
Targeting Yezo Virus Structural Proteins for Multi-Epitope Vaccine Design Using Immunoinformatics Approach

 , ,
, ,  ,
,  ,
,  and
and
Abstract
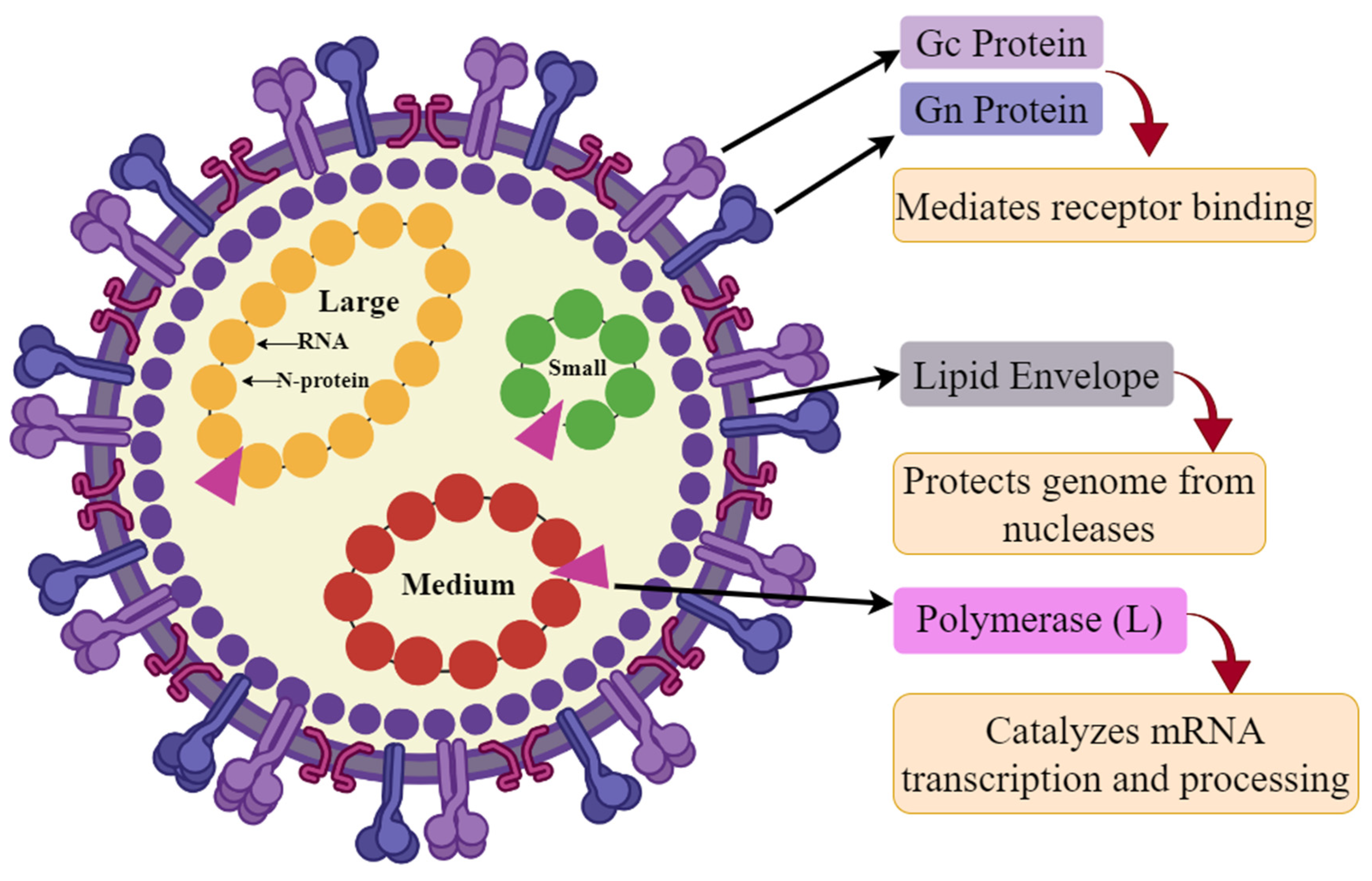
1. Introduction
2. Materials and Methods
2.1. Dataset Collection and Its Filtration
2.2. Prediction of Cytotoxic T-Lymphocyte (CTL) Epitopes
2.3. Predicting Helper T-Lymphocyte (HTL) Epitopes and Evaluating Interferon-Gamma (IFN-γ) Induction
2.4. Prediction of Linear B-Lymphocyte (LBL) Epitopes
2.5. Population Coverage Analysis of MHC Alleles
2.6. Molecular Docking Analysis of CTL and HTL Epitopes with MHC Alleles
2.7. Designing of the Multi-Epitope Vaccine Construct against YEZV
2.8. Evaluation of Physicochemical Properties of the Design Vaccine Construct
2.9. Vaccine Structures Prediction and Its Validation
2.10. Disulfide Bonds Engineering
2.11. Protein-Protein Docking
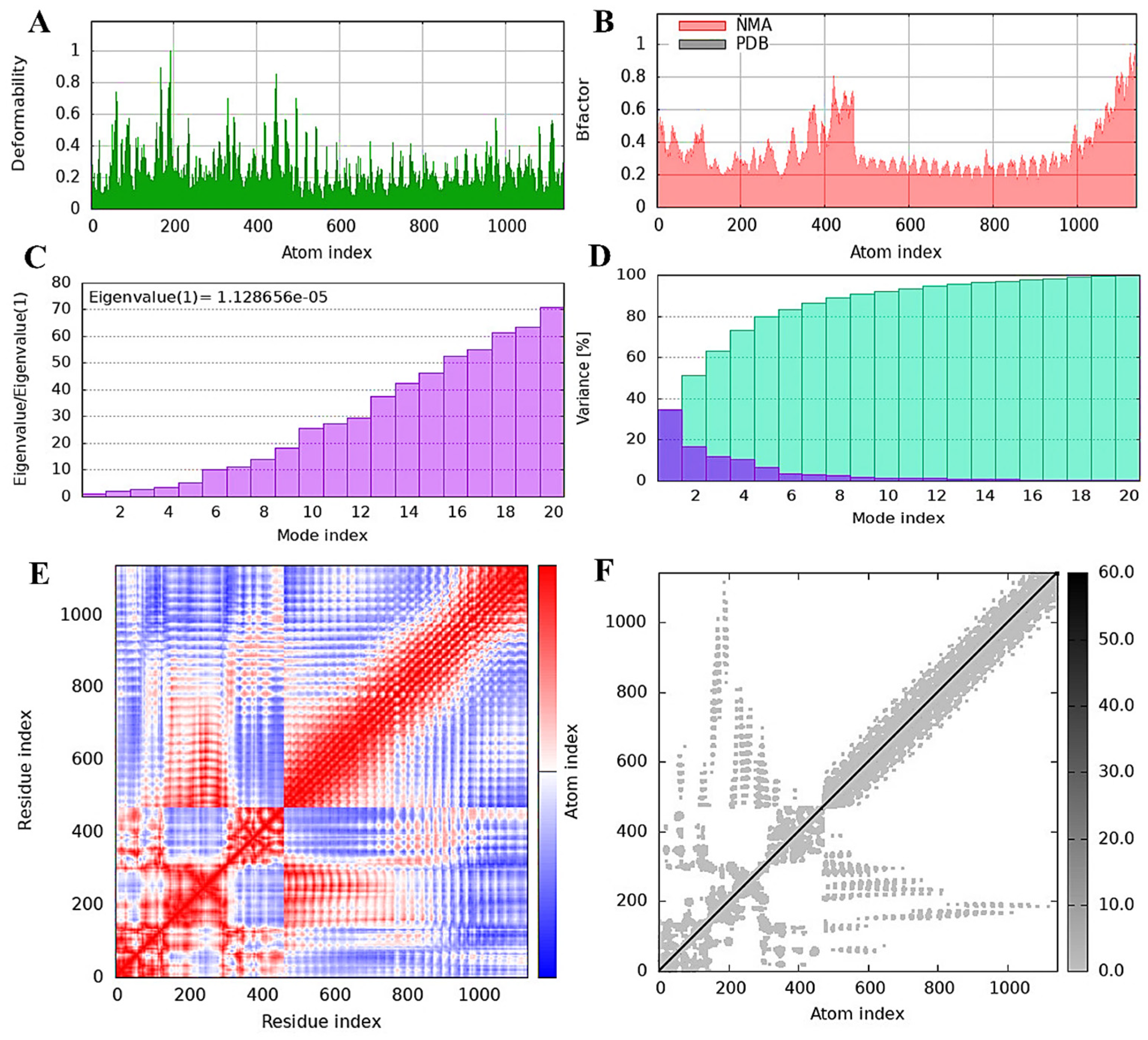
2.12. Evaluating Vaccine Construct Mobility and Stability through NMA Analysis
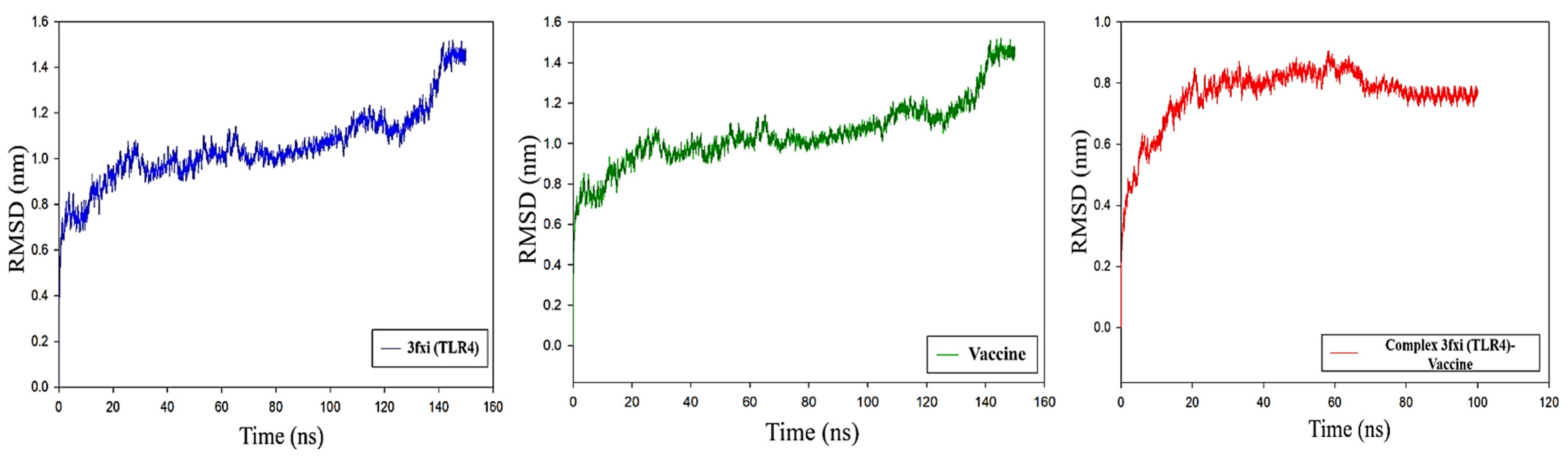
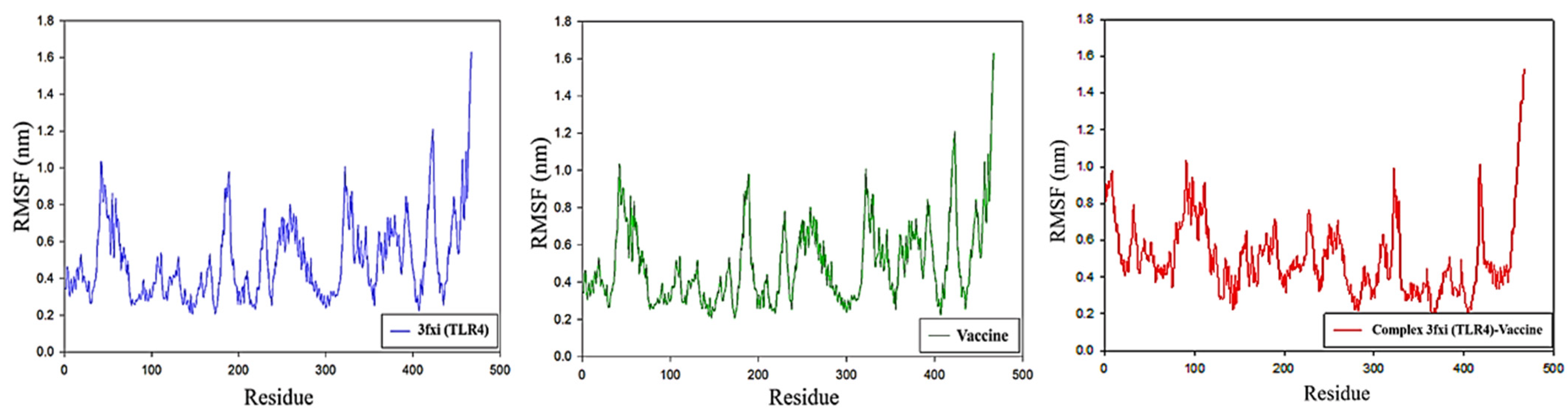
2.13. Molecular Dynamic Simulation
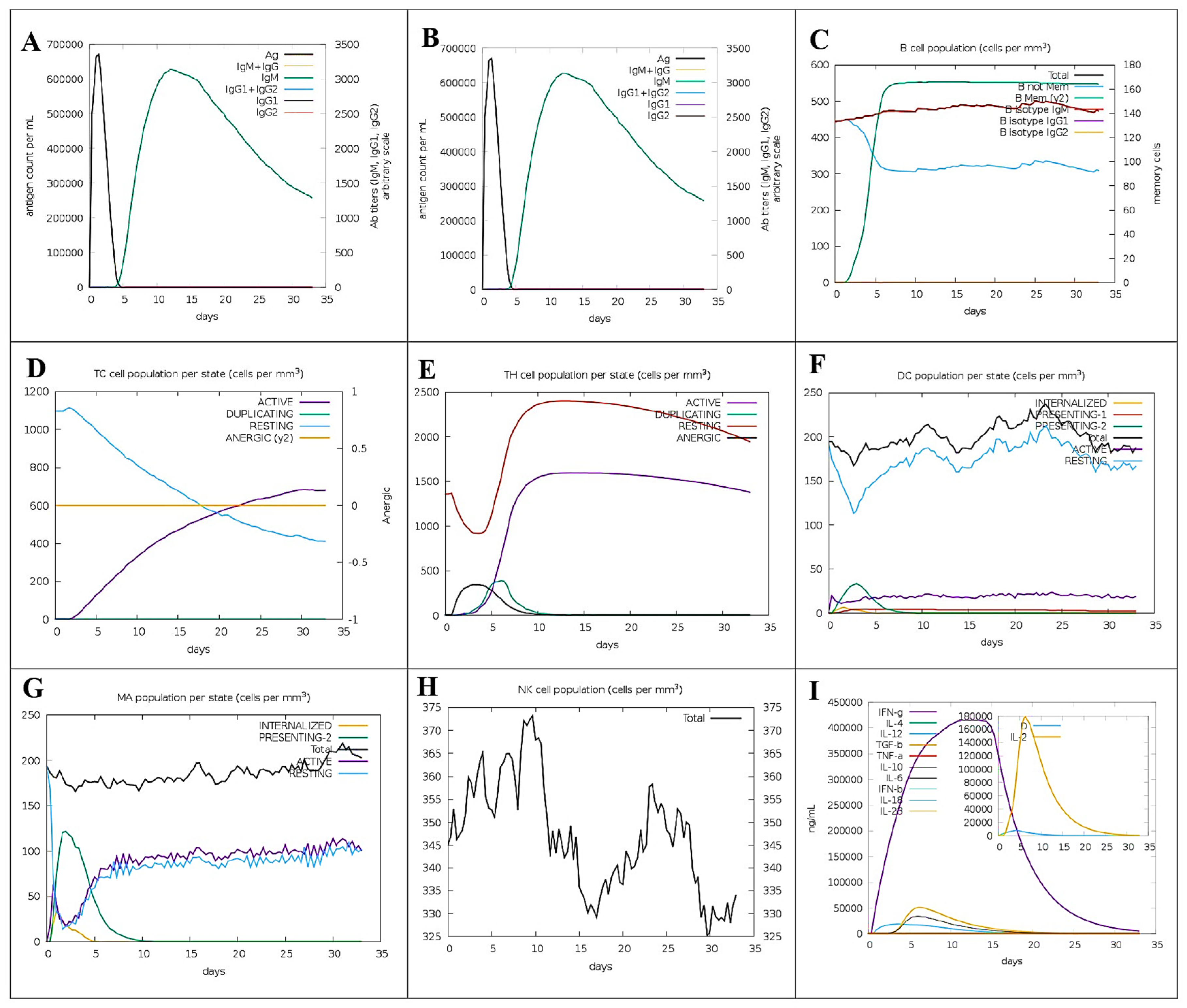
2.14. Immune Simulation
2.15. Predicting Vaccine mRNA Secondary Structure
3. Results
3.1. Proteome Retrieval and Its Screening
3.2. Screening of Epitopes
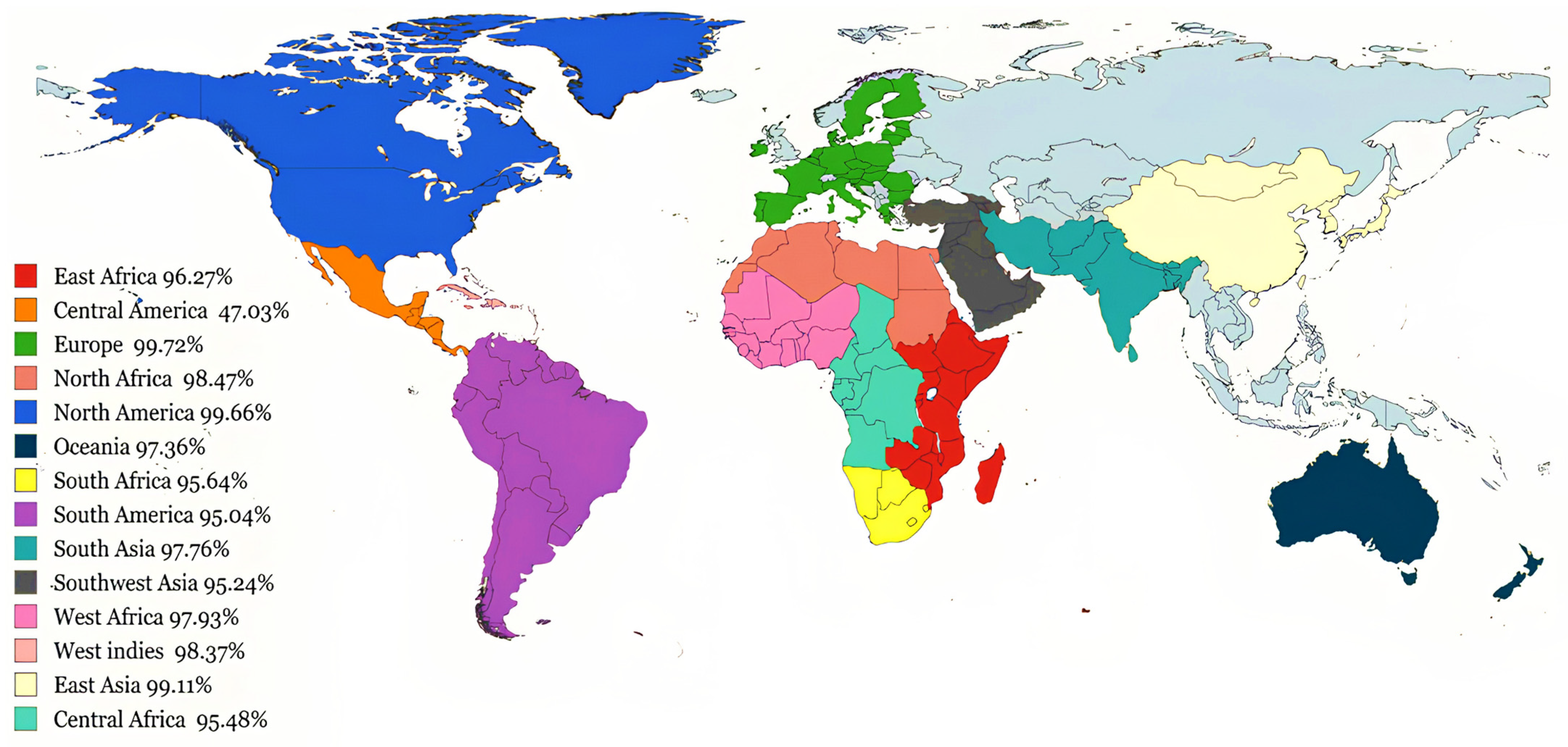
3.3. Global Coverage of Predicted Immunogenic Epitopes
3.4. Molecular Docking Analysis of CTL and HTL Epitopes with HLA Alleles
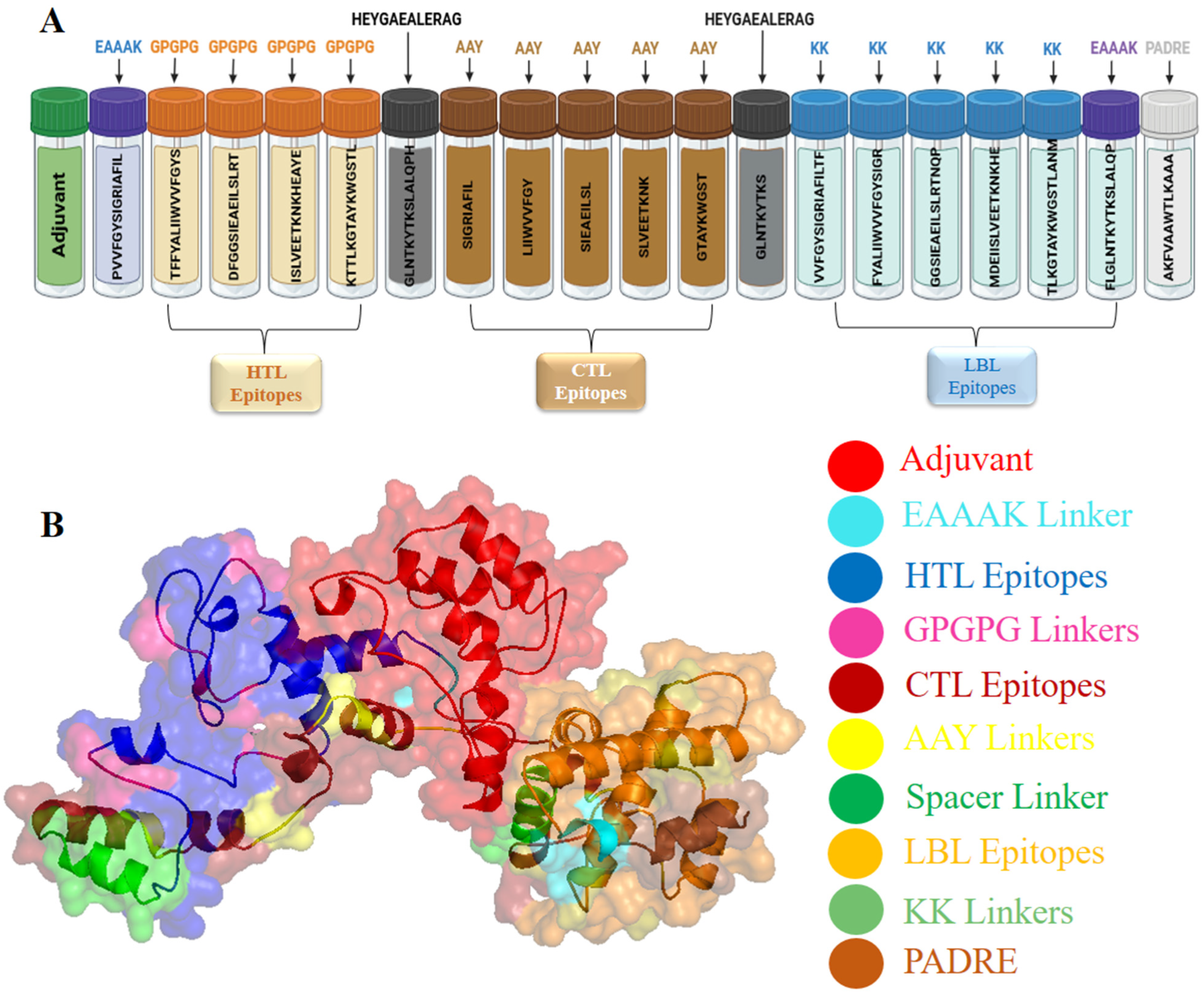
3.5. Formulation and Designing of Vaccine Construct
3.6. Physiochemical Properties of the Designed Vaccine Construct
3.7. 2D and 3D Structure Prediction, Refinement, and Its Validation
3.8. Engineering of Disulfide Bond and Evaluation of Mutant Vaccine Construct
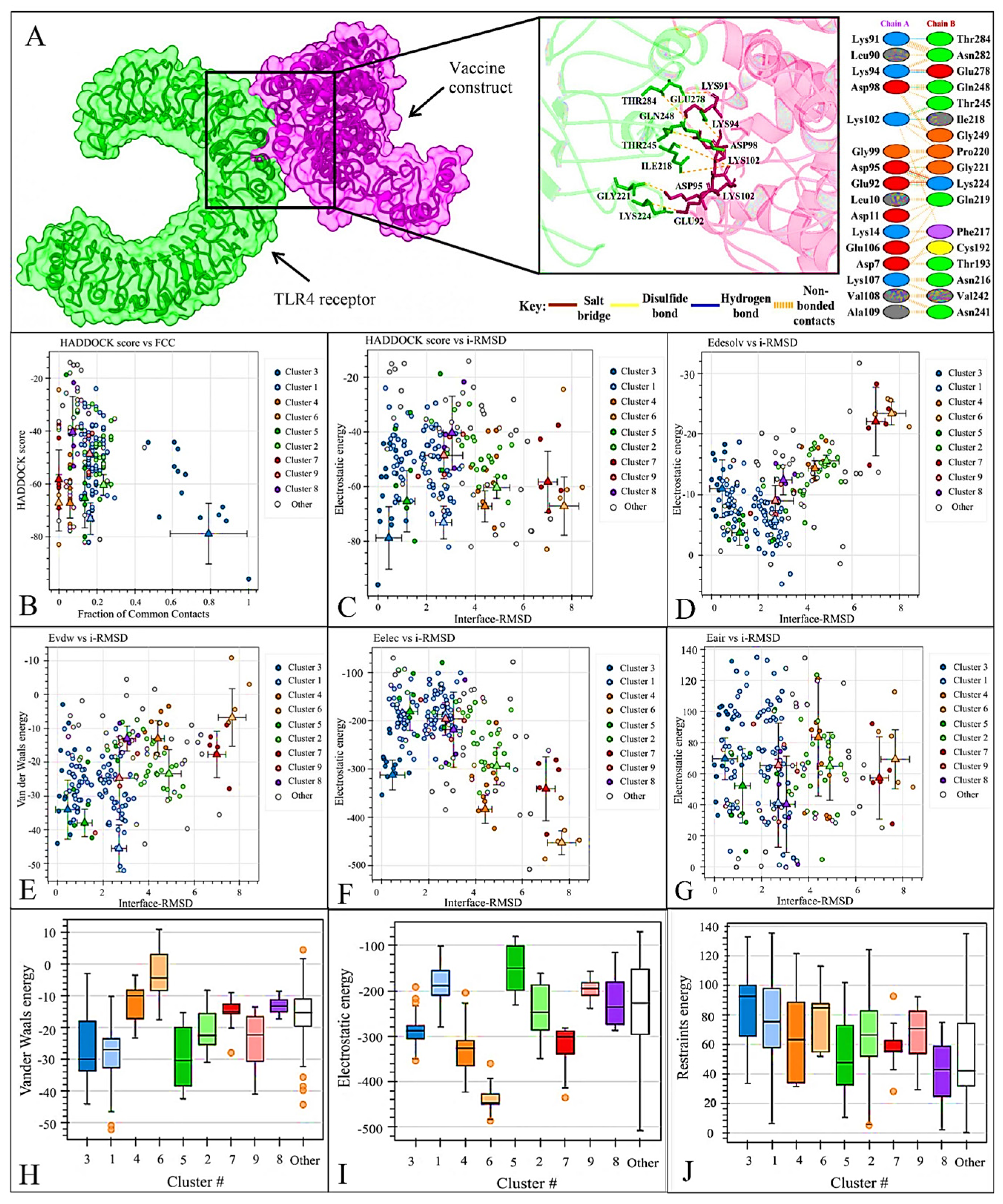
3.9. Docking of Vaccine Construct with TLR4 Receptor
3.10. TLR4 Receptor and Vaccine Construct Interactions and Stability: NMA Insights
3.11. Molecular Dynamics Analysis of Protein-Protein Interactions
RMSD Analysis
3.12. RMSF Analysis
3.13. Virtual Immunogenicity Assessment
3.14. Prediction of the Vaccine mRNA Secondary Structure
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, Y.; Pascall, D.J. Characterization of putative novel tick viruses and zoonotic risk prediction. Ecol. Evol. 2024, 14, e10814. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.H.; Alkhovsky, S.V.; Avšič-Županc, T.; Bergeron, É.; Burt, F.; Ergünay, K.; Garrison, A.R.; Marklewitz, M.; Mirazimi, A.; Papa, A.; et al. ICTV Virus Taxonomy Profile: Nairoviridae 2024. J. Gen. Virol. 2024, 105, 001974. [Google Scholar] [CrossRef]
- Hitesh, J.; Gediya Shweta, K.; Thakkar Dadhichi, K.; Mansuri Nasimabanu, Y.; Ashar Komal, M.; Pasha, T.Y. Crimean Congo Hemorrhagic Fever Virus (Cchfv). J. Pharm. Biomed. Sci. JPBMS 2011, 5. [Google Scholar]
- Crabtree, M.B.; Sang, R.; Miller, B.R. Kupe virus, a new virus in the family Bunyaviridae, genus Nairovirus, Kenya. Emerg. Infect. Dis. 2009, 15, 147. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K. Yezo virus and emerging orthonairovirus diseases. Uirusu 2021, 71, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Bratuleanu, B.E.; Răileanu, C.; Bennouna, A.; Chretien, D.; Bigot, T.; Guardado-Calvo, P.; Savuta, G.; Moutailler, S.; Eloit, M.; Temmam, S. Diversity of Viruses in Ixodes ricinus in Europe including Novel and Potential Arboviruses. Transbound. Emerg. Dis. 2023, 2023, 6661723. [Google Scholar] [CrossRef]
- Kodama, F.; Edakawa, S.; Nagasaka, A.; Matsuno, K.; Yoshii, K.; Sawa, H.; Yamagishi, A.; Furusawa, Y.; Yamaguchi, R.; Yano, K.; et al. Discovery of a tick-borne acute febrile disease caused by a novel orthonairovirus (Yezo virus) in Hokkaido. IASR 2020, 41, 11–13. [Google Scholar]
- Lv, X.; Liu, Z.; Li, L.; Xu, W.; Yuan, Y.; Liang, X.; Zhang, L.; Wei, Z.; Sui, L.; Zhao, Y.; et al. Yezo virus infection in tick-bitten patient and ticks, northeastern China. Emerg. Infect. Dis. 2023, 29, 797. [Google Scholar] [CrossRef]
- Ogata, Y.; Sato, T.; Kato, K.; Kikuchi, K.; Mitsuhashi, K.; Watari, K.; Tamiya, K.; Goto, A.; Yamaguchi, H.; Hisada, R. A case of tick-borne Yezo virus infection: Concurrent detection in the patient and tick. Int. J. Infect. Dis. 2024, 143, 107038. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, L.; Shi, W. The human-infection potential of emerging tick-borne viruses is a global public health concern. Nat. Rev. Microbiol. 2023, 21, 215–217. [Google Scholar] [CrossRef]
- Kodama, F.; Yamaguchi, H.; Park, E.; Tatemoto, K.; Sashika, M.; Nakao, R.; Terauchi, Y.; Mizuma, K.; Orba, Y.; Kariwa, H.; et al. A novel nairovirus associated with acute febrile illness in Hokkaido, Japan. Nat. Commun. 2021, 12, 5539. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Devi, Y.D.; Devi, A.; Gogoi, H.; Dehingia, B.; Doley, R.; Buragohain, A.K.; Singh, C.S.; Borah, P.P.; Rao, C.D.; Ray, P.; et al. Exploring rotavirus proteome to identify potential B-and T-cell epitope using computational immunoinformatics. Heliyon 2020, 6, e05760. [Google Scholar] [CrossRef]
- Ali, A.; Ahmad, S.; Wadood, A.; Rehman, A.U.; Zahid, H.; Qayash Khan, M.; Nawab, J.; Rahman, Z.U.; Alouffi, A.S. Modeling novel putative drugs and vaccine candidates against tick-borne pathogens: A subtractive proteomics approach. Vet. Sci. 2020, 7, 129. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Ahmad, S.; de Albuquerque, P.M.M.; Kamil, A.; Alshammari, F.A.; Alouffi, A.; da Silva Vaz, I., Jr. Prediction of novel drug targets and vaccine candidates against human lice (Insecta), Acari (Arachnida), and their associated pathogens. Vaccines 2021, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Chiou, C.C.; Ahmad, S.; Islam, Z.U.; Tanaka, T.; Alouffi, A.; Chen, C.C.; Almutairi, M.M.; Ali, A. Subtractive Proteomics and Reverse-Vaccinology Approaches for Novel Drug Target Identification and Chimeric Vaccine Development against Bartonella henselae Strain Houston-1. Bioengineering 2024, 11, 505. [Google Scholar] [CrossRef]
- Tomar, N.; De, R.K. Immunoinformatics: An integrated scenario. Immunology 2010, 131, 153–168. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Toth, I. Peptide-Based Synthetic Vaccines. Chem. Sci. 2016, 7, 842–854. [Google Scholar] [CrossRef]
- Chen, Q.; Wan, Y.; Lei, Y.; Zobel, J.; Verspoor, K. Evaluation of CD-HIT for constructing non-redundant databases. In Proceedings of the 2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Shenzhen, China, 15–18 December 2016; pp. 703–706. [Google Scholar]
- Stover, N.A.; Cavalcanti, A.R. Using NCBI BLAST. Curr. Protoc. Essent. Lab. Tech. 2017, 14, 11.1.1–11.1.34. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P.; et al. IEDB-AR: Immune epitope database—Analysis resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, P.; Kim, Y.; Haste-Andersen, P.; Beaver, J.; Bourne, P.E.; Bui, H.H.; Buus, S.; Frankild, S.; Greenbaum, J.; et al. Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Res. 2008, 36 (Suppl. S2), W513–W518. [Google Scholar] [CrossRef]
- Anwar, S.; Mourosi, J.T.; Khan, M.F.; Hosen, M.J. Prediction of epitope-based peptide vaccine against the Chikungunya virus by immuno-informatics approach. Curr. Pharm. Biotechnol. 2020, 21, 325–340. [Google Scholar] [CrossRef]
- Paul, S.; Croft, N.P.; Purcell, A.W.; Tscharke, D.C.; Sette, A.; Nielsen, M.; Peters, B. Benchmarking predictions of MHC class I restricted T cell epitopes in a comprehensively studied model system. PLoS Comput. Biol. 2020, 16, e1007757. [Google Scholar] [CrossRef]
- Chatterjee, R.; Mahapatra, S.R.; Dey, J.; Raj Takur, K.; Raina, V.; Misra, N.; Suar, M. An immunoinformatics and structural vaccinology study to design a multi-epitope vaccine against Staphylococcus aureus infection. J. Mol. Recognit. 2023, 36, e3007. [Google Scholar] [CrossRef] [PubMed]
- Bui, H.H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006, 7, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Dong, X.; Li, X.; Liu, Z.; Liu, H. Prediction of disulfide bond engineering sites using a machine learning method. Sci. Rep. 2020, 10, 10330. [Google Scholar] [CrossRef] [PubMed]
- Bhowmik, R.; Nath, R.; Sharma, S.; Roy, R.; Biswas, R. High-throughput screening and dynamic studies of selected compounds against SARS-CoV-2. Int. J. Appl. Pharm. 2022, 14, 251–260. [Google Scholar] [CrossRef]
- Rehman, A.; Ahmad, S.; Shahid, F.; Albutti, A.; Alwashmi, A.S.; Aljasir, M.A.; Alhumeed, N.; Qasim, M.; Ashfaq, U.A.; Tahir ul Qamar, M. Integrated core proteomics, subtractive proteomics, and immunoinformatics investigation to unveil a potential multi-epitope vaccine against schistosomiasis. Vaccines 2021, 9, 658. [Google Scholar] [CrossRef]
- Moro-García, M.A.; Mayo, J.C.; Sainz, R.M.; Alonso-Arias, R. Influence of inflammation in the process of T lymphocyte differentiation: Proliferative, metabolic, and oxidative changes. Front. Immunol. 2018, 9, 339. [Google Scholar] [CrossRef]
- Wherry, E.J.; Masopust, D. Adaptive immunity: Neutralizing, eliminating, and remembering for the next time. In Viral Pathogenesis; Academic Press: Cambridge, MA, USA, 2016; pp. 57–69. [Google Scholar]
- Verhoef, J.; van Kessel, K.; Snippe, H. Immune response in human pathology: Infections caused by bacteria, viruses, fungi, and parasites. In Nijkamp and Parnham’s Principles of Immunopharmacology; Springer: Berlin/Heidelberg, Germany, 2019; pp. 165–178. [Google Scholar]
- Singh, A. Eliciting B cell immunity against infectious diseases using nanovaccines. Nat. Nanotechnol. 2021, 16, 16–24. [Google Scholar] [CrossRef]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dombkowski, A.A.; Sultana, K.Z.; Craig, D.B. Protein disulfide engineering. FEBS Lett. 2014, 588, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Compton, J.R.; Legler, P.M.; Clingan, B.V.; Olson, M.A.; Millard, C.B. Introduction of a disulfide bond leads to stabilization and crystallization of a ricin immunogen. Proteins Struct. Funct. Bioinform. 2011, 79, 1048–1060. [Google Scholar] [CrossRef]
- Tan, C.; Zhu, F.; Pan, P.; Wu, A.; Li, C. Development of multi-epitope vaccines against the monkeypox virus based on envelope proteins using immunoinformatics approaches. Front. Immunol. 2023, 14, 1112816. [Google Scholar] [CrossRef] [PubMed]
- Dar, H.A.; Almajhdi, F.N.; Aziz, S.; Waheed, Y. Immunoinformatics-aided analysis of RSV fusion and attachment glycoproteins to design a potent multi-epitope vaccine. Vaccines 2022, 10, 1381. [Google Scholar] [CrossRef]
- Suleman, M.; Ul Qamar, M.T.; Kiran Rasool, S.; Rasool, A.; Albutti, A.; Alsowayeh, N.; Alwashmi, A.S.; Aljasir, M.A.; Ahmad, S.; Hussain, Z. Immunoinformatics and immunogenetics-based design of immunogenic peptides vaccine against the emerging tick-borne encephalitis virus (Tbev) and its validation through in silico cloning and immune simulation. Vaccines 2021, 9, 1210. [Google Scholar] [CrossRef]
- Tandon, A.; Pathak, M.; Harioudh, M.K.; Ahmad, S.; Sayeed, M.; Afshan, T.; Siddiqi, M.I.; Mitra, K.; Bhattacharya, S.M.; Ghosh, J.K. A TLR4-derived non-cytotoxic, self-assembling peptide functions as a vaccine adjuvant in mice. J. Biol. Chem. 2018, 293, 19874–19885. [Google Scholar] [CrossRef]
- Duan, T.; Du, Y.; Xing, C.; Wang, H.Y.; Wang, R.F. Toll-like receptor signaling and its role in cell-mediated immunity. Front. Immunol. 2022, 13, 812774. [Google Scholar] [CrossRef]
- Li, Q.; Guo, Z. Recent advances in toll like receptor-targeting glycoconjugate vaccines. Molecules 2018, 23, 1583. [Google Scholar] [CrossRef]
- Zhang, L. Multi-epitope vaccines: A promising strategy against tumors and viral infections. Cell. Mol. Immunol. 2018, 15, 182–184. [Google Scholar] [CrossRef]
- Vetter, V.; Denizer, G.; Friedland, L.R.; Krishnan, J.; Shapiro, M. Understanding modern-day vaccines: What you need to know. Ann. Med. 2018, 50, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Waqas, M.; Aziz, S.; Liò, P.; Khan, Y.; Ali, A.; Iqbal, A.; Khan, F.; Almajhdi, F.N. Immunoinformatics design of multivalent epitope vaccine against monkeypox virus and its variants using membrane-bound, enveloped, and extracellular proteins as targets. Front. Immunol. 2023, 14, 1091941. [Google Scholar] [CrossRef] [PubMed]
- Gandhamaneni, B.S.; Krishnamoorthy, H.R.; Veerappapillai, S.; Mohapatra, S.R.; Karuppasamy, R. Envelope Glycoprotein based multi-epitope vaccine against a co-infection of Human Herpesvirus 5 and Human Herpesvirus 6 using in silico strategies. Glycoconj. J. 2022, 39, 711–724. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein ID | Protein Name | Abbreviation | Antigenicity | Allergenicity | Toxicity |
|---|---|---|---|---|---|
| YP_010840879.1 | Envelope glycoprotein | GP | 0.4510 | Non-allergic | Non-toxic |
| YP_010840880.1 | RNA-directed RNA polymerase | L | 0.4327 | Non-allergic | Non-toxic |
| YP_010840881.1 | Nucleoprotein | NP | 0.4087 | Non-allergic | Non-toxic |
| Protein ID | LBL Epitopes | Score | Antigenicity/ Allergenicity/Toxicity | CTL Epitopes | Alleles | IC50 | Antigenicity/ Allergenicity/Toxicity | HTL Epitopes | Alleles | Antigenicity/ Allergenicity/Toxicity | IC50 | IFN-γ | IL-4 | IL-10 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| YP_010840879.1 | VVFGYSIGRIAFILTF | 0.59 | 0.7885/NA/NT * | SIGRIAFIL | HLA-A*32:01, HLA-A*02:01, HLA-C*15:02, HLA-A*02:06, HLA-B*08:01 | 5 | 0.8896/NA/NT | PVVFGYSIGRIAFIL | HLA-DRB1*15:01, HLA-DRB1*13:02, HLA-DRB1*12:01 | 0.9977/NA/NT | 59 | + | + | + |
| FYALIIWVVFGYSIGR | 0.57 | 0.4648/NA/NT | LIIWVVFGY | HLA-A*29:02, HLA-A*26:01, HLA-B*15:02, HLA-A*30:02, HLA-A*25:01, HLA-B*35:01 | 1.4 | 0.8744/NA/NT | TFFYALIIWVVFGYS | HLA-DRB1*15:01 | 0.4551/NA/NT | 88 | + | + | + | |
| YP_010840880.1 | GGSIEAEILSLRTNQP | 0.91 | 0.6088/NA/NT | SIEAEILSL | HLA-C*08:02, HLA-C*05:01, HLA-A*02:06, HLA-A*02:01 | 8.1 | 0.8887/NA/NT | DFGGSIEAEILSLRT | HLA-DRB1*12:01, HLA-DRB1*04:01 | 0.7948/NA/NT | 71 | + | + | + |
| MDEIISLVEETKNKHE | 0.85 | 0.7700/NA/NT | SLVEETKNK | HLA-A*03:01, HLA-A*11:01, HLA-A*68:01, HLA-A*30:01, HLA-A*31:01, HLA-A*30:02, HLA-B*46:01, HLA-B*15:01, HLA-A*02:01, HLA-A*26:01, HLA-A*32:01 | 7.9 | 0.8892/NA/NT | ISLVEETKNKHEAYE | HLA-DRB1*15:01, HLA-DRB1*12:01 | 1.1296/NA/NT | 37 | + | + | + | |
| YP_010840881.1 | TLKGTAYKWGSTLANM | 0.91 | 0.4680/NA/NT | GTAYKWGST | HLA-A*30:01, HLA-A*30:02, HLA-B*15:01 | 10 | 0.5146/NA/NT | KTTLKGTAYKWGSTL | HLA-DRB1*08:02, HLA-DRB1*11:01, HLA-DRB1*15:01, HLA-DRB1*07:01 | 0.4351/NA/NT | 70 | + | + | + |
| FLGLNTKYTKSLALQP | 0.67 | 1.2806/NA/NT | GLNTKYTKS | HLA-A*02:01 | 9.1 | 1.3243/NA/NT | GLNTKYTKSLALQPH | HLA-DRB5*01:01, HLA-DRB1*04:01, HLA-DRB1*01:01 | 1.0808/NA/NT | 26 | + | + | + |
| S.No | Physicochemical Properties | Results |
|---|---|---|
| 1 | Total amino acids residue | 467 |
| 2 | Molecular weight | 49,737.41 |
| 3 | Extinction coefficients (at 280 nm in water) | 69,790 M−1 cm−1 |
| 4 | Theoretical pI | 8.80 |
| 5 | Formula | C2290H3596N570O656S4 |
| 6 | Instability index | 24.22 (Stable) |
| 7 | Aliphatic index | 92.72 |
| 8 | GRAVY value | 0.045 |
| 9 | Estimated half-life (mammalian reticulocytes, in vitro), (E. coli in vivo), (yeast in vivo) | >30 h, >10 h, >20 h |
| 10 | Antigenicity (AntigenPRO) | 0.6255 |
| 11 | Antigenicity (Vaxijen) | 0.5904 |
| 12 | Allergenicity (AllerTOP) | Non-Allergic |
| 13 | Solubility (Solpro) | 0.964 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, S.; Chiou, C.-C.; Almutairi, M.M.; Ajmal, A.; Batool, S.; Javed, B.; Tanaka, T.; Chen, C.-C.; Alouffi, A.; Ali, A. Targeting Yezo Virus Structural Proteins for Multi-Epitope Vaccine Design Using Immunoinformatics Approach. Viruses 2024, 16, 1408. https://doi.org/10.3390/v16091408
Rahman S, Chiou C-C, Almutairi MM, Ajmal A, Batool S, Javed B, Tanaka T, Chen C-C, Alouffi A, Ali A. Targeting Yezo Virus Structural Proteins for Multi-Epitope Vaccine Design Using Immunoinformatics Approach. Viruses. 2024; 16(9):1408. https://doi.org/10.3390/v16091408
Chicago/Turabian StyleRahman, Sudais, Chien-Chun Chiou, Mashal M. Almutairi, Amar Ajmal, Sidra Batool, Bushra Javed, Tetsuya Tanaka, Chien-Chin Chen, Abdulaziz Alouffi, and Abid Ali. 2024. "Targeting Yezo Virus Structural Proteins for Multi-Epitope Vaccine Design Using Immunoinformatics Approach" Viruses 16, no. 9: 1408. https://doi.org/10.3390/v16091408
APA StyleRahman, S., Chiou, C.-C., Almutairi, M. M., Ajmal, A., Batool, S., Javed, B., Tanaka, T., Chen, C.-C., Alouffi, A., & Ali, A. (2024). Targeting Yezo Virus Structural Proteins for Multi-Epitope Vaccine Design Using Immunoinformatics Approach. Viruses, 16(9), 1408. https://doi.org/10.3390/v16091408