
Role of PQBP1 in Pathogen Recognition—Impact on Innate Immunity



{kind=link}
{kind=link}
Abstract
1. Introduction
2. PQBP1 Structure and Functions
2.1. Mapped PQBP1–Protein Interactions
2.2. Additional Protein Interactions
3. PQBP1 Role in Innate Immunity
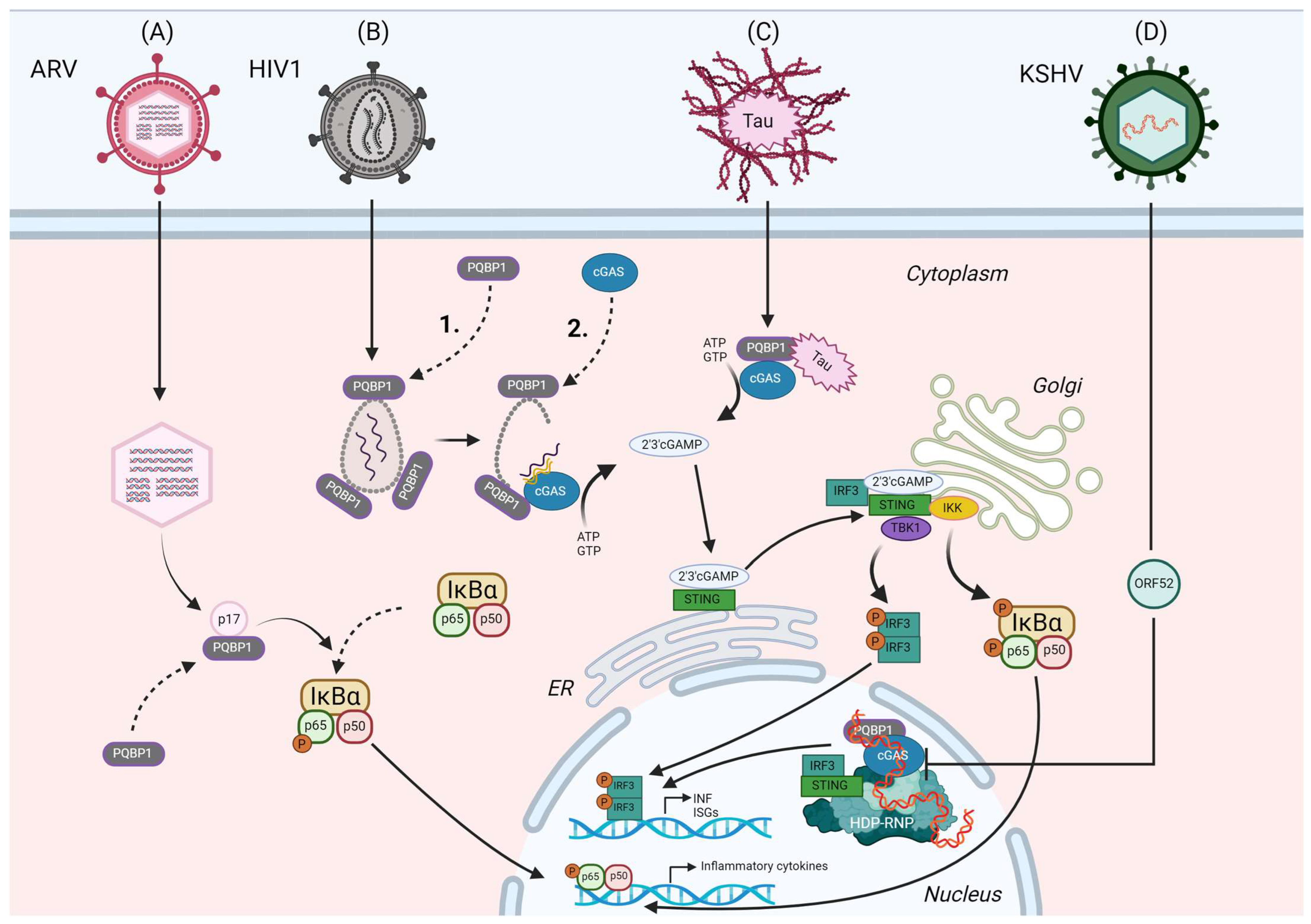
4. PQBP1 in Neurodegenerative Disorders
5. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Imafuku, I.; Waragai, M.; Takeuchi, S.; Kanazawa, I.; Kawabata, M.; Mouradian, M.M.; Okazawa, H. Polar amino acid-rich sequences bind to polyglutamine tracts. Biochem. Biophys. Res. Commun. 1998, 253, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Waragai, M.; Junn, E.; Kajikawa, M.; Takeuchi, S.; Kanazawa, I.; Shibata, M.; Mouradian, M.M.; Okazawa, H. PQBP-1/Npw38, a nuclear protein binding to the polyglutamine tract, interacts with U5-15kD/dim1p via the carboxyl-terminal domain. Biochem. Biophys. Res. Commun. 2000, 273, 592–595. [Google Scholar] [CrossRef]
- Komuro, A.; Saeki, M.; Kato, S. Npw38, a novel nuclear protein possessing a WW domain capable of activating basal transcription. Nucleic Acids Res. 1999, 27, 1957–1965. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhang, Z.C.; Cheng, S.; Liu, A.; Zuo, J.; Xia, S.; Liu, X.; Liu, W.; Jia, Z.; Xie, W.; et al. PQBP1 promotes translational elongation and regulates hippocampal mGluR-LTD by suppressing eEF2 phosphorylation. Mol. Cell 2021, 81, 1425–1438.e10. [Google Scholar] [CrossRef]
- Qi, Y.; Hoshino, M.; Wada, Y.; Marubuchi, S.; Yoshimura, N.; Kanazawa, I.; Shinomiya, K.; Okazawa, H. PQBP-1 is expressed predominantly in the central nervous system during development. Eur. J. Neurosci. 2005, 22, 1277–1286. [Google Scholar] [CrossRef]
- Waragai, M.; Lammers, C.H.; Takeuchi, S.; Imafuku, I.; Udagawa, Y.; Kanazawa, I.; Kawabata, M.; Mouradian, M.M.; Okazawa, H. PQBP-1, a novel polyglutamine tract-binding protein, inhibits transcription activation by Brn-2 and affects cell survival. Hum. Mol. Genet. 1999, 8, 977–987. [Google Scholar] [CrossRef]
- Yoh, S.M.; Mamede, J.I.; Lau, D.; Ahn, N.; Sánchez-Aparicio, M.T.; Temple, J.; Tuckwell, A.; Fuchs, N.V.; Cianci, G.C.; Riva, L.; et al. Recognition of HIV-1 capsid by PQBP1 licenses an innate immune sensing of nascent HIV-1 DNA. Mol Cell. 2022, 82, 2871–2884.e6. [Google Scholar] [CrossRef] [PubMed]
- Germanaud, D.; Rossi, M.; Bussy, G.; Gérard, D.; Hertz-Pannier, L.; Blanchet, P.; Dollfus, H.; Giuliano, F.; Bennouna-Greene, V.; Sarda, P.; et al. The Renpenning syndrome spectrum: New clinical insights supported by 13 new PQBP1-mutated males. Clin. Genet. 2011, 79, 225–235. [Google Scholar] [CrossRef]
- Lubs, H.; Abidi, F.E.; Echeverri, R.; Holloway, L.; Meindl, A.; Stevenson, R.E.; Schwartz, C.E. Golabi-Ito-Hall syndrome results from a missense mutation in the WW domain of the PQBP1 gene. J. Med. Genet. 2006, 43, e30. [Google Scholar] [CrossRef]
- Kalscheuer, V.M.; Freude, K.; Musante, L.; Jensen, L.R.; Yntema, H.G.; Gécz, J.; Sefiani, A.; Hoffmann, K.; Moser, B.; Haas, S.; et al. Mutations in the polyglutamine binding protein 1 gene cause X-linked mental retardation. Nat. Genet. 2003, 35, 313–315. [Google Scholar] [CrossRef]
- Stevenson, R.E.; Bennett, C.W.; Abidi, F.; Kleefstra, T.; Porteous, M.; Simensen, R.J.; Lubs, H.A.; Hamel, B.C.J.; Schwartz, C.E. Renpenning syndrome comes into focus. Am. J. Med. Genet. A 2005, 134, 415–421. [Google Scholar] [CrossRef]
- Okazawa, H.; Rich, T.; Chang, A.; Lin, X.; Waragai, M.; Kajikawa, M.; Enokido, Y.; Komuro, A.; Kato, S.; Shibata, M.; et al. Interaction between mutant ataxin-1 and PQBP-1 affects transcription and cell death. Neuron 2002, 34, 701–713. [Google Scholar] [CrossRef]
- Busch, A.; Engemann, S.; Lurz, R.; Okazawa, H.; Lehrach, H.; Wanker, E.E. Mutant huntingtin promotes the fibrillogenesis of wild-type huntingtin: A potential mechanism for loss of huntingtin function in Huntington’s disease. J. Biol. Chem. 2003, 278, 41452–41461. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Shiwaku, H.; Tanaka, H.; Obita, T.; Ohuchi, S.; Yoshioka, Y.; Jin, X.; Kondo, K.; Fujita, K.; Homma, H.; et al. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 2021, 12, 6565. [Google Scholar] [CrossRef]
- Syed, P.; Gupta, S.; Choudhary, S.; Pandala, N.G.; Atak, A.; Richharia, A.; K P, M.; Zhu, H.; Epari, S.; Noronha, S.B.; et al. Autoantibody Profiling of Glioma Serum Samples to Identify Biomarkers Using Human Proteome Arrays. Sci. Rep. 2015, 5, 13895. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yoshina, S.; Masashi, M.; Ito, W.; Inoue, T.; Shiwaku, H.; Arai, H.; Mitani, S.; Okazawa, H. Nematode homologue of PQBP1, a mental retardation causative gene, is involved in lipid metabolism. PLoS ONE 2009, 4, e4104. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, H.A.; Foroughi Asl, H.; Jain, R.K.; Ermel, R.; Ruusalepp, A.; Franzén, O.; Kidd, B.A.; Readhead, B.; Giannarelli, C.; Kovacic, J.C.; et al. Cross-Tissue Regulatory Gene Networks in Coronary Artery Disease. Cell Syst. 2016, 2, 196–208. [Google Scholar] [CrossRef]
- Frades, I.; Readhead, B.; Amadori, L.; Koplev, S.; Talukdar, H.A.; Crane, H.M.; Crane, P.K.; Kovacic, J.C.; Dudley, J.T.; Giannarelli, C.; et al. Systems Pharmacology Identifies an Arterial Wall Regulatory Gene Network Mediating Coronary Artery Disease Side Effects of Antiretroviral Therapy. Circ. Genom. Precis. Med. 2019, 12, e002390. [Google Scholar] [CrossRef]
- Rees, M.; Gorba, C.; de Chiara, C.; Bui, T.T.T.; Garcia-Maya, M.; Drake, A.F.; Okazawa, H.; Pastore, A.; Svergun, D.; Chen, Y.W. Solution model of the intrinsically disordered polyglutamine tract-binding protein-1. Biophys. J. 2012, 102, 1608–1616. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradović, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradović, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Takahashi, M.; Mizuguchi, M.; Shinoda, H.; Aizawa, T.; Demura, M.; Okazawa, H.; Kawano, K. Polyglutamine tract binding protein-1 is an intrinsically unstructured protein. Biochim. Biophys. Acta 2009, 1794, 936–943. [Google Scholar] [CrossRef]
- Macias, M.J.; Gervais, V.; Civera, C.; Oschkinat, H. Structural analysis of WW domains and design of a WW prototype. Nat. Struct. Biol. 2000, 7, 375–379. [Google Scholar] [CrossRef]
- Hu, H.; Columbus, J.; Zhang, Y.; Wu, D.; Lian, L.; Yang, S.; Goodwin, J.; Luczak, C.; Carter, M.; Chen, L.; et al. A map of WW domain family interactions. Proteomics 2004, 4, 643–655. [Google Scholar] [CrossRef]
- Macias, M.J.; Wiesner, S.; Sudol, M. WW and SH3 domains, two different scaffolds to recognize proline-rich ligands. FEBS Lett. 2002, 513, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Komuro, A.; Saeki, M.; Kato, S. Association of two nuclear proteins, Npw38 and NpwBP, via the interaction between the WW domain and a novel proline-rich motif containing glycine and arginine. J. Biol. Chem. 1999, 274, 36513–36519. [Google Scholar] [CrossRef]
- Park, E.M.; Scott, P.M.; Clutario, K.; Cassidy, K.B.; Zhan, K.; Gerber, S.A.; Holland, A.J. WBP11 is required for splicing the TUBGCP6 pre-mRNA to promote centriole duplication. J. Cell Biol. 2020, 219, e201904203. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Thomsen, G.H. The splicing factor PQBP1 regulates mesodermal and neural development through FGF signaling. Development 2014, 141, 3740–3751. [Google Scholar] [CrossRef]
- Llorian, M.; Beullens, M.; Lesage, B.; Nicolaescu, E.; Beke, L.; Landuyt, W.; Ortiz, J.-M.; Bollen, M. Nucleocytoplasmic shuttling of the splicing factor SIPP1. J. Biol. Chem. 2005, 280, 38862–38869. [Google Scholar] [CrossRef]
- Wang, Q.; Moore, M.J.; Adelmant, G.; Marto, J.A.; Silver, P.A. PQBP1, a factor linked to intellectual disability, affects alternative splicing associated with neurite outgrowth. Genes Dev. 2013, 27, 615–626. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Obita, T.; Serita, T.; Kojima, R.; Nabeshima, Y.; Okazawa, H. Mutations in the PQBP1 gene prevent its interaction with the spliceosomal protein U5-15 kD. Nat. Commun. 2014, 5, 3822. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Manley, J.L. RNA polymerase II and the integration of nuclear events. Genes Dev. 2000, 14, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Yoh, S.M.; Schneider, M.; Seifried, J.; Soonthornvacharin, S.; Akleh, R.E.; Olivieri, K.C.; de Jesus, P.D.; Ruan, C.; de Castro, E.; Ruiz, P.A.; et al. PQBP1 Is a Proximal Sensor of the cGAS-Dependent Innate Response to HIV-1. Cell 2015, 161, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, Y.; de La Torre-Ubieta, L.; Matsuda, T.; Steen, H.; Okazawa, H.; Bonni, A. The XLID protein PQBP1 and the GTPase Dynamin 2 define a signaling link that orchestrates ciliary morphogenesis in postmitotic neurons. Cell Rep. 2013, 4, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Zhang, C.; Hu, J.; Wang, X.; Wang, Y.; Guo, M.; Zhang, X.; Wu, Y. Avian reovirus infection activate the cellular unfold protein response and induced apoptosis via ATF6-dependent mechanism. Virus Res. 2021, 297, 198346. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Zhao, F.; Zhang, Q.; Zuo, W.; Guo, M.; Zhang, X.; Wu, Y. Identification and Functional Analyses of Host Proteins Interacting with the p17 Protein of Avian Reovirus. Viruses 2022, 14, 892. [Google Scholar] [CrossRef]
- Zhou, L.; Li, J.; Haiyilati, A.; Li, X.; Gao, L.; Cao, H.; Wang, Y.; Zheng, S.J. Gga-miR-29a-3p suppresses avian reovirus-induced apoptosis and viral replication via targeting Caspase-3. Vet. Microbiol. 2022, 264, 109294. [Google Scholar] [CrossRef]
- Liu, H.-J.; Lin, P.-Y.; Lee, J.-W.; Hsu, H.-Y.; Shih, W.-L. Retardation of cell growth by avian reovirus p17 through the activation of p53 pathway. Biochem. Biophys. Res. Commun. 2005, 336, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, K.; Yamanaka, M.; Ueda, S. POU3F2 participates in cognitive function and adult hippocampal neurogenesis via mammalian-characteristic amino acid repeats. Genes Brain Behav. 2018, 17, 118–125. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Shakkottai, V.G.; Albin, R.L. Polyglutamine Repeats in Neurodegenerative Diseases. Annu. Rev. Pathol. 2019, 14, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lyu, J.; Li, X.; Guo, G.; Zhou, X.; Chen, T.; Lin, Y.; Li, T. Phase separation as a possible mechanism for dosage sensitivity. Genome Biol. 2024, 25, 17. [Google Scholar] [CrossRef]
- Lee, B.J.; Cansizoglu, A.E.; Süel, K.E.; Louis, T.H.; Zhang, Z.; Chook, Y.M. Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell 2006, 126, 543–558. [Google Scholar] [CrossRef]
- Liu, X.; Dou, L.-X.; Han, J.; Zhang, Z.C. The Renpenning syndrome-associated protein PQBP1 facilitates the nuclear import of splicing factor TXNL4A through the karyopherin β2 receptor. J. Biol. Chem. 2020, 295, 4093–4100. [Google Scholar] [CrossRef]
- Kunde, S.A.; Musante, L.; Grimme, A.; Fischer, U.; Müller, E.; Wanker, E.E.; Kalscheuer, V.M. The X-chromosome-linked intellectual disability protein PQBP1 is a component of neuronal RNA granules and regulates the appearance of stress granules. Hum. Mol. Genet. 2011, 20, 4916–4931. [Google Scholar] [CrossRef]
- Takahashi, M.; Mizuguchi, M.; Shinoda, H.; Aizawa, T.; Demura, M.; Okazawa, H.; Kawano, K. Polyglutamine tract-binding protein-1 binds to U5-15kD via a continuous 23-residue segment of the C-terminal domain. Biochim. Biophys. Acta 2010, 1804, 1500–1507. [Google Scholar] [CrossRef]
- Courraud, J.; Engel, C.; Quartier, A.; Drouot, N.; Houessou, U.; Plassard, D.; Sorlin, A.; Brischoux-Boucher, E.; Gouy, E.; van Maldergem, L.; et al. Molecular consequences of PQBP1 deficiency, involved in the X-linked Renpenning syndrome. Mol. Psychiatry 2023, 29, 287–296. [Google Scholar] [CrossRef]
- Kurt Colak, F.; Eyerci, N.; Aytekin, C.; Eksioglu, A.S. Renpenning Syndrome in a Turkish Patient: De novo Variant c.607CT in PACS1 and Hypogammaglobulinemia Phenotype. Mol. Syndromol. 2020, 11, 157–161. [Google Scholar] [CrossRef] [PubMed]
- He, T.-S.; Dang, L.; Zhang, J.; Zhang, J.; Wang, G.; Wang, E.; Xia, H.; Zhou, W.; Wu, S.; Liu, X. The Hippo signaling component LATS2 enhances innate immunity to inhibit HIV-1 infection through PQBP1-cGAS pathway. Cell Death Differ. 2022, 29, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhou, R.; Xu, P. The Hippo Pathway in Innate Anti-microbial Immunity and Anti-tumor Immunity. Front. Immunol. 2020, 11, 1473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Meng, F.; Chen, S.; Plouffe, S.W.; Wu, S.; Liu, S.; Li, X.; Zhou, R.; Wang, J.; Zhao, B.; et al. Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade. Nat. Cell Biol. 2017, 19, 362–374. [Google Scholar] [CrossRef]
- Makarova, O.V.; Makarov, E.M.; Urlaub, H.; Will, C.L.; Gentzel, M.; Wilm, M.; Lührmann, R. A subset of human 35S U5 proteins, including Prp19, function prior to catalytic step 1 of splicing. EMBO J. 2004, 23, 2381–2391. [Google Scholar] [CrossRef]
- Goh, J.J.; Manahan-Vaughan, D. Endogenous hippocampal LTD that is enabled by spatial object recognition requires activation of NMDA receptors and the metabotropic glutamate receptor, mGlu5. Hippocampus 2013, 23, 129–138. [Google Scholar] [CrossRef]
- Wilkerson, J.R.; Albanesi, J.P.; Huber, K.M. Roles for Arc in metabotropic glutamate receptor-dependent LTD and synapse elimination: Implications in health and disease. Semin. Cell Dev. Biol. 2018, 77, 51–62. [Google Scholar] [CrossRef]
- Tamura, T.; Horiuchi, D.; Chen, Y.-C.; Sone, M.; Miyashita, T.; Saitoe, M.; Yoshimura, N.; Chiang, A.-S.; Okazawa, H. Drosophila PQBP1 regulates learning acquisition at projection neurons in aversive olfactory conditioning. J. Neurosci. 2010, 30, 14091–14101. [Google Scholar] [CrossRef]
- Elefteriou, F. Regulation of bone remodeling by the central and peripheral nervous system. Arch. Biochem. Biophys. 2008, 473, 231–236. [Google Scholar] [CrossRef]
- Yang, S.-S.; Ishida, T.; Fujita, K.; Nakai, Y.; Ono, T.; Okazawa, H. PQBP1, an intellectual disability causative gene, affects bone development and growth. Biochem. Biophys. Res. Commun. 2020, 523, 894–899. [Google Scholar] [CrossRef]
- Ito, H.; Shiwaku, H.; Yoshida, C.; Homma, H.; Luo, H.; Chen, X.; Fujita, K.; Musante, L.; Fischer, U.; Frints, S.G.M.; et al. In utero gene therapy rescues microcephaly caused by Pqbp1-hypofunction in neural stem progenitor cells. Mol. Psychiatry 2015, 20, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M.; Hartmann, G. Discriminating self from non-self in nucleic acid sensing. Nat. Rev. Immunol. 2016, 16, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shu, C.; Yi, G.; Chaton, C.T.; Shelton, C.L.; Diao, J.; Zuo, X.; Kao, C.C.; Herr, A.B.; Li, P. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity 2013, 39, 1019–1031. [Google Scholar] [CrossRef]
- Gao, D.; Wu, J.; Wu, Y.-T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef]
- Ablasser, A.; Chen, Z.J. cGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363, eaat8657. [Google Scholar] [CrossRef]
- Piacentini, J.; Allen, D.S.; Ganser-Pornillos, B.K.; Chanda, S.K.; Yoh, S.M.; Pornillos, O. Molecular Determinants of PQBP1 Binding to the HIV-1 Capsid Lattice. J. Mol. Biol. 2023, 436, 168409. [Google Scholar] [CrossRef]
- Shannon, J.L.; Murphy, M.S.; Kantheti, U.; Burnett, J.M.; Hahn, M.G.; Dorrity, T.J.; Bacas, C.J.; Mattice, E.B.; Corpuz, K.D.; Barker, B.R. Polyglutamine binding protein 1 (PQBP1) inhibits innate immune responses to cytosolic DNA. Mol. Immunol. 2018, 99, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Spandidos, D.A.; Graham, A.F. Physical and chemical characterization of an avian reovirus. J. Virol. 1976, 19, 968–976. [Google Scholar] [CrossRef]
- Glass, S.E.; Naqi, S.A.; Hall, C.F.; Kerr, K.M. Isolation and Characterization of a Virus Associated with Arthritis of Chickens. Avian Dis. 1973, 17, 415. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Zhang, Q.; Sun, J.; Zhang, X.; Wu, Y. PQBP1 regulates the cellular inflammation induced by avian reovirus and interacts with the viral p17 protein. Virus Res. 2023, 332, 199119. [Google Scholar] [CrossRef]
- Morchikh, M.; Cribier, A.; Raffel, R.; Amraoui, S.; Cau, J.; Severac, D.; Dubois, E.; Schwartz, O.; Bennasser, Y.; Benkirane, M. HEXIM1 and NEAT1 Long Non-coding RNA Form a Multi-subunit Complex that Regulates DNA-Mediated Innate Immune Response. Mol. Cell 2017, 67, 387–399.e5. [Google Scholar] [CrossRef]
- Ferguson, B.J.; Mansur, D.S.; Peters, N.E.; Ren, H.; Smith, G.L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 2012, 1, e00047. [Google Scholar] [CrossRef]
- Stempel, M.; Chan, B.; Brinkmann, M.M. Coevolution pays off: Herpesviruses have the license to escape the DNA sensing pathway. Med. Microbiol. Immunol. 2019, 208, 495–512. [Google Scholar] [CrossRef]
- Du, M.; Chen, Z.J. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 2018, 361, 704–709. [Google Scholar] [CrossRef]
- Xu, G.; Liu, C.; Zhou, S.; Li, Q.; Feng, Y.; Sun, P.; Feng, H.; Gao, Y.; Zhu, J.; Luo, X.; et al. Viral tegument proteins restrict cGAS-DNA phase separation to mediate immune evasion. Mol. Cell 2021, 81, 2823–2837.e9. [Google Scholar] [CrossRef] [PubMed]
- Hew, K.; Dahlroth, S.-L.; Pan, L.X.; Cornvik, T.; Nordlund, P. VP22 core domain from Herpes simplex virus 1 reveals a surprising structural conservation in both the Alpha- and Gammaherpesvirinae subfamilies. J. Gen. Virol. 2015, 96, 1436–1445. [Google Scholar] [CrossRef] [PubMed]
- Roizman, R.; Knipe, D.M.; Whitley, R.J. Herpes Simplex Viruses. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: New York, NY, USA, 2007; Volume 2, pp. 2501–2601. [Google Scholar]
- Paludan, S.R.; Bowie, A.G.; Horan, K.A.; Fitzgerald, K.A. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 2011, 11, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Reinert, L.S.; Lopušná, K.; Winther, H.; Sun, C.; Thomsen, M.K.; Nandakumar, R.; Mogensen, T.H.; Meyer, M.; Vægter, C.; Nyengaard, J.R.; et al. Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat. Commun. 2016, 7, 13348. [Google Scholar] [CrossRef]
- Patrycy, M.; Chodkowski, M.; Krzyzowska, M. Role of Microglia in Herpesvirus-Related Neuroinflammation and Neurodegeneration. Pathogens 2022, 11, 809. [Google Scholar] [CrossRef]
- Feng, S.; Liu, Y.; Zhou, Y.; Shu, Z.; Cheng, Z.; Brenner, C.; Feng, P. Mechanistic insights into the role of herpes simplex virus 1 in Alzheimer’s disease. Front. Aging Neurosci. 2023, 15, 1245904. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Joh, T.H. Microglia, major player in the brain inflammation: Their roles in the pathogenesis of Parkinson’s disease. Exp. Mol. Med. 2006, 38, 333–347. [Google Scholar] [CrossRef]
- Zheng, C.; Zhou, X.-W.; Wang, J.-Z. The dual roles of cytokines in Alzheimer’s disease: Update on interleukins, TNF-α, TGF-β and IFN-γ. Transl. Neurodegener. 2016, 5, 7. [Google Scholar] [CrossRef]
- Blank, T.; Prinz, M. Type I interferon pathway in CNS homeostasis and neurological disorders. Glia 2017, 65, 1397–1406. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Burgaletto, C.; Munafò, A.; Di Benedetto, G.; de Francisci, C.; Caraci, F.; Di Mauro, R.; Bucolo, C.; Bernardini, R.; Cantarella, G. The immune system on the TRAIL of Alzheimer’s disease. J. Neuroinflamm. 2020, 17, 298. [Google Scholar] [CrossRef] [PubMed]
- Paludan, S.R.; Mogensen, T.H. Constitutive and latent immune mechanisms exert s‘ilent’ control of virus infections in the central nervous system. Curr. Opin. Immunol. 2021, 72, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Zayyad, Z.; Spudich, S. Neuropathogenesis of HIV: From initial neuroinvasion to HIV-associated neurocognitive disorder (HAND). Curr. HIV/AIDS Rep. 2015, 12, 16–24. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, B.; Sinha, S.C.; Amin, S.; Gan, L. Mechanism and therapeutic potential of targeting cGAS-STING signaling in neurological disorders. Mol. Neurodegener. 2023, 18, 79. [Google Scholar] [CrossRef]
- Tanaka, H.; Okazawa, H. PQBP1: The Key to Intellectual Disability, Neurodegenerative Diseases, and Innate Immunity. Int. J. Mol. Sci. 2022, 23, 6227. [Google Scholar] [CrossRef]
- Gao, Y.-L.; Wang, N.; Sun, F.-R.; Cao, X.-P.; Zhang, W.; Yu, J.-T. Tau in neurodegenerative disease. Ann. Transl. Med. 2018, 6, 175. [Google Scholar] [CrossRef]
- Zhao, J.; Wu, H.; Tang, X.-Q. Tau internalization: A complex step in tau propagation. Ageing Res. Rev. 2021, 67, 101272. [Google Scholar] [CrossRef] [PubMed]
- Tenchov, R.; Sasso, J.M.; Zhou, Q.A. Polyglutamine (PolyQ) Diseases: Navigating the Landscape of Neurodegeneration. ACS Chem. Neurosci. 2024, 15, 2665–2694. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef]
- Gulen, M.F.; Samson, N.; Keller, A.; Schwabenland, M.; Liu, C.; Glück, S.; Thacker, V.V.; Favre, L.; Mangeat, B.; Kroese, L.J.; et al. cGAS-STING drives ageing-related inflammation and neurodegeneration. Nature 2023, 620, 374–380. [Google Scholar] [CrossRef]
- The Tabula Muris Consortium. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 2020, 583, 590–595. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiench, L.; Rizzo, D.; Sinay, Z.; Nacsa, Z.; Fuchs, N.V.; König, R. Role of PQBP1 in Pathogen Recognition—Impact on Innate Immunity. Viruses 2024, 16, 1340. https://doi.org/10.3390/v16081340
Wiench L, Rizzo D, Sinay Z, Nacsa Z, Fuchs NV, König R. Role of PQBP1 in Pathogen Recognition—Impact on Innate Immunity. Viruses. 2024; 16(8):1340. https://doi.org/10.3390/v16081340
Chicago/Turabian StyleWiench, Lukas, Domenico Rizzo, Zora Sinay, Zsófia Nacsa, Nina V. Fuchs, and Renate König. 2024. "Role of PQBP1 in Pathogen Recognition—Impact on Innate Immunity" Viruses 16, no. 8: 1340. https://doi.org/10.3390/v16081340
APA StyleWiench, L., Rizzo, D., Sinay, Z., Nacsa, Z., Fuchs, N. V., & König, R. (2024). Role of PQBP1 in Pathogen Recognition—Impact on Innate Immunity. Viruses, 16(8), 1340. https://doi.org/10.3390/v16081340