods
2.1. Study Domain and Observed Incidence
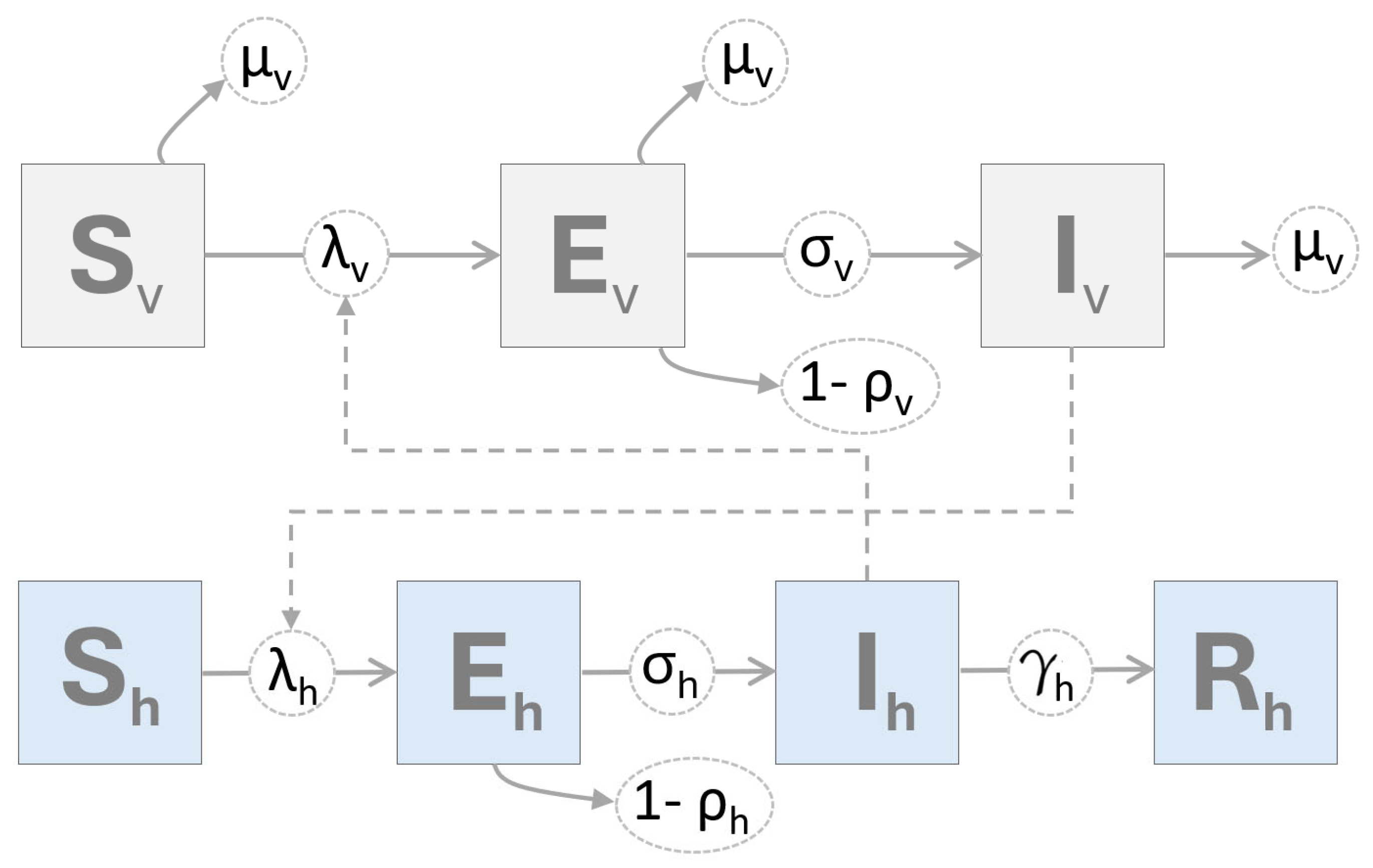
2.2. Mathematical Model
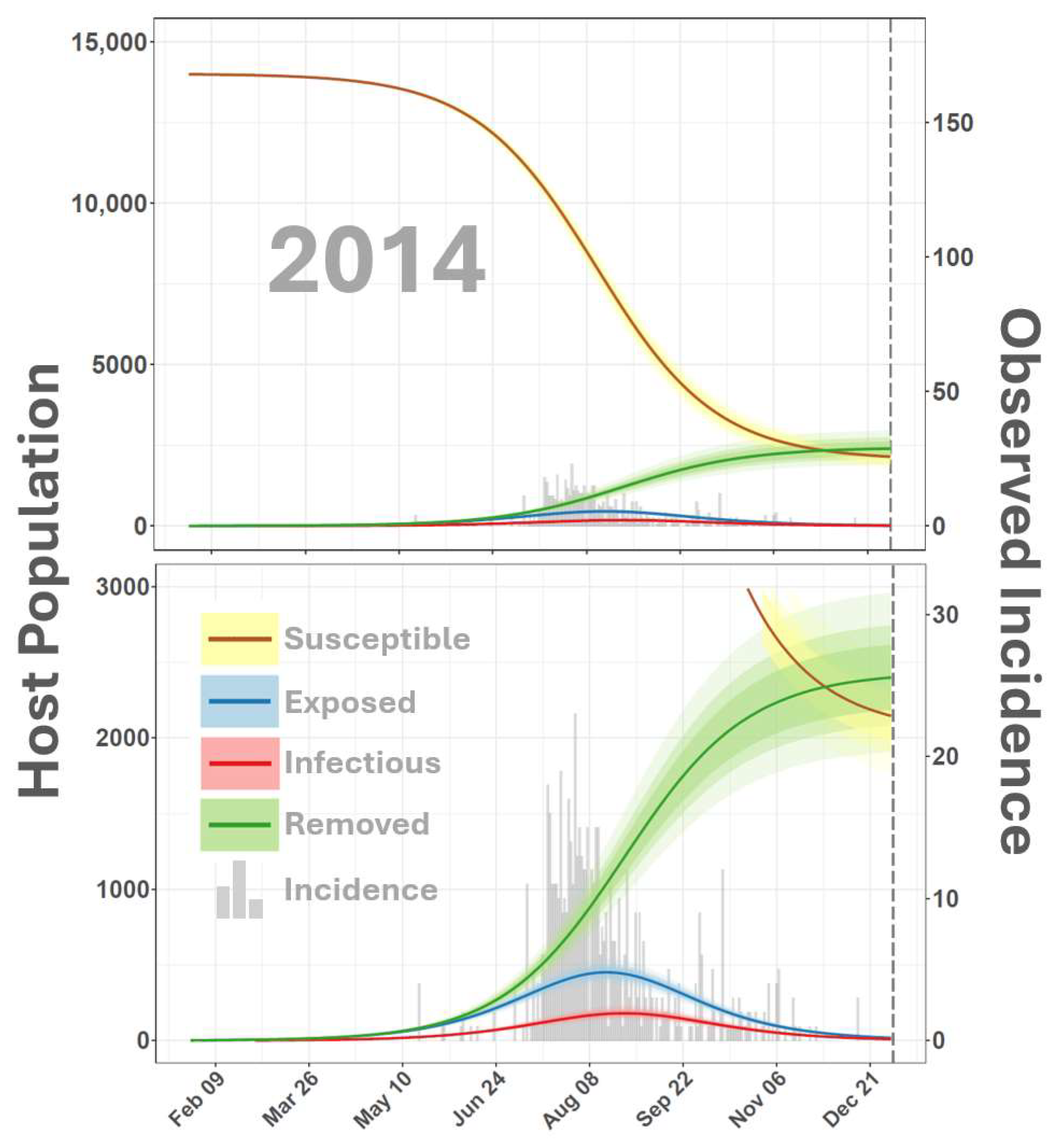
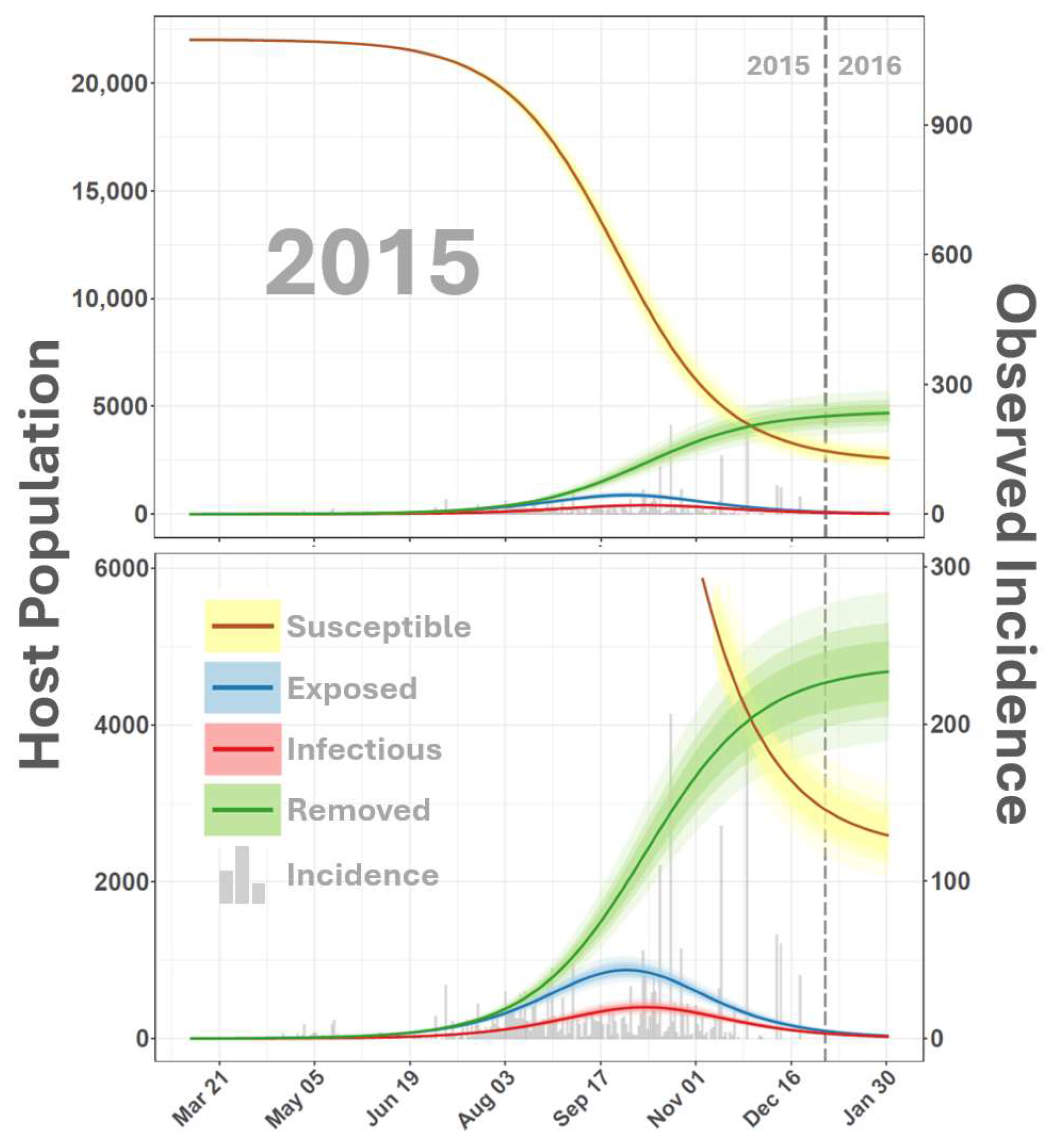
3. Results and Discussion
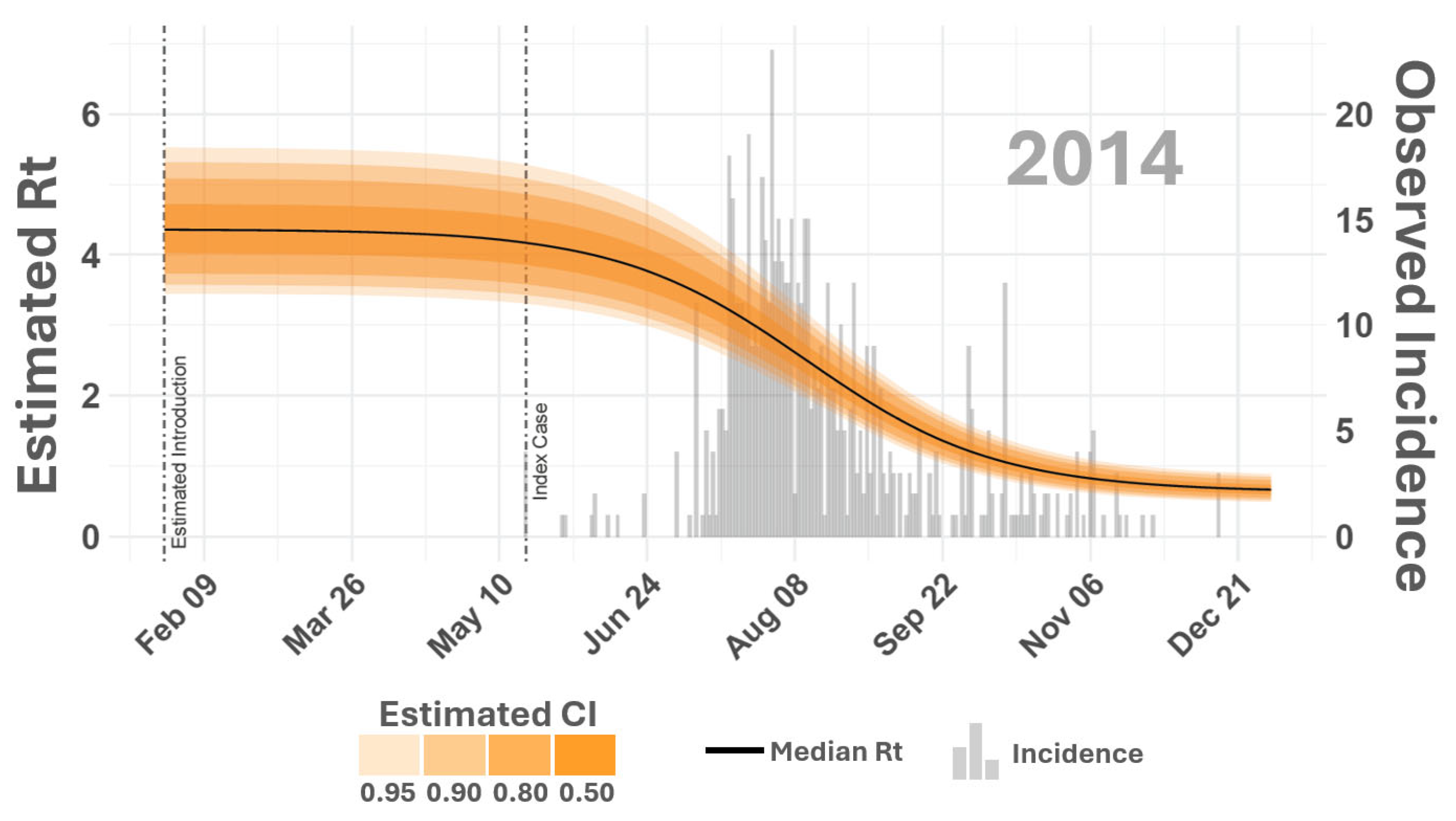
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APHIS | Animal and Plant Health Inspection Service |
| ARS | Agricultural Research Service |
| EIP | extrinsic incubation period |
| FOI | force of infection |
| FMD | foot and mouth disease |
| IIP | intrinsic incubation period |
| US | United States |
| USDA | United States Department of Agriculture |
| VS | vesicular stomatitis disease |
| VSNJV | vesicular stomatitis virus |
| VSIV | vesicular stomatitis Indiana virus |
| VSNJV | vesicular stomatitis New Jersey virus |
References
- Letchworth, G.; Rodriguez, L.; Del Cbarrera, J. Vesicular Stomatitis. Vet. J. 1999, 157, 239–260. [Google Scholar] [CrossRef]
- Pelzel-McCluskey, A.; Christensen, B.; Humphreys, J.; Bertram, M.; Keener, R.; Ewing, R.; Cohnstaedt, L.W.; Tell, R.; Peters, D.P.; Rodriguez, L. Review of Vesicular Stomatitis in the United States with Focus on 2019 and 2020 Outbreaks. Pathogens 2021, 10, 993. [Google Scholar] [CrossRef] [PubMed]
- APHIS. Final Situation Reports 2014–2015 VS Outbreaks; Technical Repor; Animal and Plant Health Inspection Service, USDA: Riverdale Park, MD, USA, 2016.
- Goodger, W.; Thurmond, M.; Nehay, J.; Mitchell, J.; Smith, P. Economic impact of an epizootic of bovine vesicular stomatitis in California. J. Am. Vet. Med. Assoc. 1985, 186, 370–373. [Google Scholar] [PubMed]
- Hayek, A.; McCluskey, B.; Chavez, G.; Salman, M. Financial impact of the 1995 outbreak of vesicular stomatitis on 16 beef ranches in Colorado. J. Am. Vet. Med. Assoc. 1998, 212, 820–823. [Google Scholar] [CrossRef]
- Urie, N.J.; Lombard, J.E.; Marshall, K.L.; Digianantonio, R.; Pelzel-McCluskey, A.M.; McCluskey, B.J.; Traub-Dargatz, J.L.; Kopral, C.A.; Swenson, S.L.; Schiltz, J.J. Risk factors associated with clinical signs of vesicular stomatitis and seroconversion without clinical disease in Colorado horses during the 2014 outbreak. Prev. Vet. Med. 2018, 156, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Perez, A.M.; Pauszek, S.J.; Jimenez, D.; Kelley, W.N.; Whedbee, Z.; Rodriguez, L.L. Spatial and phylogenetic analysis of vesicular stomatitis virus over-wintering in the United States. Prev. Vet. Med. 2010, 93, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Berlin, E.R.; Kinney, M.E.; Howard, L.L.; Perrin, K.L.; Rodriguez, P.; Kubiski, S.V.; Phair, K.A. Vesicular stomatitis virus in two species of rhinoceros at a California zoological park. Am. J. Vet. Res. 2024, 85, Ajvr.23.12.0284. [Google Scholar] [CrossRef]
- Mead, D.G.; Ramberg, F.B.; Besselsen, D.G.; Maré, C.J. Transmission of Vesicular Stomatitis Virus from Infected to Noninfected Black Flies Co-Feeding on Nonviremic Deer Mice. Science 2000, 287, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Stallknecht, D.E.; Greer, J.B.; Murphy, M.D.; Mead, D.G.; Howerth, E.W. Effect of strain and serotype of vesicular stomatitis virus on viral shedding, vesicular lesion development, and contact transmission in pigs. Am. J. Vet. Res. 2004, 65, 1233–1239. [Google Scholar] [CrossRef]
- Duarte, P.C.; Morley, P.S.; Traub-Dargatz, J.L.; Creekmore, L.H. Factors associated with vesicular stomatitis in animals in the western United States. J. Am. Vet. Med. Assoc. 2008, 232, 249–256. [Google Scholar] [CrossRef]
- Smith, P.F.; Howerth, E.W.; Carter, D.; Gray, E.W.; Noblet, R.; Smoliga, G.; Rodriguez, L.L.; Mead, D.G. Domestic cattle as a non-conventional amplifying host of vesicular stomatitis New Jersey virus. Med. Vet. Entomol. 2011, 25, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Mohler, J.R. Vesicular Stomatitis of Horses and Cattle; Number 662 in 1; US Department of Agriculture: Washington, DC, USA, 1918.
- Hanson, R.P. The natural history of vesicular stomatitis. Bacteriol. Rev. 1952, 16, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Nichol, S.T. Genetic diversity of enzootic isolates of vesicular stomatitis virus New Jersey. J. Virol. 1988, 62, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Francy, D.B.; Moore, C.G.; Smith, G.C.; Jakob, W.L.; Taylor, S.A.; Calisher, C.H. Epizoötic Vesicular Stomatitis in Colorado, 1982: Isolation of Virus from Insects Collected along the Northern Colorado Rocky Mountain Front Range. J. Med. Entomol. 1988, 25, 343–347. [Google Scholar] [CrossRef]
- Drolet, B.S.; Reeves, W.K.; Bennett, K.E.; Pauszek, S.J.; Bertram, M.R.; Rodriguez, L.L. Identical viral genetic sequence found in black flies (Simulium bivittatum) and the equine index case of the 2006 U.S. vesicular stomatitis outbreak. Pathogens 2021, 10, 929. [Google Scholar] [CrossRef]
- Rodrıguez, L.L. Emergence and re-emergence of vesicular stomatitis in the United States. Virus Res. 2002, 85, 211–219. [Google Scholar] [CrossRef]
- Rainwater-Lovett, K.; Pauszek, S.J.; Kelley, W.N.; Rodriguez, L.L. Molecular epidemiology of vesicular stomatitis New Jersey virus from the 2004–2005 US outbreak indicates a common origin with Mexican strains. J. Gen. Virol. 2007, 88, 2042–2051. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L.; Pauszek, S.J.; Zarate, S.; Basurto-Alcantara, F.J.; Verdugo-Rodriguez, A.; Perez, A.M.; Rodriguez, L.L. Phylogeographic characteristics of vesicular stomatitis New Jersey viruses circulating in Mexico from 2005 to 2011 and their relationship to epidemics in the United States. Virology 2014, 449, 17–24. [Google Scholar] [CrossRef]
- Palinski, R.; Pauszek, S.J.; Humphreys, J.M.; Peters, D.P.; McVey, D.S.; Pelzel-McCluskey, A.M.; Derner, J.D.; Burruss, N.D.; Arzt, J.; Rodriguez, L.L. Evolution and expansion dynamics of a vector-borne virus: 2004–2006 vesicular stomatitis outbreak in the western USA. Ecosphere 2021, 12, 2004–2006. [Google Scholar] [CrossRef]
- Rodríguez, L.L.; Fitch, W.M.; Nichol, S.T. Ecological factors rather than temporal factors dominate the evolution of vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 1996, 93, 13030–13035. [Google Scholar] [CrossRef]
- Rodriguez, L.L.; Bunch, T.A.; Fraire, M.; Llewellyn, Z.N. Re-emergence of Vesicular Stomatitis in the Western United States Is Associated with Distinct Viral Genetic Lineages. Virology 2000, 271, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Rozo-Lopez, P.; Drolet, B.S.; Londoño-Renteria, B. Vesicular stomatitis virus transmission: A comparison of incriminated vectors. Insects 2018, 9, 190. [Google Scholar] [CrossRef] [PubMed]
- Bertoldo, A.; Cobelli, C. Chapter Fourteen—Data modeling and simulation. In Biomedical Information Technology, 2nd ed.; Feng, D.D., Ed.; Biomedical Engineering; Academic Press: Cambridge, MA, USA, 2020; pp. 415–460. [Google Scholar] [CrossRef]
- Ross, R. Report on the Prevention of Malaria in Mauritius; J. & A. Churchill: London, UK, 1908. [Google Scholar]
- Sharpe, F.R.; Lokta, A. Contributions to the analysis of malaria epidemiology. Am. J. Hyg. 1923, 3, 96–112. [Google Scholar]
- Macdonald. G, M.G. The Epidemiology and Control of Malaria; Oxford University Press: London, UK; New York, NY, USA, 1957; pp. 1–201. [Google Scholar]
- Roosa, K.; Chowell, G. Assessing parameter identifiability in compartmental dynamic models using a computational approach: Application to infectious disease transmission models. Theor. Biol. Med. Model. 2019, 16, 1. [Google Scholar] [CrossRef]
- Rahman, M.; Bekele-Maxwell, K.; Cates, L.L.; Banks, H.; Vaidya, N.K. Modeling Zika Virus Transmission Dynamics: Parameter Estimates, Disease Characteristics, and Prevention. Sci. Rep. 2019, 9, 10575. [Google Scholar] [CrossRef] [PubMed]
- Bowman, C.; Gumel, A.; van den Driessche, P.; Wu, J.; Zhu, H. A mathematical model for assessing control strategies against West Nile virus. Bull. Math. Biol. 2005, 67, 1107–1133. [Google Scholar] [CrossRef] [PubMed]
- Alhaj, M.S. Mathematical Model for Malaria Disease Transmission. J. Math. Anal. Model. 2023, 4, 1–16. [Google Scholar] [CrossRef]
- de Wit, M.M.; Dimas Martins, A.; Delecroix, C.; Heesterbeek, H.; ten Bosch, Q.A. Mechanistic models for West Nile virus transmission: A systematic review of features, aims and parametrization. Proc. R. Soc. B Biol. Sci. 2024, 291, 20232432. [Google Scholar] [CrossRef]
- Macdonald, G. The Measurement of Malaria Transmission. Proc. R. Soc. Med. 1955, 48, 295–302. [Google Scholar] [CrossRef]
- Suparit, P.; Wiratsudakul, A.; Modchang, C. A mathematical model for Zika virus transmission dynamics with a time-dependent mosquito biting rate. Theor. Biol. Med. Model. 2018, 15, 1–11. [Google Scholar] [CrossRef]
- Esteva, L.; Vargas, C. Analysis of a dengue disease transmission model. Math. Biosci. 1998, 150, 131–151. [Google Scholar] [CrossRef] [PubMed]
- Otero, M.; Barmak, D.H.; Dorso, C.O.; Solari, H.G.; Natiello, M.A. Modeling dengue outbreaks. Math. Biosci. 2011, 232, 87–95. [Google Scholar] [CrossRef]
- Hardy, J.L.; Houk, E.J.; Kramer, L.D.; Reeves, W.C. Intrinsic Factors Affecting Vector Competence of Mosquitoes for Arboviruses. Annu. Rev. Entomol. 1983, 28, 229–262. [Google Scholar] [CrossRef]
- Wu, V.Y.; Chen, B.; Christofferson, R.; Ebel, G.; Fagre, A.C.; Gallichotte, E.N.; Sweeny, A.R.; Carlson, C.J.; Ryan, S.J. A minimum data standard for vector competence experiments. Sci. Data 2022, 9, 634. [Google Scholar] [CrossRef]
- Drolet, B.S.; Campbell, C.L.; Stuart, M.A.; Wilson, W.C. Vector Competence of Culicoides sonorensis (Diptera: Ceratopogonidae) for Vesicular Stomatitis Virus. J. Med. Entomol. 2005, 42, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.F.; Howerth, E.W.; Carter, D.; Gray, E.W.; Noblet, R.; Berghaus, R.D.; Stallknecht, D.E.; Mead, D.G. Host predilection and transmissibility of vesicular stomatitis New Jersey virus strains in domestic cattle (Bos taurus) and swine (Sus scrofa). BMC Vet. Res. 2012, 8, 183. [Google Scholar] [CrossRef] [PubMed]
- Mörk, M.; Lindberg, A.; Alenius, S.; Vågsholm, I.; Egenvall, A. Comparison between dairy cow disease incidence in data registered by farmers and in data from a disease-recording system based on veterinary reporting. Prev. Vet. Med. 2009, 88, 298–307. [Google Scholar] [CrossRef]
- Vergne, T.; Del Rio Vilas, V.J.; Cameron, A.; Dufour, B.; Grosbois, V. Capture–recapture approaches and the surveillance of livestock diseases: A review. Prev. Vet. Med. 2015, 120, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Carabali, M.; Jaramillo-Ramirez, G.I.; Rivera, V.A.; Mina Possu, N.J.; Restrepo, B.N.; Zinszer, K. Assessing the reporting of Dengue, Chikungunya and Zika to the National Surveillance System in Colombia from 2014–2017: A Capture-recapture analysis accounting for misclassification of arboviral diagnostics. PLoS Negl. Trop. Dis. 2021, 15, e0009014. [Google Scholar] [CrossRef]
- Danforth, M.E.; Snyder, R.E.; Lonstrup, E.T.N.; Barker, C.M.; Kramer, V.L. Evaluation of the effectiveness of the California mosquito-borne virus surveillance and response plan, 2009–2018. PLoS Negl. Trop. Dis. 2022, 16, e0010375. [Google Scholar] [CrossRef]
- Gabry, J.; Simpson, D.; Vehtari, A.; Betancourt, M.; Gelman, A. Visualization in Bayesian Workflow. J. R. Stat. Soc. Ser. A Stat. Soc. 2019, 182, 389–402. [Google Scholar] [CrossRef]
- Grinsztajn, L.; Semenova, E.; Margossian, C.C.; Riou, J. Bayesian workflow for disease transmission modeling in Stan. Stat. Med. 2021, 40, 6209–6234. [Google Scholar] [CrossRef]
- Stan Development Team. Stan Modeling Language Users Guide and Reference Manual, version 2.28. Comput. Softw. 2024. Available online: https://mc-stan.org (accessed on 16 August 2024).
- Szmaragd, C.; Wilson, A.J.; Carpenter, S.; Wood, J.L.N.; Mellor, P.S.; Gubbins, S. A Modeling Framework to Describe the Transmission of Bluetongue Virus within and between Farms in Great Britain. PLoS ONE 2009, 4, e7741. [Google Scholar] [CrossRef]
- Carpenter, S.; Wilson, A.; Barber, J.; Veronesi, E.; Mellor, P.; Venter, G.; Gubbins, S. Temperature Dependence of the Extrinsic Incubation Period of Orbiviruses in Culicoides Biting Midges. PLoS ONE 2011, 6, e27987. [Google Scholar] [CrossRef]
- Fairbanks, E.L.; Brennan, M.L.; Mertens, P.P.C.; Tildesley, M.J.; Daly, J.M. Re-parameterization of a mathematical model of African horse sickness virus using data from a systematic literature search. Transbound. Emerg. Dis. 2022, 69, e671–e681. [Google Scholar] [CrossRef] [PubMed]
- Brand, S.P.C.; Rock, K.S.; Keeling, M.J. The Interaction between Vector Life History and Short Vector Life in Vector-Borne Disease Transmission and Control. PLoS Comput. Biol. 2016, 12, e1004837. [Google Scholar] [CrossRef]
- Rozo-Lopez, P.; Park, Y.; Drolet, B.S. Effect of Constant Temperatures on Culicoides sonorensis Midge Physiology and Vesicular Stomatitis Virus Infection. Insects 2022, 13, 372. [Google Scholar] [CrossRef] [PubMed]
- Gubbins, S.; Carpenter, S.; Baylis, M.; Wood, J.L.; Mellor, P.S. Assessing the risk of bluetongue to UK livestock: Uncertainty and sensitivity analyses of a temperature-dependent model for the basic reproduction number. J. R. Soc. Interface 2008, 5, 363–371. [Google Scholar] [CrossRef]
- Turner, J.; Bowers, R.G.; Baylis, M. Two-Host, Two-Vector Basic Reproduction Ratio (R0) for Bluetongue. PLoS ONE 2013, 8, e53128. [Google Scholar] [CrossRef]
- Howerth, E.W.; Mead, D.G.; Mueller, P.O.; Duncan, L.; Murphy, M.D.; Stallknecht, D.E. Experimental Vesicular Stomatitis Virus Infection in Horses: Effect of Route of Inoculation and Virus Serotype. Vet. Pathol. 2006, 43, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Killmaster, L.F.; Stallknecht, D.E.; Howerth, E.W.; Moulton, J.K.; Smith, P.F.; Mead, D.G. Apparent disappearance of vesicular stomatitis New Jersey virus from Ossabaw Island, Georgia. Vector-Borne Zoonotic Dis. 2011, 11, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Rozo-Lopez, P.; Londono-Renteria, B.; Drolet, B.S. Impacts of Infectious Dose, Feeding Behavior, and Age of Culicoides sonorensis Biting Midges on Infection Dynamics of Vesicular Stomatitis Virus. Pathogens 2021, 10, 816. [Google Scholar] [CrossRef]
- Backer, J.A.; Nodelijk, G. Transmission and Control of African Horse Sickness in The Netherlands: A Model Analysis. PLoS ONE 2011, 6, e23066. [Google Scholar] [CrossRef]
- Gerry, A.C.; Mullens, B.A. Seasonal Abundance and Survivorship of Culicoides sonorensis (Diptera: Ceratopogonidae) at a Southern California Dairy, with Reference to Potential Bluetongue Virus Transmission and Persistence. J. Med. Entomol. 2000, 37, 675–688. [Google Scholar] [CrossRef]
- Mullens, B.; Gerry, A.; Lysyk, T.; Schmidtmann, E. Environmental effects on vector competence and virogenesis of bluetongue virus in Culicoides: Interpreting laboratory data in a field context. Vet. Ital. 2004, 40, 160–166. [Google Scholar] [PubMed]
- Mills, M.K.; Ruder, M.G.; Nayduch, D.; Michel, K.; Drolet, B.S. Dynamics of epizootic hemorrhagic disease virus infection within the vector, Culicoides sonorensis (Diptera: Ceratopogonidae). PLoS ONE 2017, 12, e0188865. [Google Scholar] [CrossRef]
- Andrade, J.; Duggan, J. Anchoring the mean generation time in the SEIR to mitigate biases in R0 estimates due to uncertainty in the distribution of the epidemiological delays. R. Soc. Open Sci. 2023, 10, 230515. [Google Scholar] [CrossRef]
- Bretó, C. Modeling and Inference for Infectious Disease Dynamics: A Likelihood-Based Approach. Stat. Sci. 2018, 33, 57–69. [Google Scholar] [CrossRef]
- Stan Development Team. RStan: The R Interface to Stan, R package version 2.32.6. 2024. Available online: https://CRAN.R-project.org/package=rstan (accessed on 16 August 2024).
- Gabry, J.; Češnovar, R. cmdstanr: R Interface to ‘CmdStan’. 2022. Available online: https://mc-stan.org/cmdstanr/ (accessed on 16 August 2024).
- Hoffman, M.D.; Gelman, A. The No-U-Turn sampler: Adaptively setting path lengths in Hamiltonian Monte Carlo. J. Mach. Learn. Res. 2014, 15, 1593–1623. [Google Scholar]
- Vehtari, A.; Gelman, A.; Gabry, J. Practical Bayesian model evaluation using leave-one-out cross-validation and WAIC. Stat. Comput. 2017, 27, 1413–1432. [Google Scholar] [CrossRef]
- SENASICA. Weekly Reports on Diseases and Pests of Mandatory Immediate Reporting (Week 53, 2013); Technical Report; National Service of Agrifood Health and Quality, Directorate of Epidemiology and Risk Analysis, National Epidemiological Surveillance System, 2013. Available online: https://www.gob.mx/senasica/acciones-y-programas/sistema-nacional-de-vigilancia-epidemiologica-sive (accessed on 16 August 2024).
- Humphreys, J.; Bertram, M.R.; Shults, P.; Velazquez-Salinas, L.; Peters, D.P.; Rodriguez, L.L. Overwintering or Re-Invasion? Interrogating Genomes and Geography to Unravel Multiyear Vesicular Stomatitis Epizootics. Viruses 2024, 16, 1118. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, A.M.; Randolph, S.E. Drivers, dynamics, and control of emerging vector-borne zoonotic diseases. Lancet 2012, 380, 1946–1955. [Google Scholar] [CrossRef] [PubMed]
- Parham, P.E.; Michael, E. Modeling the Effects of Weather and Climate Change on Malaria Transmission. Environ. Health Perspect. 2010, 118, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Nichol, S.T.; Rowe, J.E.; Fitch, W.M. Punctuated equilibrium and positive Darwinian evolution in vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 1993, 90, 10424–10428. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, J.M. Amplification in Time and Dilution in Space: Partitioning Spatiotemporal Processes to Assess the Role of Avian-Host Phylodiversity in Shaping Eastern Equine Encephalitis Virus Distribution. Geographies 2022, 2, 419–434. [Google Scholar] [CrossRef]
- Keesing, F.; Holt, R.D.; Ostfeld, R.S. Effects of species diversity on disease risk. Ecol. Lett. 2006, 9, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Stallknecht, D.E.; Perzak, D.E.; Bauer, L.D.; Murphy, M.D.; Howerth, E.W. Contact transmission of vesicular stomatitis virus New Jersey in pigs. Am. J. Vet. Res. 2001, 62, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Trainer, D.; Hanson, R. Serologic evidence of arbovirus infections in wild ruminants. Am. J. Epidemiol. 1969, 90, 354–358. [Google Scholar] [CrossRef]
- Fletcher, W.; Stallknecht, D.; Jenney, E. Serologic surveillance for vesicular stomatitis virus on Ossabaw Island, Georgia. J. Wildl. Dis. 1985, 21, 100–104. [Google Scholar] [CrossRef]
- Webb, P.A.; McLean, R.G.; Smith, G.C.; Ellenberger, J.H.; Francy, D.B.; Walton, T.E.; Monath, T.P. Epizootic Vesicular Stomatitis in Colorado, 1982: Some Observations on the Possible Role of Wildlife Populations in an Enzootic Mainetance Cycle. J. Wildl. Dis. 1987, 23, 192–198. [Google Scholar] [CrossRef]
- Fletcher, W.; Stallknecht, D.; Kearney, M.; Eernisse, K. Antibodies to vesicular stomatitis New Jersey type virus in white-tailed deer on Ossabaw Island, Georgia, 1985 to 1989. J. Wildl. Dis. 1991, 27, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Tesh, R.B.; Peralta, P.H.; Johnson, K.M. Ecologic studies of vesicular stomatitis virus: i. prevalence of infection among animals and humans living in an area of endemic vsv activity1. Am. J. Epidemiol. 1969, 90, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Reis, J.L.; Mead, D.; Rodriguez, L.L.; Brown, C.C. Transmission and pathogenesis of vesicular stomatitis viruses. Braz. J. Vet. Pathol. 2009, 2, 49–58. [Google Scholar]
- Lewis, J.; Corn, J.; Mayer, J.; Jordan, T.; Farnsworth, M.; Burdett, C.; VerCauteren, K.; Sweeney, S.; Miller, R. Historical, current, and potential population size estimates of invasive wild pigs (Sus scrofa) in the United States. Biol. Invasions 2019, 21, 2373–2384. [Google Scholar] [CrossRef]
- Brown, V.R.; Miller, R.S.; Pepin, K.M.; Carlisle, K.M.; Cook, M.A.; Vanicek, C.F.; Holmstrom, L.K.; Rochette, L.T.; Smyser, T.J. African swine fever at the wildlife-livestock interface: Challenges for management and outbreak response within invasive wild pigs in the United States. Front. Vet. Sci. 2024, 11, 1348123. [Google Scholar] [CrossRef] [PubMed]
- Alexander, K.A.; Carlson, C.J.; Lewis, B.L.; Getz, W.M.; Marathe, M.V.; Eubank, S.G.; Sanderson, C.E.; Blackburn, J.K. The Ecology of Pathogen Spillover and Disease Emergence at the Human-Wildlife-Environment Interface. In The Connections Between Ecology and Infectious Disease; Springer International Publishing: Cham, Switzerland, 2018; pp. 267–298. [Google Scholar] [CrossRef]
- Blumberg, S.; Lloyd-Smith, J.O. Inference of R0 and Transmission Heterogeneity from the Size Distribution of Stuttering Chains. PLoS Comput. Biol. 2013, 9, e1002993. [Google Scholar] [CrossRef]
- Humphreys, J.M.; Young, K.I.; Cohnstaedt, L.W.; Hanley, K.A.; Peters, D.P.C. Vector Surveillance, Host Species Richness, and Demographic Factors as West Nile Disease Risk Indicators. Viruses 2021, 13, 934. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, J.M.; Pelzel-McCluskey, A.M.; Cohnstaedt, L.W.; McGregor, B.L.; Hanley, K.A.; Hudson, A.R.; Young, K.I.; Peck, D.; Rodriguez, L.L.; Peters, D.P.C. Integrating Spatiotemporal Epidemiology, Eco-Phylogenetics, and Distributional Ecology to Assess West Nile Disease Risk in Horses. Viruses 2021, 13, 1811. [Google Scholar] [CrossRef] [PubMed]
- USDA. USDA National Agricultural Statistics Service. 2024. Available online: https://www.nass.usda.gov/ (accessed on 16 August 2024).
- Council, A.H. US Horse Population—Statistics. 2024. Available online: https://horsecouncil.org/economic-impact-study/ (accessed on 16 August 2024).
- Brand, S.P.C.; Keeling, M.J. The impact of temperature changes on vector-borne disease transmission: Culicoides midges and bluetongue virus. J. R. Soc. Interface 2017, 14, 20160481. [Google Scholar] [CrossRef]
- Tabachnick, W.J. Culicoides Variipennis and Bluetongue-Virus Epidemiology in the United States. Annu. Rev. Entomol. 1996, 41, 23–43. [Google Scholar] [CrossRef]
- Holbrook, F.R.; Tabachnick, W.J.; Schmidtmann, E.T.; McKinnon, C.N.; Bobian, R.J.; Grogan, W.L. Sympatry in the Culicoides variipennis Complex (Diptera: Ceratopogonidae): A Taxonomic Reassessment. J. Med. Entomol. 2000, 37, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Shults, P.; Hopken, M.; Eyer, P.A.; Blumenfeld, A.; Mateos, M.; Cohnstaedt, L.W.; Vargo, E.L. Species delimitation and mitonuclear discordance within a species complex of biting midges. Sci. Rep. 2022, 12, 1730. [Google Scholar] [CrossRef] [PubMed]
- Mullens, B.A.; Schmidtmann, E.T. The Gonotrophic Cycle of Culicoides Variipennis (Diptera: Ceratopogonidae) and its Implications in Age-Grading Field Populations in New York State, USA1. J. Med. Entomol. 1982, 19, 340–349. [Google Scholar] [CrossRef]
- Mullens, B.; Holbrook, F. Temperature effects on the gonotrophic cycle of Culicoides variipennis (Diptera: Ceratopogonidae). J. Am. Mosq. Control Assoc. 1991, 7, 588–591. [Google Scholar]
- Peters, D.P.; McVey, D.S.; Elias, E.H.; Pelzel-McCluskey, A.M.; Derner, J.D.; Burruss, N.D.; Schrader, T.S.; Yao, J.; Pauszek, S.J.; Lombard, J.; et al. Big data–model integration and AI for vector-borne disease prediction. Ecosphere 2020, 11, e03157. [Google Scholar] [CrossRef]
- Stenkamp-Strahm, C.; Patyk, K.; McCool-Eye, M.; Fox, A.; Humphreys, J.; James, A.; South, D.; Magzamen, S. Using geospatial methods to measure the risk of environmental persistence of avian influenza virus in South Carolina. Spat. Spatio-Temporal Epidemiol. 2020, 34, 100342. [Google Scholar] [CrossRef]
- Mellor, P.S.; Boorman, J.; Baylis, M. Culicoides Biting Midges: Their Role as Arbovirus Vectors. Annu. Rev. Entomol. 2000, 45, 307–340. [Google Scholar] [CrossRef]
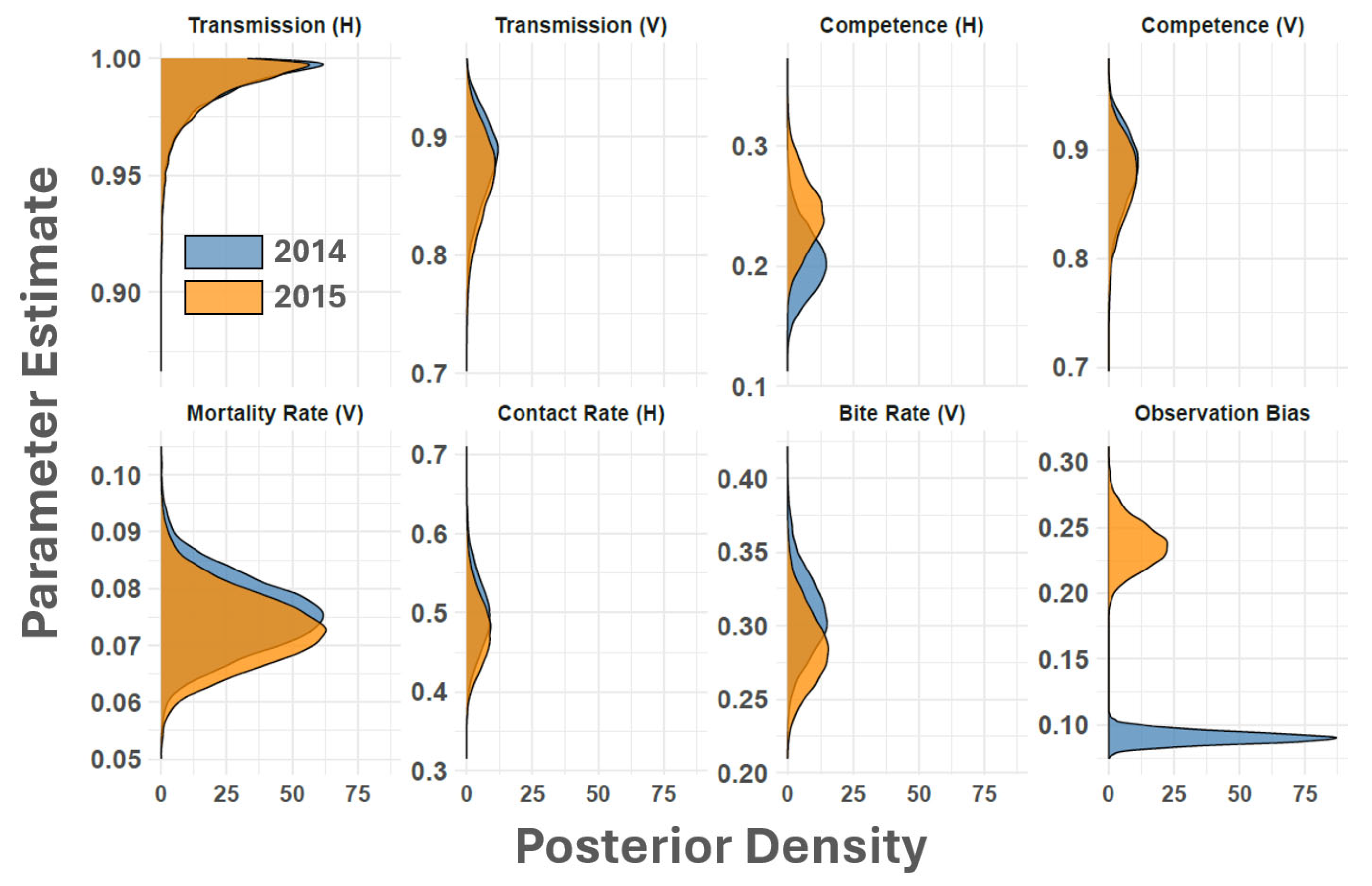
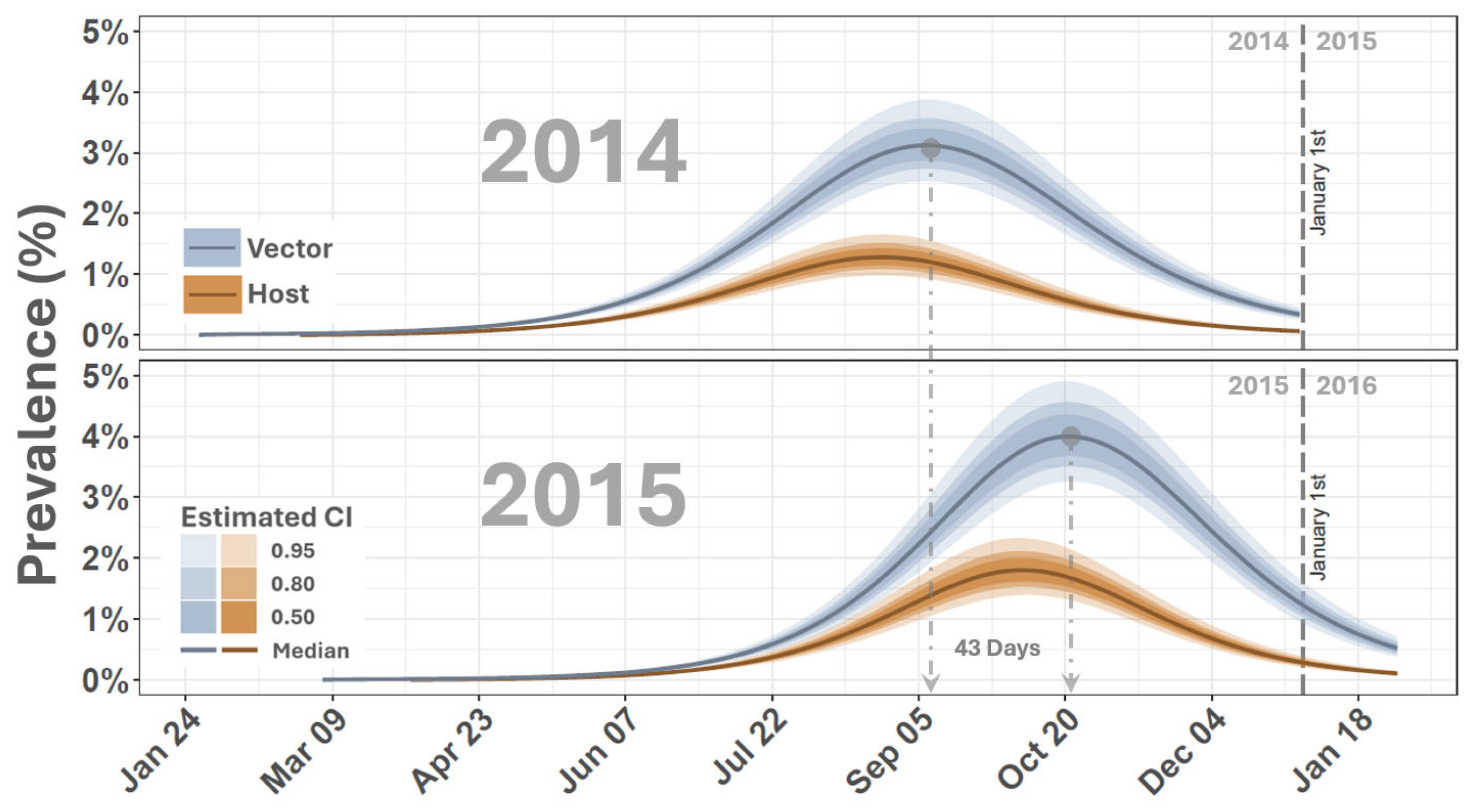

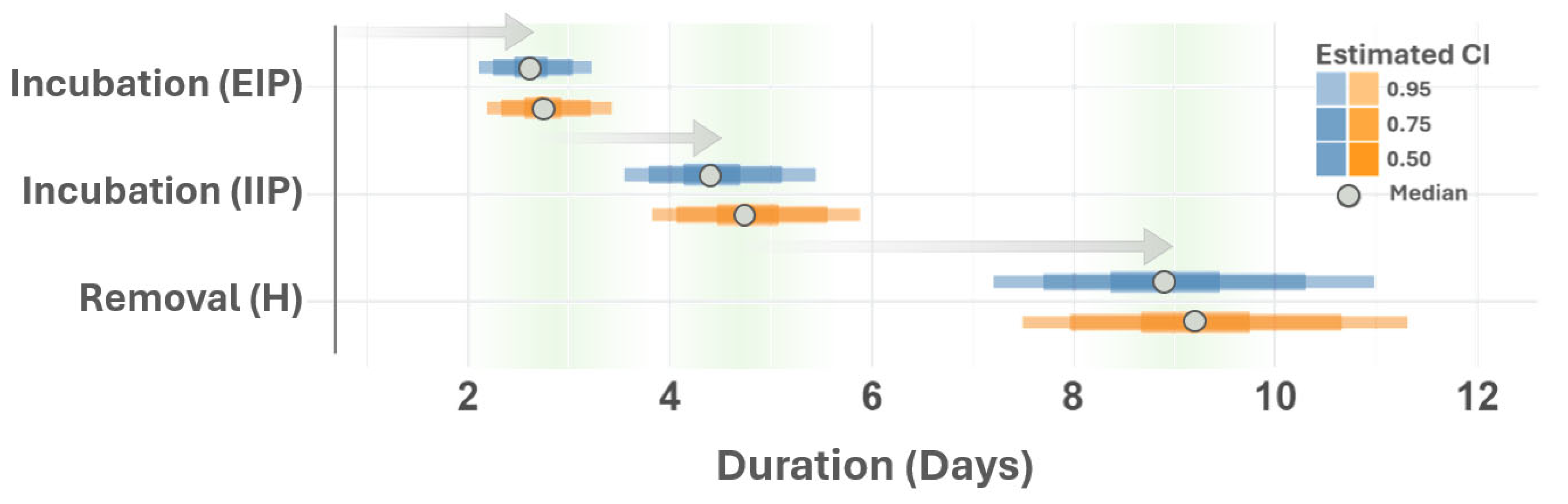


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Description | Evaluated Range | Reference/Notes |
|---|---|---|---|
| Total number of vertebrate hosts | 3200–40,000 | Data a | |
| Total number of insect vectors | 1–2 × | [49,50,51] b | |
| Contact rate, bites sustained by host | 0.25–0.33/vector×day | [49,52,53] c | |
| Contact rate, vector biting rate | 0.25–0.33/day | [49,52,53] | |
| Vector-to-host transmission probability | 0.05–0.50/bite | [49,51,54,55] | |
| Host-to-vector transmission probability | 0.75–1.00/bite | [49,51,54,55] | |
| Intrinsic Incubation Period (IIP) | 0.05–0.50/day | [41,53,56] | |
| Extrinsic Incubation Period (EIP) | 0.10–0.60/day | [51,53,54,55] | |
| Host removal rate (recovery or quarantine) | 0.05–0.20/day | [54,55] | |
| Host heterogeneous competency | 0.01–1.00 (∝/day) | unlimited d | |
| Vector heterogeneous competency | 0.01–1.00 (∝/day) | unlimited d | |
| Vector background mortality rate | 0.05–0.50/day | [49,52,54,55] | |
| Observation bias (proportion observed) | 0.01–1.00 (∝/day) | unlimited e |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Humphreys, J.M.; Pelzel-McCluskey, A.M.; Shults, P.T.; Velazquez-Salinas, L.; Bertram, M.R.; McGregor, B.L.; Cohnstaedt, L.W.; Swanson, D.A.; Scroggs, S.L.P.; Fautt, C.; et al. Modeling the 2014–2015 Vesicular Stomatitis Outbreak in the United States Using an SEIR-SEI Approach. Viruses 2024, 16, 1315. https://doi.org/10.3390/v16081315
Humphreys JM, Pelzel-McCluskey AM, Shults PT, Velazquez-Salinas L, Bertram MR, McGregor BL, Cohnstaedt LW, Swanson DA, Scroggs SLP, Fautt C, et al. Modeling the 2014–2015 Vesicular Stomatitis Outbreak in the United States Using an SEIR-SEI Approach. Viruses. 2024; 16(8):1315. https://doi.org/10.3390/v16081315
Chicago/Turabian StyleHumphreys, John M., Angela M. Pelzel-McCluskey, Phillip T. Shults, Lauro Velazquez-Salinas, Miranda R. Bertram, Bethany L. McGregor, Lee W. Cohnstaedt, Dustin A. Swanson, Stacey L. P. Scroggs, Chad Fautt, and et al. 2024. "Modeling the 2014–2015 Vesicular Stomatitis Outbreak in the United States Using an SEIR-SEI Approach" Viruses 16, no. 8: 1315. https://doi.org/10.3390/v16081315