


Towards the Development of a Minigenome Assay for Species A Rotaviruses

 and
and
Abstract
:1. Introduction
2. Materials and Methods
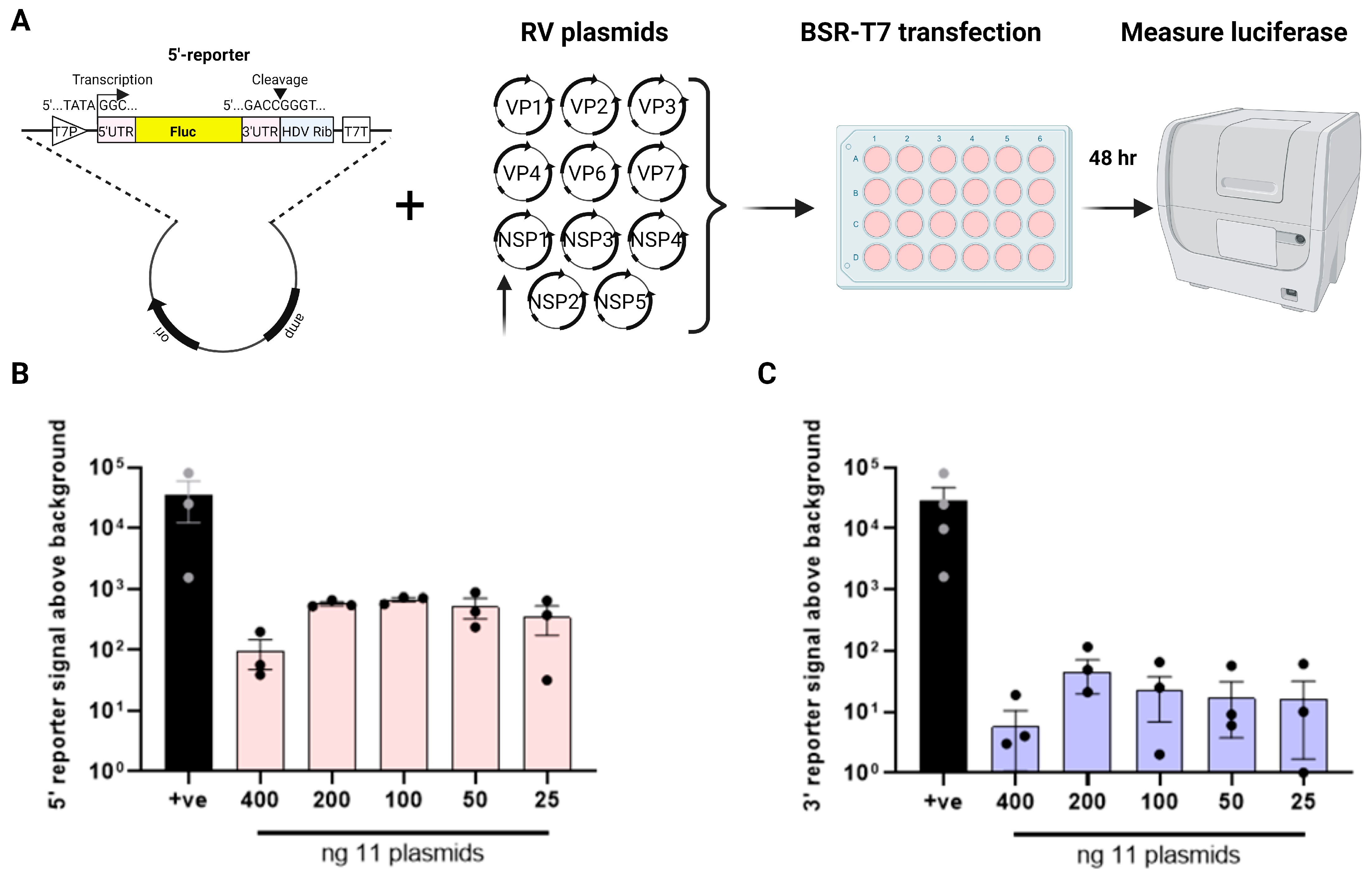
3. Results
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Troeger, C.; Khalil, I.A.; Rao, P.C.; Cao, S.; Blacker, B.F.; Ahmed, T.; Armah, G.; Bines, J.E.; Brewer, T.G.; Colombara, D.V.; et al. Rotavirus Vaccination and the Global Burden of Rotavirus Diarrhea among Children Younger than 5 Years. JAMA Pediatr. 2018, 172, 958–965. [Google Scholar] [CrossRef]
- Bányai, K.; Kemenesi, G.; Budinski, I.; Földes, F.; Zana, B.; Marton, S.; Varga-Kugler, R.; Oldal, M.; Kurucz, K.; Jakab, F. Candidate new rotavirus species in Schreiber’s bats, Serbia. Infect. Genet. Evol. 2017, 48, 19–26. [Google Scholar] [CrossRef]
- Johne, R.; Schilling-Loeffler, K.; Ulrich, R.G.; Tausch, S.H. Whole Genome Sequence Analysis of a Prototype Strain of the Novel Putative Rotavirus Species L. Viruses 2022, 14, 462. [Google Scholar] [CrossRef]
- Johne, R.; Tausch, S.H.; Ulrich, R.G.; Schilling-Loeffler, K. Genome analysis of the novel putative rotavirus species K. Virus Res. 2023, 334, 199171. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Otto, P.H.; Ciarlet, M.; Desselberger, U.; Van Ranst, M.; Johne, R. VP6-sequence-based cutoff values as a criterion for rotavirus species demarcation. Arch. Virol. 2012, 157, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Mihalov-Kovács, E.; Gellért, Á.; Marton, S.; Farkas, S.L.; Fehér, E.; Oldal, M.; Jakab, F.; Martella, V.; Bányai, K. Candidate new rotavirus species in sheltered dogs, Hungary. Emerg. Infect. Dis. 2015, 21, 660–663. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Baker, M.L.; Jiang, W.; Estes, M.K.; Prasad, B.V. Rotavirus architecture at subnanometer resolution. J. Virol. 2009, 83, 1754–1766. [Google Scholar] [CrossRef]
- Settembre, E.C.; Chen, J.Z.; Dormitzer, P.R.; Grigorieff, N.; Harrison, S.C. Atomic model of an infectious rotavirus particle. Embo J. 2011, 30, 408–416. [Google Scholar] [CrossRef]
- McClain, B.; Settembre, E.; Temple, B.R.; Bellamy, A.R.; Harrison, S.C. X-ray crystal structure of the rotavirus inner capsid particle at 3.8 A resolution. J. Mol. Biol. 2010, 397, 587–599. [Google Scholar] [CrossRef]
- Estrozi, L.F.; Settembre, E.C.; Goret, G.; McClain, B.; Zhang, X.; Chen, J.Z.; Grigorieff, N.; Harrison, S.C. Location of the dsRNA-dependent polymerase, VP1, in rotavirus particles. J. Mol. Biol. 2013, 425, 124–132. [Google Scholar] [CrossRef]
- Prasad, B.V.V.; Rothnagel, R.; Zeng, C.Q.Y.; Jakana, J.; Lawton, J.A.; Chiu, W.; Estes, M.K. Visualization of ordered genomic RNA and localization of transcriptional complexes in rotavirus. Nature 1996, 382, 471–473. [Google Scholar] [CrossRef]
- Guglielmi, K.M.; McDonald, S.M.; Patton, J.T. Mechanism of intraparticle synthesis of the rotavirus double-stranded RNA genome. J. Biol. Chem. 2010, 285, 18123–18128. [Google Scholar] [CrossRef] [PubMed]
- Jenni, S.; Salgado, E.N.; Herrmann, T.; Li, Z.; Grant, T.; Grigorieff, N.; Trapani, S.; Estrozi, L.F.; Harrison, S.C. In situ Structure of Rotavirus VP1 RNA-Dependent RNA Polymerase. J. Mol. Biol. 2019, 431, 3124–3138. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Ding, S.D.; Greenberg, H.B.; Estes, M.K. Rotaviruses. In Fields Virology: RNA Viruses; Wolters Kluwer: Alphen aan den Rijn, The Netherlands, 2023. [Google Scholar]
- Periz, J.; Celma, C.; Jing, B.; Pinkney, J.N.; Roy, P.; Kapanidis, A.N. Rotavirus mRNAS are released by transcript-specific channels in the double-layered viral capsid. Proc. Natl. Acad. Sci. USA 2013, 110, 12042–12047. [Google Scholar] [CrossRef] [PubMed]
- Tortorici, M.A.; Broering, T.J.; Nibert, M.L.; Patton, J.T. Template recognition and formation of initiation complexes by the replicase of a segmented double-stranded RNA virus. J. Biol. Chem. 2003, 278, 32673–32682. [Google Scholar] [CrossRef]
- Tortorici, M.A.; Shapiro, B.A.; Patton, J.T. A base-specific recognition signal in the 5′ consensus sequence of rotavirus plus-strand RNAs promotes replication of the double-stranded RNA genome segments. RNA 2006, 12, 133–146. [Google Scholar] [CrossRef]
- Ding, K.; Celma, C.C.; Zhang, X.; Chang, T.; Shen, W.; Atanasov, I.; Roy, P.; Zhou, Z.H. In situ structures of rotavirus polymerase in action and mechanism of mRNA transcription and release. Nat. Commun. 2019, 10, 2216. [Google Scholar] [CrossRef]
- Lu, X.; McDonald, S.M.; Tortorici, M.A.; Tao, Y.J.; Vasquez-Del Carpio, R.; Nibert, M.L.; Patton, J.T.; Harrison, S.C. Mechanism for coordinated RNA packaging and genome replication by rotavirus polymerase VP1. Structure 2008, 16, 1678–1688. [Google Scholar] [CrossRef]
- Barro, M.; Mandiola, P.; Chen, D.; Patton, J.T.; Spencer, E. Identification of sequences in rotavirus mRNAs important for minus strand synthesis using antisense oligonucleotides. Virology 2001, 288, 71–80. [Google Scholar] [CrossRef]
- Chen, D.; Barros, M.; Spencer, E.; Patton, J.T. Features of the 3′-consensus sequence of rotavirus mRNAs critical to minus strand synthesis. Virology 2001, 282, 221–229. [Google Scholar] [CrossRef]
- Navarro, A.; Trask, S.D.; Patton, J.T. Generation of genetically stable recombinant rotaviruses containing novel genome rearrangements and heterologous sequences by reverse genetics. J. Virol. 2013, 87, 6211–6220. [Google Scholar] [CrossRef] [PubMed]
- Patton, J.T.; Chnaiderman, J.; Spencer, E. Open reading frame in rotavirus mRNA specifically promotes synthesis of double-stranded RNA: Template size also affects replication efficiency. Virology 1999, 264, 167–180. [Google Scholar] [CrossRef]
- Chen, D.; Patton, J.T. Rotavirus RNA replication requires a single-stranded 3′ end for efficient minus-strand synthesis. J. Virol. 1998, 72, 7387–7396. [Google Scholar] [CrossRef]
- Li, W.; Manktelow, E.; von Kirchbach, J.C.; Gog, J.R.; Desselberger, U.; Lever, A.M. Genomic analysis of codon, sequence and structural conservation with selective biochemical-structure mapping reveals highly conserved and dynamic structures in rotavirus RNAs with potential cis-acting functions. Nucleic Acids Res. 2010, 38, 7718–7735. [Google Scholar] [CrossRef] [PubMed]
- Ogden, K.M.; Ramanathan, H.N.; Patton, J.T. Mutational analysis of residues involved in nucleotide and divalent cation stabilization in the rotavirus RNA-dependent RNA polymerase catalytic pocket. Virology 2012, 431, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Steger, C.; Brown, M.; Sullivan, O.; Boudreaux, C.; Cohen, C.; LaConte, L.; McDonald, S. In Vitro Double-Stranded RNA Synthesis by Rotavirus Polymerase Mutants with Lesions at Core Shell Contact Sites. J. Virol. 2019, 93, 10–1128. [Google Scholar] [CrossRef]
- Tao, Y.; Farsetta, D.L.; Nibert, M.L.; Harrison, S.C. RNA synthesis in a cage--structural studies of reovirus polymerase lambda3. Cell 2002, 111, 733–745. [Google Scholar] [CrossRef]
- Zeng, C.Q.; Estes, M.K.; Charpilienne, A.; Cohen, J. The N terminus of rotavirus VP2 is necessary for encapsidation of VP1 and VP3. J. Virol. 1998, 72, 201–208. [Google Scholar] [CrossRef]
- Gridley, C.L.; Patton, J.T. Regulation of rotavirus polymerase activity by inner capsid proteins. Curr. Opin. Virol. 2014, 9, 31–38. [Google Scholar] [CrossRef]
- Patton, J.T.; Jones, M.T.; Kalbach, A.N.; He, Y.W.; Xiaobo, J. Rotavirus RNA polymerase requires the core shell protein to synthesize the double-stranded RNA genome. J. Virol. 1997, 71, 9618–9626. [Google Scholar] [CrossRef]
- Diebold, O.; Gonzalez, V.; Venditti, L.; Sharp, C.; Blake, R.A.; Tan, W.S.; Stevens, J.; Caddy, S.; Digard, P.; Borodavka, A.; et al. Using Species a Rotavirus Reverse Genetics to Engineer Chimeric Viruses Expressing SARS-CoV-2 Spike Epitopes. J. Virol. 2022, 96, e0048822. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Komoto, S.; Kawagishi, T.; Nouda, R.; Nagasawa, N.; Onishi, M.; Matsuura, Y.; Taniguchi, K.; Kobayashi, T. Entirely plasmid-based reverse genetics system for rotaviruses. Proc. Natl. Acad. Sci. USA 2017, 114, 2349. [Google Scholar] [CrossRef]
- Komoto, S.; Fukuda, S.; Ide, T.; Ito, N.; Sugiyama, M.; Yoshikawa, T.; Murata, T.; Taniguchi, K. Generation of Recombinant Rotaviruses Expressing Fluorescent Proteins by Using an Optimized Reverse Genetics System. J. Virol. 2018, 92, e00588-18. [Google Scholar] [CrossRef] [PubMed]
- Komoto, S.; Kanai, Y.; Fukuda, S.; Kugita, M.; Kawagishi, T.; Ito, N.; Sugiyama, M.; Matsuura, Y.; Kobayashi, T.; Taniguchi, K. Reverse Genetics System Demonstrates that Rotavirus Nonstructural Protein NSP6 Is Not Essential for Viral Replication in Cell Culture. J. Virol. 2017, 91, e00695-17. [Google Scholar] [CrossRef]
- Percy, N.; Barclay, W.S.; Sullivan, M.; Almond, J.W. A poliovirus replicon containing the chloramphenicol acetyltransferase gene can be used to study the replication and encapsidation of poliovirus RNA. J. Virol. 1992, 66, 5040–5046. [Google Scholar] [CrossRef]
- Sidhu, M.S.; Chan, J.; Kaelin, K.; Spielhofer, P.; Radecke, F.; Schneider, H.; Masurekar, M.; Dowling, P.C.; Billeter, M.A.; Udem, S.A. Rescue of Synthetic Measles Virus Minireplicons: Measles Genomic Termini Direct Efficient Expression and Propagation of a Reporter Gene. Virology 1995, 208, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Groseth, A.; Feldmann, H.; Theriault, S.; Mehmetoglu, G.; Flick, R. RNA Polymerase I-Driven Minigenome System for Ebola Viruses. J. Virol. 2005, 79, 4425–4433. [Google Scholar] [CrossRef]
- Lutz, A.; Dyall, J.; Olivo, P.D.; Pekosz, A. Virus-inducible reporter genes as a tool for detecting and quantifying influenza A virus replication. J. Virol. Methods 2005, 126, 13–20. [Google Scholar] [CrossRef]
- Simmonds, P. SSE: A nucleotide and amino acid sequence analysis platform. BMC Res. Notes 2012, 5, 50. [Google Scholar] [CrossRef]
- Matrosovich, M.; Matrosovich, T.; Garten, W.; Klenk, H.-D. New low-viscosity overlay medium for viral plaque assays. Virol. J. 2006, 3, 63. [Google Scholar] [CrossRef]
- Arnold, M.; Patton, J.T.; McDonald, S.M. Culturing, Storage, and Quantification of Rotaviruses. Curr. Protoc. Microbiol. 2009, 15, 15C-3. [Google Scholar] [CrossRef] [PubMed]
- Wise, H.M.; Foeglein, A.; Sun, J.; Dalton Rosa, M.; Patel, S.; Howard, W.; Anderson, E.C.; Barclay, W.S.; Digard, P. A Complicated Message: Identification of a Novel PB1-Related Protein Translated from Influenza A Virus Segment 2 mRNA. J. Virol. 2009, 83, 8021–8031. [Google Scholar] [CrossRef]
- Roner, M.R.; Joklik, W.K. Reovirus reverse genetics: Incorporation of the CAT gene into the reovirus genome. Proc. Natl. Acad. Sci. USA 2001, 98, 8036–8041. [Google Scholar] [CrossRef] [PubMed]
- Desselberger, U. Rotaviruses. Virus Res. 2014, 190, 75–96. [Google Scholar] [CrossRef] [PubMed]
- te Velthuis, A.; Long, J.; Barclay, W. Assays to Measure the Activity of Influenza Virus Polymerase. Influenza Virus Methods Protoc. 2018, 1836, 343–374. [Google Scholar]
- Noton, S.L.; Cowton, V.M.; Zack, C.R.; McGivern, D.R.; Fearns, R. Evidence that the polymerase of respiratory syncytial virus initiates RNA replication in a nontemplated fashion. Proc. Natl. Acad. Sci. USA 2010, 107, 10226–10231. [Google Scholar] [CrossRef]
- Paul, A.V.; Rieder, E.; Kim, D.W.; Boom, J.H.v.; Wimmer, E. Identification of an RNA Hairpin in Poliovirus RNA That Serves as the Primary Template in the In Vitro Uridylylation of VPg. J. Virol. 2000, 74, 10359–10370. [Google Scholar] [CrossRef]
- Wehrfritz, J.M.; Boyce, M.; Mirza, S.; Roy, P. Reconstitution of bluetongue virus polymerase activity from isolated domains based on a three-dimensional structural model. Biopolymers 2007, 86, 83–94. [Google Scholar] [CrossRef]
- Vende, P.; Piron, M.; Castagné, N.; Poncet, D. Efficient Translation of Rotavirus mRNA Requires Simultaneous Interaction of NSP3 with the Eukaryotic Translation Initiation Factor eIF4G and the mRNA 3′ End. J. Virol. 2000, 74, 7064–7071. [Google Scholar] [CrossRef]
- Sánchez-Tacuba, L.; Feng, N.; Meade, N.J.; Mellits, K.H.; Jaïs, P.H.; Yasukawa, L.L.; Resch, T.K.; Jiang, B.; López, S.; Ding, S.; et al. An Optimized Reverse Genetics System Suitable for Efficient Recovery of Simian, Human, and Murine-Like Rotaviruses. J. Virol. 2020, 94, 10–1128. [Google Scholar] [CrossRef]
- Zheng, H.; Palese, P.; García-Sastre, A. Nonconserved nucleotides at the 3′ and 5′ ends of an influenza A virus RNA play an important role in viral RNA replication. Virology 1996, 217, 242–251. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target gene | Sequence (5′ to 3′) | Use |
|---|---|---|
| Fluc in pMA plasmid | TAATACGACTCACTATAGGG TCGTCCACTCGGATGGCTA | Sequence 5′- and 3′-plasmids containing Fluc gene |
| VP1 plasmid | GGAAGGAGAGATGTACCAGGA | Sequence mutations of GDD motif in VP1 plasmid |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diebold, O.; Zhou, S.; Sharp, C.P.; Tesla, B.; Chook, H.W.; Digard, P.; Gaunt, E.R. Towards the Development of a Minigenome Assay for Species A Rotaviruses. Viruses 2024, 16, 1396. https://doi.org/10.3390/v16091396
Diebold O, Zhou S, Sharp CP, Tesla B, Chook HW, Digard P, Gaunt ER. Towards the Development of a Minigenome Assay for Species A Rotaviruses. Viruses. 2024; 16(9):1396. https://doi.org/10.3390/v16091396
Chicago/Turabian StyleDiebold, Ola, Shu Zhou, Colin Peter Sharp, Blanka Tesla, Hou Wei Chook, Paul Digard, and Eleanor R. Gaunt. 2024. "Towards the Development of a Minigenome Assay for Species A Rotaviruses" Viruses 16, no. 9: 1396. https://doi.org/10.3390/v16091396