
Exploring HIV-1 Maturation: A New Frontier in Antiviral Development

 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Viral Replication Cycle
3. Molecular Mechanisms of HIV-1 Maturation
3.1. Fundamentals in HIV-1 Maturation
3.2. Host Proteins in HIV-1 Maturation and Core Association
3.3. Host Metabolites in HIV-1 Maturation and Core Association
3.4. Host Membrane Lipid Rafts in HIV-1 Maturation
3.5. Environmental Factor in HIV-1 Maturation
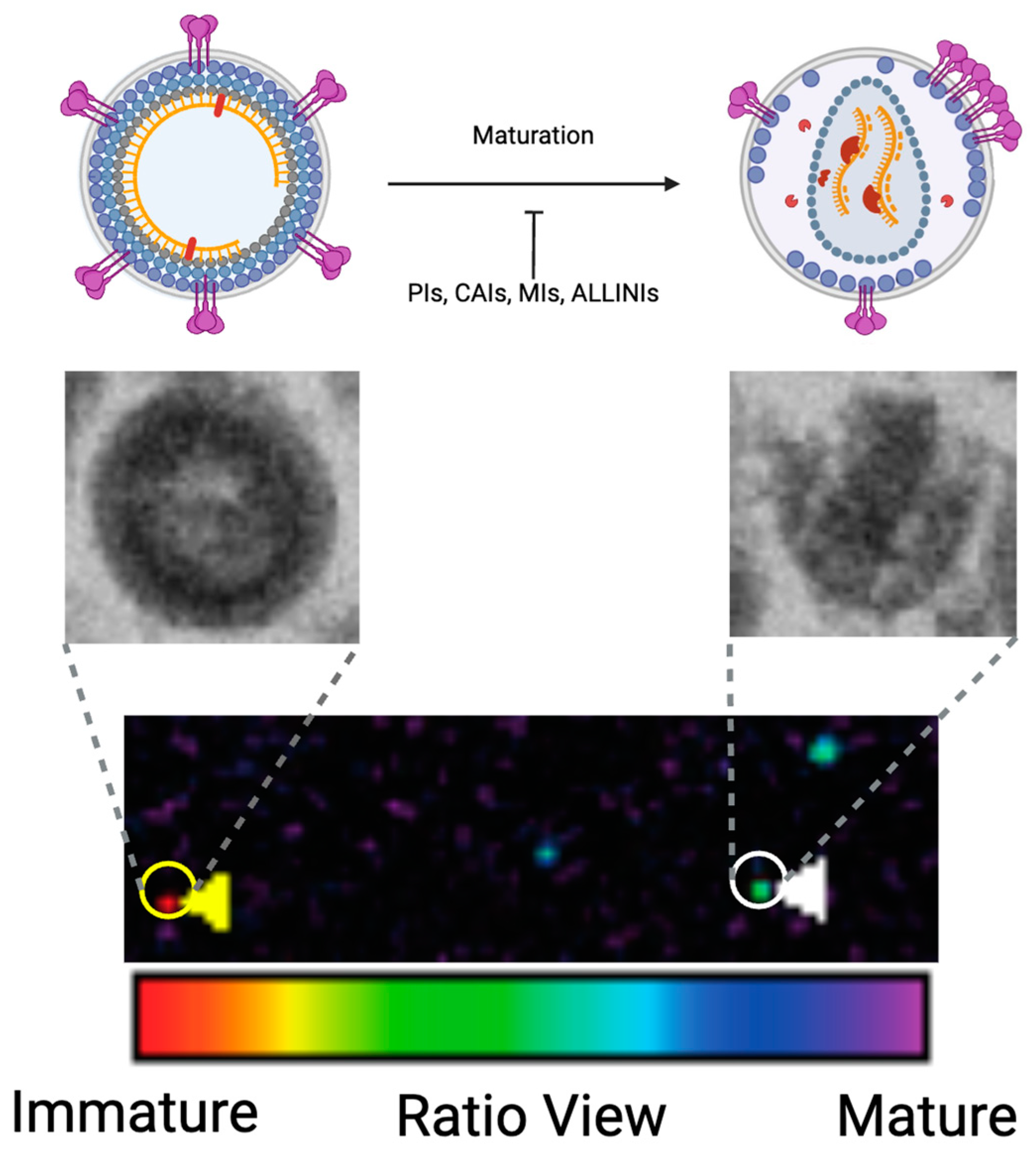
4. Tools for Assessing Virion Maturation
4.1. Electron Microscopy
4.2. Fluorescence Microscopy
5. Influence of Virion Maturation on HIV-1 Env Function
6. Targeting Maturation for Therapeutic Intervention: Small Molecule Discovery, Mode of Action, and Clinical Development
6.1. Protease Inhibitors
6.2. Maturation Inhibitors
6.3. Other Classes
6.3.1. Capsid Inhibitors
6.3.2. Allosteric Integrase Inhibitors
7. Conclusions
Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef] [PubMed]
- Ta, T.M.; Malik, S.; Anderson, E.M.; Jones, A.D.; Perchik, J.; Freylikh, M.; Sardo, L.; Klase, Z.A.; Izumi, T. Insights into Persistent HIV-1 Infection and Functional Cure: Novel Capabilities and Strategies. Front. Microbiol. 2022, 13, 862270. [Google Scholar] [CrossRef] [PubMed]
- Sardo, L.; Parolin, C.; Yoshida, T.; Garzino-Demo, A.; Izumi, T. Editorial: Novel Insights into a Functional HIV Cure. Front. Microbiol. 2021, 12, 797570. [Google Scholar] [CrossRef] [PubMed]
- Briz, V.; Poveda, E.; Soriano, V. HIV entry inhibitors: Mechanisms of action and resistance pathways. J. Antimicrob. Chemother. 2006, 57, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A. In PrEP: Long-acting antivirals for HIV prevention. Cell Host Microbe 2022, 30, 148–150. [Google Scholar] [CrossRef]
- Goebel, M.C.; Guajardo, E.; Giordano, T.P.; Patel, S.M. The New Era of Long-Acting Antiretroviral Therapy: When and Why to Make the Switch. Curr. HIV/AIDS Rep. 2023, 20, 271–285. [Google Scholar] [CrossRef]
- Sension, M.G.; Brunet, L.; Hsu, R.K.; Fusco, J.S.; Cochran, Q.; Uranaka, C.; Sridhar, G.; Vannappagari, V.; Van Wyk, J.; McCurdy, L.; et al. Cabotegravir + Rilpivirine Long-Acting Injections for HIV Treatment in the US: Real World Data from the OPERA Cohort. Infect. Dis. Ther. 2023, 12, 2807–2817. [Google Scholar] [CrossRef]
- Dvory-Sobol, H.; Shaik, N.; Callebaut, C.; Rhee, M.S. Lenacapavir: A first-in-class HIV-1 capsid inhibitor. Curr. Opin. HIV AIDS 2022, 17, 15–21. [Google Scholar] [CrossRef]
- Novikova, M.; Zhang, Y.; Freed, E.O.; Peng, K. Multiple Roles of HIV-1 Capsid during the Virus Replication Cycle. Virol. Sin. 2019, 34, 119–134. [Google Scholar] [CrossRef]
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 2015, 13, 484–496. [Google Scholar] [CrossRef]
- Klingler, J.; Anton, H.; Real, E.; Zeiger, M.; Moog, C.; Mely, Y.; Boutant, E. How HIV-1 Gag Manipulates Its Host Cell Proteins: A Focus on Interactors of the Nucleocapsid Domain. Viruses 2020, 12, 888. [Google Scholar] [CrossRef] [PubMed]
- Olety, B.; Ono, A. Roles played by acidic lipids in HIV-1 Gag membrane binding. Virus Res. 2014, 193, 108–115. [Google Scholar] [CrossRef]
- Rossi, E.; Meuser, M.E.; Cunanan, C.J.; Cocklin, S. Structure, Function, and Interactions of the HIV-1 Capsid Protein. Life 2021, 11, 100. [Google Scholar] [CrossRef]
- Yu, K.L.; Lee, S.H.; Lee, E.S.; You, J.C. HIV-1 nucleocapsid protein localizes efficiently to the nucleus and nucleolus. Virology 2016, 492, 204–212. [Google Scholar] [CrossRef]
- Levin, J.G.; Mitra, M.; Mascarenhas, A.; Musier-Forsyth, K. Role of HIV-1 nucleocapsid protein in HIV-1 reverse transcription. RNA Biol. 2010, 7, 754–774. [Google Scholar] [CrossRef]
- Thomas, J.A.; Gorelick, R.J. Nucleocapsid protein function in early infection processes. Virus Res 2008, 134, 39–63. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Krausslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef]
- Meulendyke, K.A.; Croteau, J.D.; Zink, M.C. HIV life cycle, innate immunity and autophagy in the central nervous system. Curr. Opin. HIV AIDS 2014, 9, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.S.A.; Hassan, S.S.; Ahemad, N. Evolution of HIV-1 reverse transcriptase and integrase dual inhibitors: Recent advances and developments. Eur. J. Med. Chem. 2019, 179, 423–448. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Burdick, R.C.; Nagashima, K.; Hu, W.S.; Pathak, V.K. HIV-1 cores retain their integrity until minutes before uncoating in the nucleus. Proc. Natl. Acad. Sci. USA 2021, 118, e2019467118. [Google Scholar] [CrossRef]
- Burdick, R.C.; Duchon, A.; Hu, W.S.; Pathak, V.K. Imaging HIV-1 Nuclear Import, Uncoating, and Proviral Transcription. Methods Mol. Biol. 2024, 2807, 15–30. [Google Scholar] [PubMed]
- Ajasin, D.; Eugenin, E.A. HIV-1 Tat: Role in Bystander Toxicity. Front. Cell. Infect. Microbiol. 2020, 10, 517857. [Google Scholar] [CrossRef] [PubMed]
- Reeder, J.E.; Kwak, Y.T.; McNamara, R.P.; Forst, C.V.; D’Orso, I. HIV Tat controls RNA Polymerase II and the epigenetic landscape to transcriptionally reprogram target immune cells. Elife 2015, 4, e08955. [Google Scholar] [CrossRef] [PubMed]
- Beemon, K.L. Retroviral RNA Processing. Viruses 2022, 14, 1113. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, J.; Zhang, M.; Bai, B.; Ma, W.; Lin, Y.; Guo, X.; Wang, X.F.; Wang, X. A Novel, Fully Spliced, Accessory Gene in Equine Lentivirus with Distinct Rev-Responsive Element. J. Virol. 2022, 96, e0098622. [Google Scholar] [CrossRef]
- Kuzembayeva, M.; Dilley, K.; Sardo, L.; Hu, W.S. Life of psi: How full-length HIV-1 RNAs become packaged genomes in the viral particles. Virology 2014, 454, 362–370. [Google Scholar] [CrossRef]
- Coffin, J.M.; Hughes, S.H.; Varmus, H.E. Retroviruses; Cold Spring Harbor: New York, NY, USA, 1997. [Google Scholar]
- Chen, J.; Liu, Y.; Wu, B.; Nikolaitchik, O.A.; Mohan, P.R.; Chen, J.; Pathak, V.K.; Hu, W.S. Visualizing the translation and packaging of HIV-1 full-length RNA. Proc. Natl. Acad. Sci. USA 2020, 117, 6145–6155. [Google Scholar] [CrossRef]
- Cullen, B.R. Nuclear mRNA export: Insights from virology. Trends Biochem. Sci. 2003, 28, 419–424. [Google Scholar] [CrossRef]
- Affranchino, J.L.; Gonzalez, S.A. Understanding the process of envelope glycoprotein incorporation into virions in simian and feline immunodeficiency viruses. Viruses 2014, 6, 264–283. [Google Scholar] [CrossRef]
- McGraw, A.; Hillmer, G.; Choi, J.; Narayan, K.; Mehedincu, S.M.; Marquez, D.; Tibebe, H.; DeCicco-Skinner, K.L.; Izumi, T. Evaluating HIV-1 Infectivity and Virion Maturation across Varied Producer Cells with a Novel FRET-Based Detection and Quantification Assay. Int. J. Mol. Sci. 2024, 25, 6396. [Google Scholar] [CrossRef]
- Kleinpeter, A.B.; Freed, E.O. HIV-1 Maturation: Lessons Learned from Inhibitors. Viruses 2020, 12, 940. [Google Scholar] [CrossRef]
- Biswas, P.; Jiang, X.; Pacchia, A.L.; Dougherty, J.P.; Peltz, S.W. The human immunodeficiency virus type 1 ribosomal frameshifting site is an invariant sequence determinant and an important target for antiviral therapy. J. Virol. 2004, 78, 2082–2087. [Google Scholar] [CrossRef]
- Korniy, N.; Goyal, A.; Hoffmann, M.; Samatova, E.; Peske, F.; Pohlmann, S.; Rodnina, M.V. Modulation of HIV-1 Gag/Gag-Pol frameshifting by tRNA abundance. Nucleic Acids Res. 2019, 47, 5210–5222. [Google Scholar] [CrossRef]
- Balvay, L.; Lopez Lastra, M.; Sargueil, B.; Darlix, J.L.; Ohlmann, T. Translational control of retroviruses. Nat. Rev. Microbiol. 2007, 5, 128–140. [Google Scholar] [CrossRef]
- Das, A.T.; Harwig, A.; Berkhout, B. The HIV-1 Tat protein has a versatile role in activating viral transcription. J. Virol. 2011, 85, 9506–9516. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.D.; Wu, J.; Shao, R.; Xue, Y.H. Mechanism and factors that control HIV-1 transcription and latency activation. J. Zhejiang Univ. Sci. B 2014, 15, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Pornillos, O.; Ganser-Pornillos, B.K. Maturation of retroviruses. Curr. Opin. Virol. 2019, 36, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Jacks, T.; Power, M.D.; Masiarz, F.R.; Luciw, P.A.; Barr, P.J.; Varmus, H.E. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature 1988, 331, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.R.; Chu, S.M.; Yu, F.H.; Huang, K.J.; Wang, C.T. Effects of reduced gag cleavage efficiency on HIV-1 Gag-Pol package. BMC Microbiol. 2022, 22, 94. [Google Scholar] [CrossRef]
- Centazzo, M.; Manganaro, L.; Alvisi, G. Cellular Targets of HIV-1 Protease: Just the Tip of the Iceberg? Viruses 2023, 15, 712. [Google Scholar] [CrossRef]
- Qu, K.; Ke, Z.; Zila, V.; Anders-Osswein, M.; Glass, B.; Mucksch, F.; Muller, R.; Schultz, C.; Muller, B.; Krausslich, H.G.; et al. Maturation of the matrix and viral membrane of HIV-1. Science 2021, 373, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Perilla, J.R.; Yufenyuy, E.L.; Meng, X.; Chen, B.; Ning, J.; Ahn, J.; Gronenborn, A.M.; Schulten, K.; Aiken, C.; et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 2013, 497, 643–646. [Google Scholar] [CrossRef]
- Nicastro, G.; Lucci, M.; Oregioni, A.; Kelly, G.; Frenkiel, T.A.; Taylor, I.A. CP-MAS and Solution NMR Studies of Allosteric Communication in CA-assemblies of HIV-1. J. Mol. Biol. 2022, 434, 167691. [Google Scholar] [CrossRef] [PubMed]
- Schirra, R.T.; Dos Santos, N.F.B.; Zadrozny, K.K.; Kucharska, I.; Ganser-Pornillos, B.K.; Pornillos, O. A molecular switch modulates assembly and host factor binding of the HIV-1 capsid. Nat. Struct. Mol. Biol. 2023, 30, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Mallery, D.L.; Faysal, K.M.R.; Kleinpeter, A.; Wilson, M.S.C.; Vaysburd, M.; Fletcher, A.J.; Novikova, M.; Bocking, T.; Freed, E.O.; Saiardi, A.; et al. Cellular IP(6) Levels Limit HIV Production while Viruses that Cannot Efficiently Package IP(6) Are Attenuated for Infection and Replication. Cell. Rep. 2019, 29, 3983–3996.e4. [Google Scholar] [CrossRef]
- Price, A.J.; Jacques, D.A.; McEwan, W.A.; Fletcher, A.J.; Essig, S.; Chin, J.W.; Halambage, U.D.; Aiken, C.; James, L.C. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 2014, 10, e1004459. [Google Scholar] [CrossRef]
- Li, S.; Patel, J.S.; Yang, J.; Crabtree, A.M.; Rubenstein, B.M.; Lund-Andersen, P.K.; Ytreberg, F.M.; Rowley, P.A. Defining the HIV Capsid Binding Site of Nucleoporin 153. mSphere 2022, 7, e0031022. [Google Scholar] [CrossRef]
- Link, J.O.; Rhee, M.S.; Tse, W.C.; Zheng, J.; Somoza, J.R.; Rowe, W.; Begley, R.; Chiu, A.; Mulato, A.; Hansen, D.; et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020, 584, 614–618. [Google Scholar] [CrossRef]
- Noser, J.A.; Towers, G.J.; Sakuma, R.; Dumont, J.M.; Collins, M.K.; Ikeda, Y. Cyclosporine increases human immunodeficiency virus type 1 vector transduction of primary mouse cells. J. Virol. 2006, 80, 7769–7774. [Google Scholar] [CrossRef]
- Yoo, S.; Myszka, D.G.; Yeh, C.; McMurray, M.; Hill, C.P.; Sundquist, W.I. Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J. Mol. Biol. 1997, 269, 780–795. [Google Scholar] [CrossRef]
- Gamble, T.R.; Vajdos, F.F.; Yoo, S.; Worthylake, D.K.; Houseweart, M.; Sundquist, W.I.; Hill, C.P. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 1996, 87, 1285–1294. [Google Scholar] [CrossRef]
- Luban, J. Absconding with the chaperone: Essential cyclophilin-Gag interaction in HIV-1 virions. Cell 1996, 87, 1157–1159. [Google Scholar] [CrossRef]
- Braaten, D.; Franke, E.K.; Luban, J. Cyclophilin A is required for an early step in the life cycle of human immunodeficiency virus type 1 before the initiation of reverse transcription. J. Virol. 1996, 70, 3551–3560. [Google Scholar] [CrossRef] [PubMed]
- Burdick, R.C.; Li, C.; Munshi, M.; Rawson, J.M.O.; Nagashima, K.; Hu, W.S.; Pathak, V.K. HIV-1 uncoats in the nucleus near sites of integration. Proc. Natl. Acad. Sci. USA 2020, 117, 5486–5493. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, R.; Prakash, P.; Kumbhar, B.V.; Balasubramaniam, M.; Dash, C. HIV-1 capsid and viral DNA integration. mBio 2024, 15, e0021222. [Google Scholar] [CrossRef] [PubMed]
- Sokolskaja, E.; Sayah, D.M.; Luban, J. Target cell cyclophilin A modulates human immunodeficiency virus type 1 infectivity. J. Virol. 2004, 78, 12800–12808. [Google Scholar] [CrossRef]
- Kim, K.; Dauphin, A.; Komurlu, S.; McCauley, S.M.; Yurkovetskiy, L.; Carbone, C.; Diehl, W.E.; Strambio-De-Castillia, C.; Campbell, E.M.; Luban, J. Cyclophilin A protects HIV-1 from restriction by human TRIM5alpha. Nat. Microbiol. 2019, 4, 2044–2051. [Google Scholar] [CrossRef]
- Li, Y.; Kar, A.K.; Sodroski, J. Target cell type-dependent modulation of human immunodeficiency virus type 1 capsid disassembly by cyclophilin A. J. Virol. 2009, 83, 10951–10962. [Google Scholar] [CrossRef]
- Shah, V.B.; Shi, J.; Hout, D.R.; Oztop, I.; Krishnan, L.; Ahn, J.; Shotwell, M.S.; Engelman, A.; Aiken, C. The host proteins transportin SR2/TNPO3 and cyclophilin A exert opposing effects on HIV-1 uncoating. J. Virol. 2013, 87, 422–432. [Google Scholar] [CrossRef]
- Rasaiyaah, J.; Tan, C.P.; Fletcher, A.J.; Price, A.J.; Blondeau, C.; Hilditch, L.; Jacques, D.A.; Selwood, D.L.; James, L.C.; Noursadeghi, M.; et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 2013, 503, 402–405. [Google Scholar] [CrossRef]
- Lu, M.; Hou, G.; Zhang, H.; Suiter, C.L.; Ahn, J.; Byeon, I.J.; Perilla, J.R.; Langmead, C.J.; Hung, I.; Gor’kov, P.L.; et al. Dynamic allostery governs cyclophilin A-HIV capsid interplay. Proc. Natl. Acad. Sci. USA 2015, 112, 14617–14622. [Google Scholar] [CrossRef] [PubMed]
- Luban, J. TRIM5 and the Regulation of HIV-1 Infectivity. Mol. Biol. Int. 2012, 2012, 426840. [Google Scholar] [CrossRef]
- Grutter, M.G.; Luban, J. TRIM5 structure, HIV-1 capsid recognition, and innate immune signaling. Curr. Opin. Virol. 2012, 2, 142–150. [Google Scholar] [CrossRef]
- Kono, K.; Song, H.; Shingai, Y.; Shioda, T.; Nakayama, E.E. Comparison of anti-viral activity of rhesus monkey and cynomolgus monkey TRIM5alphas against human immunodeficiency virus type 2 infection. Virology 2008, 373, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Ke, D.; Vu, T.; Ahn, J.; Shah, V.B.; Yang, R.; Aiken, C.; Charlton, L.M.; Gronenborn, A.M.; Zhang, P. Rhesus TRIM5alpha disrupts the HIV-1 capsid at the inter-hexamer interfaces. PLoS Pathog. 2011, 7, e1002009. [Google Scholar] [CrossRef]
- Sakuma, R.; Noser, J.A.; Ohmine, S.; Ikeda, Y. Rhesus monkey TRIM5alpha restricts HIV-1 production through rapid degradation of viral Gag polyproteins. Nat. Med. 2007, 13, 631–635. [Google Scholar] [CrossRef]
- Soll, S.J.; Wilson, S.J.; Kutluay, S.B.; Hatziioannou, T.; Bieniasz, P.D. Assisted evolution enables HIV-1 to overcome a high TRIM5alpha-imposed genetic barrier to rhesus macaque tropism. PLoS Pathog. 2013, 9, e1003667. [Google Scholar] [CrossRef] [PubMed]
- Santelli, E.; Bankston, L.A.; Leppla, S.H.; Liddington, R.C. Crystal structure of a complex between anthrax toxin and its host cell receptor. Nature 2004, 430, 905–908. [Google Scholar] [CrossRef]
- Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin A and TRIM5alpha independently regulate human immunodeficiency virus type 1 infectivity in human cells. J. Virol. 2006, 80, 2855–2862. [Google Scholar] [CrossRef]
- Strebel, K.; Luban, J.; Jeang, K.T. Human cellular restriction factors that target HIV-1 replication. BMC Med. 2009, 7, 48. [Google Scholar] [CrossRef]
- Sayah, D.M.; Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 2004, 430, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Moyano, E.; Ruiz, A.; Kloverpris, H.N.; Rodriguez-Plata, M.T.; Pena, R.; Blondeau, C.; Selwood, D.L.; Izquierdo-Useros, N.; Moris, A.; Clotet, B.; et al. Nonhuman TRIM5 Variants Enhance Recognition of HIV-1-Infected Cells by CD8+ T Cells. J. Virol. 2016, 90, 8552–8562. [Google Scholar] [CrossRef] [PubMed]
- Cloherty, A.P.M.; Rader, A.G.; Compeer, B.; Ribeiro, C.M.S. Human TRIM5alpha: Autophagy Connects Cell-Intrinsic HIV-1 Restriction and Innate Immune Sensor Functioning. Viruses 2021, 13, 320. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Pornillos, O. Restriction of HIV-1 and other retroviruses by TRIM5. Nat. Rev. Microbiol. 2019, 17, 546–556. [Google Scholar] [CrossRef]
- Achuthan, V.; Perreira, J.M.; Sowd, G.A.; Puray-Chavez, M.; McDougall, W.M.; Paulucci-Holthauzen, A.; Wu, X.; Fadel, H.J.; Poeschla, E.M.; Multani, A.S.; et al. Capsid-CPSF6 Interaction Licenses Nuclear HIV-1 Trafficking to Sites of Viral DNA Integration. Cell Host Microbe 2018, 24, 392–404.e8. [Google Scholar] [CrossRef]
- McDonald, D.; Vodicka, M.A.; Lucero, G.; Svitkina, T.M.; Borisy, G.G.; Emerman, M.; Hope, T.J. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 2002, 159, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Ning, J.; Boggs, E.A.; Jang, S.; Wallace, C.; Telmer, C.; Bruchez, M.P.; Ahn, J.; Engelman, A.N.; Zhang, P.; et al. Cytoplasmic CPSF6 Regulates HIV-1 Capsid Trafficking and Infection in a Cyclophilin A-Dependent Manner. mBio 2021, 12, e03142-20. [Google Scholar] [CrossRef]
- Lukic, Z.; Dharan, A.; Fricke, T.; Diaz-Griffero, F.; Campbell, E.M. HIV-1 uncoating is facilitated by dynein and kinesin 1. J. Virol. 2014, 88, 13613–13625. [Google Scholar] [CrossRef]
- Malikov, V.; Naghavi, M.H. Localized Phosphorylation of a Kinesin-1 Adaptor by a Capsid-Associated Kinase Regulates HIV-1 Motility and Uncoating. Cell Rep. 2017, 20, 2792–2799. [Google Scholar] [CrossRef]
- Fernandez, J.; Portilho, D.M.; Danckaert, A.; Munier, S.; Becker, A.; Roux, P.; Zambo, A.; Shorte, S.; Jacob, Y.; Vidalain, P.O.; et al. Microtubule-associated proteins 1 (MAP1) promote human immunodeficiency virus type I (HIV-1) intracytoplasmic routing to the nucleus. J. Biol. Chem. 2015, 290, 4631–4646. [Google Scholar] [CrossRef]
- Carnes, S.K.; Zhou, J.; Aiken, C. HIV-1 Engages a Dynein-Dynactin-BICD2 Complex for Infection and Transport to the Nucleus. J. Virol. 2018, 92, e00358-18. [Google Scholar] [CrossRef]
- Dharan, A.; Opp, S.; Abdel-Rahim, O.; Keceli, S.K.; Imam, S.; Diaz-Griffero, F.; Campbell, E.M. Bicaudal D2 facilitates the cytoplasmic trafficking and nuclear import of HIV-1 genomes during infection. Proc. Natl. Acad. Sci. USA 2017, 114, E10707–E10716. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.T.; Summers, B.J.; Xu, C.; Perilla, J.R.; Malikov, V.; Naghavi, M.H.; Xiong, Y. FEZ1 Is Recruited to a Conserved Cofactor Site on Capsid to Promote HIV-1 Trafficking. Cell Rep. 2019, 28, 2373–2385.e7. [Google Scholar] [CrossRef]
- Malikov, V.; da Silva, E.S.; Jovasevic, V.; Bennett, G.; de Souza Aranha Vieira, D.A.; Schulte, B.; Diaz-Griffero, F.; Walsh, D.; Naghavi, M.H. HIV-1 capsids bind and exploit the kinesin-1 adaptor FEZ1 for inward movement to the nucleus. Nat. Commun. 2015, 6, 6660. [Google Scholar] [CrossRef]
- Dharan, A.; Talley, S.; Tripathi, A.; Mamede, J.I.; Majetschak, M.; Hope, T.J.; Campbell, E.M. KIF5B and Nup358 Cooperatively Mediate the Nuclear Import of HIV-1 during Infection. PLoS Pathog. 2016, 12, e1005700. [Google Scholar] [CrossRef]
- Schaller, T.; Ocwieja, K.E.; Rasaiyaah, J.; Price, A.J.; Brady, T.L.; Roth, S.L.; Hue, S.; Fletcher, A.J.; Lee, K.; KewalRamani, V.N.; et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011, 7, e1002439. [Google Scholar] [CrossRef]
- Bichel, K.; Price, A.J.; Schaller, T.; Towers, G.J.; Freund, S.M.; James, L.C. HIV-1 capsid undergoes coupled binding and isomerization by the nuclear pore protein NUP358. Retrovirology 2013, 10, 81. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Iqbal, N.; Courouble, V.V.; Francis, A.C.; Singh, P.K.; Hudait, A.; Annamalai, A.S.; Bester, S.; Huang, S.W.; Shkriabai, N.; et al. Prion-like low complexity regions enable avid virus-host interactions during HIV-1 infection. Nat. Commun. 2022, 13, 5879. [Google Scholar] [CrossRef]
- Shen, Q.; Kumari, S.; Xu, C.; Jang, S.; Shi, J.; Burdick, R.C.; Levintov, L.; Xiong, Q.; Wu, C.; Devarkar, S.C.; et al. The capsid lattice engages a bipartite NUP153 motif to mediate nuclear entry of HIV-1 cores. Proc. Natl. Acad. Sci. USA 2023, 120, e2202815120. [Google Scholar] [CrossRef] [PubMed]
- Matreyek, K.A.; Yucel, S.S.; Li, X.; Engelman, A. Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog. 2013, 9, e1003693. [Google Scholar] [CrossRef]
- Bejarano, D.A.; Peng, K.; Laketa, V.; Borner, K.; Jost, K.L.; Lucic, B.; Glass, B.; Lusic, M.; Muller, B.; Krausslich, H.G. HIV-1 nuclear import in macrophages is regulated by CPSF6-capsid interactions at the nuclear pore complex. Elife 2019, 8, e41800. [Google Scholar] [CrossRef] [PubMed]
- King, M.C.; Raposo, G.; Lemmon, M.A. Inhibition of nuclear import and cell-cycle progression by mutated forms of the dynamin-like GTPase MxB. Proc. Natl. Acad. Sci. USA 2004, 101, 8957–8962. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Zhao, J.; Yang, Z.; Ma, L.; Mi, Z.; Wu, Y.; Guo, J.; Zhou, J.; Li, X.; Guo, Y.; et al. Microbial Natural Product Alternariol 5-O-Methyl Ether Inhibits HIV-1 Integration by Blocking Nuclear Import of the Pre-Integration Complex. Viruses 2017, 9, 105. [Google Scholar] [CrossRef]
- Raghavendra, N.K.; Shkriabai, N.; Graham, R.; Hess, S.; Kvaratskhelia, M.; Wu, L. Identification of host proteins associated with HIV-1 preintegration complexes isolated from infected CD4+ cells. Retrovirology 2010, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Fricke, T.; White, T.E.; Schulte, B.; de Souza Aranha Vieira, D.A.; Dharan, A.; Campbell, E.M.; Brandariz-Nunez, A.; Diaz-Griffero, F. MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology 2014, 11, 68. [Google Scholar] [CrossRef]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef]
- Goujon, C.; Moncorge, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hue, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef]
- Liu, Z.; Pan, Q.; Ding, S.; Qian, J.; Xu, F.; Zhou, J.; Cen, S.; Guo, F.; Liang, C. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 2013, 14, 398–410. [Google Scholar] [CrossRef]
- Haller, O.; Staeheli, P.; Schwemmle, M.; Kochs, G. Mx GTPases: Dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015, 23, 154–163. [Google Scholar] [CrossRef]
- Xie, L.; Chen, L.; Zhong, C.; Yu, T.; Ju, Z.; Wang, M.; Xiong, H.; Zeng, Y.; Wang, J.; Hu, H.; et al. MxB impedes the NUP358-mediated HIV-1 pre-integration complex nuclear import and viral replication cooperatively with CPSF6. Retrovirology 2020, 17, 16. [Google Scholar] [CrossRef]
- Lu, L.; Yang, Y.; Yang, Z.; Wu, Y.; Liu, X.; Li, X.; Chen, L.; Han, Y.; Song, X.; Kong, Z.; et al. Altered plasma metabolites and inflammatory networks in HIV-1 infected patients with different immunological responses after long-term antiretroviral therapy. Front. Immunol. 2023, 14, 1254155. [Google Scholar] [CrossRef]
- Mikaeloff, F.; Svensson Akusjarvi, S.; Ikomey, G.M.; Krishnan, S.; Sperk, M.; Gupta, S.; Magdaleno, G.D.V.; Escos, A.; Lyonga, E.; Okomo, M.C.; et al. Trans cohort metabolic reprogramming towards glutaminolysis in long-term successfully treated HIV-infection. Commun. Biol. 2022, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Aiken, C.; Rousso, I. The HIV-1 capsid and reverse transcription. Retrovirology 2021, 18, 29. [Google Scholar] [CrossRef] [PubMed]
- Renner, N.; Kleinpeter, A.; Mallery, D.L.; Albecka, A.; Rifat Faysal, K.M.; Bocking, T.; Saiardi, A.; Freed, E.O.; James, L.C. HIV-1 is dependent on its immature lattice to recruit IP6 for mature capsid assembly. Nat. Struct. Mol. Biol. 2023, 30, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Pak, A.J.; Purdy, M.D.; Yeager, M.; Voth, G.A. Preservation of HIV-1 Gag Helical Bundle Symmetry by Bevirimat Is Central to Maturation Inhibition. J. Am. Chem. Soc. 2021, 143, 19137–19148. [Google Scholar] [CrossRef]
- Dick, R.A.; Zadrozny, K.K.; Xu, C.; Schur, F.K.M.; Lyddon, T.D.; Ricana, C.L.; Wagner, J.M.; Perilla, J.R.; Ganser-Pornillos, B.K.; Johnson, M.C.; et al. Inositol phosphates are assembly co-factors for HIV-1. Nature 2018, 560, 509–512. [Google Scholar] [CrossRef]
- Mallery, D.L.; Marquez, C.L.; McEwan, W.A.; Dickson, C.F.; Jacques, D.A.; Anandapadamanaban, M.; Bichel, K.; Towers, G.J.; Saiardi, A.; Bocking, T.; et al. IP6 is an HIV pocket factor that prevents capsid collapse and promotes DNA synthesis. Elife 2018, 7, e35335. [Google Scholar] [CrossRef]
- Marie, V.; Gordon, M.L. The HIV-1 Gag Protein Displays Extensive Functional and Structural Roles in Virus Replication and Infectivity. Int. J. Mol. Sci. 2022, 23, 7569. [Google Scholar] [CrossRef]
- Evilevitch, A.; Sae-Ueng, U. Mechanical Capsid Maturation Facilitates the Resolution of Conflicting Requirements for Herpesvirus Assembly. J. Virol. 2022, 96, e0183121. [Google Scholar] [CrossRef]
- Dick, R.A.; Xu, C.; Morado, D.R.; Kravchuk, V.; Ricana, C.L.; Lyddon, T.D.; Broad, A.M.; Feathers, J.R.; Johnson, M.C.; Vogt, V.M.; et al. Structures of immature EIAV Gag lattices reveal a conserved role for IP6 in lentivirus assembly. PLoS Pathog. 2020, 16, e1008277. [Google Scholar] [CrossRef]
- Sowd, G.A.; Aiken, C. Inositol phosphates promote HIV-1 assembly and maturation to facilitate viral spread in human CD4+ T cells. PLoS Pathog. 2021, 17, e1009190, Erratum in PLoS Pathog. 2021, 17, e1009389. [Google Scholar]
- Mallery, D.L.; Kleinpeter, A.B.; Renner, N.; Faysal, K.M.R.; Novikova, M.; Kiss, L.; Wilson, M.S.C.; Ahsan, B.; Ke, Z.; Briggs, J.A.G.; et al. A stable immature lattice packages IP(6) for HIV capsid maturation. Sci. Adv. 2021, 7, eabe4716. [Google Scholar] [CrossRef] [PubMed]
- Ricana, C.L.; Lyddon, T.D.; Dick, R.A.; Johnson, M.C. Primate lentiviruses require Inositol hexakisphosphate (IP6) or inositol pentakisphosphate (IP5) for the production of viral particles. PLoS Pathog. 2020, 16, e1008646. [Google Scholar] [CrossRef]
- Obr, M.; Schur, F.K.M.; Dick, R.A. A Structural Perspective of the Role of IP6 in Immature and Mature Retroviral Assembly. Viruses 2021, 13, 1853. [Google Scholar] [CrossRef] [PubMed]
- Jennings, J.; Bracey, H.; Nguyen, D.T.; Dasgupta, R.; Rivera, A.V.; Sluis-Cremer, N.; Shi, J.; Aiken, C. The HIV-1 capsid serves as a nanoscale reaction vessel for reverse transcription. PLoS Pathog. 2024, 20, e1011810. [Google Scholar] [CrossRef]
- Roncato, R.; Angelini, J.; Pani, A.; Talotta, R. Lipid rafts as viral entry routes and immune platforms: A double-edged sword in SARS-CoV-2 infection? Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159140. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.J.; Hinner, M.J. Getting across the cell membrane: An overview for small molecules, peptides, and proteins. Methods Mol. Biol. 2015, 1266, 29–53. [Google Scholar]
- Ono, A.; Ablan, S.D.; Lockett, S.J.; Nagashima, K.; Freed, E.O. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. USA 2004, 101, 14889–14894. [Google Scholar] [CrossRef]
- Shamseddine, A.A.; Airola, M.V.; Hannun, Y.A. Roles and regulation of neutral sphingomyelinase-2 in cellular and pathological processes. Adv. Biol. Regul. 2015, 57, 24–41. [Google Scholar] [CrossRef]
- Waheed, A.A.; Zhu, Y.; Agostino, E.; Naing, L.; Hikichi, Y.; Soheilian, F.; Yoo, S.W.; Song, Y.; Zhang, P.; Slusher, B.S.; et al. Neutral sphingomyelinase 2 is required for HIV-1 maturation. Proc. Natl. Acad. Sci. USA 2023, 120, e2219475120. [Google Scholar] [CrossRef]
- Yoo, S.W.; Waheed, A.A.; Deme, P.; Tohumeken, S.; Rais, R.; Smith, M.D.; DeMarino, C.; Calabresi, P.A.; Kashanchi, F.; Freed, E.O.; et al. Inhibition of neutral sphingomyelinase 2 impairs HIV-1 envelope formation and substantially delays or eliminates viral rebound. Proc. Natl. Acad. Sci. USA 2023, 120, e2219543120. [Google Scholar] [CrossRef] [PubMed]
- Konvalinka, J.; Heuser, A.M.; Hruskova-Heidingsfeldova, O.; Vogt, V.M.; Sedlacek, J.; Strop, P.; Krausslich, H.G. Proteolytic processing of particle-associated retroviral polyproteins by homologous and heterologous viral proteinases. Eur. J. Biochem. 1995, 228, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Darke, P.L.; Leu, C.T.; Davis, L.J.; Heimbach, J.C.; Diehl, R.E.; Hill, W.S.; Dixon, R.A.; Sigal, I.S. Human immunodeficiency virus protease. Bacterial expression and characterization of the purified aspartic protease. J. Biol. Chem. 1989, 264, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Hyland, L.J.; Tomaszek, T.A., Jr.; Meek, T.D. Human immunodeficiency virus-1 protease. 2. Use of pH rate studies and solvent kinetic isotope effects to elucidate details of chemical mechanism. Biochemistry 1991, 30, 8454–8463. [Google Scholar] [CrossRef]
- Perilla, J.R.; Schulten, K. Physical properties of the HIV-1 capsid from all-atom molecular dynamics simulations. Nat. Commun. 2017, 8, 15959. [Google Scholar] [CrossRef]
- Cosset, F.L.; Lavillette, D. Cell entry of enveloped viruses. Adv. Genet. 2011, 73, 121–183. [Google Scholar]
- Roche, S.; Gaudin, Y. Characterization of the equilibrium between the native and fusion-inactive conformation of rabies virus glycoprotein indicates that the fusion complex is made of several trimers. Virology 2002, 297, 128–135. [Google Scholar] [CrossRef]
- Romero-Brey, I.; Bartenschlager, R. Viral Infection at High Magnification: 3D Electron Microscopy Methods to Analyze the Architecture of Infected Cells. Viruses 2015, 7, 6316–6345. [Google Scholar] [CrossRef]
- Roingeard, P.; Raynal, P.I.; Eymieux, S.; Blanchard, E. Virus detection by transmission electron microscopy: Still useful for diagnosis and a plus for biosafety. Rev. Med. Virol. 2019, 29, e2019. [Google Scholar] [CrossRef]
- Richert-Poggeler, K.R.; Franzke, K.; Hipp, K.; Kleespies, R.G. Electron Microscopy Methods for Virus Diagnosis and High Resolution Analysis of Viruses. Front. Microbiol. 2018, 9, 3255. [Google Scholar] [CrossRef]
- Graham, L.; Orenstein, J.M. Processing tissue and cells for transmission electron microscopy in diagnostic pathology and research. Nat. Protoc. 2007, 2, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Rey, J.S.; Li, W.; Bryer, A.J.; Beatson, H.; Lantz, C.; Engelman, A.N.; Perilla, J.R. Deep-learning in situ classification of HIV-1 virion morphology. Comput. Struct. Biotechnol. J. 2021, 19, 5688–5700. [Google Scholar] [CrossRef]
- Sarca, A.D.; Sardo, L.; Fukuda, H.; Matsui, H.; Shirakawa, K.; Horikawa, K.; Takaori-Kondo, A.; Izumi, T. FRET-Based Detection and Quantification of HIV-1 Virion Maturation. Front. Microbiol. 2021, 12, 647452. [Google Scholar] [CrossRef] [PubMed]
- Benjin, X.; Ling, L. Developments, applications, and prospects of cryo-electron microscopy. Protein Sci. 2020, 29, 872–882. [Google Scholar] [CrossRef]
- Mendonca, L.; Sun, D.; Ning, J.; Liu, J.; Kotecha, A.; Olek, M.; Frosio, T.; Fu, X.; Himes, B.A.; Kleinpeter, A.B.; et al. CryoET structures of immature HIV Gag reveal six-helix bundle. Commun. Biol. 2021, 4, 481. [Google Scholar] [CrossRef]
- Schoehn, G.; Chenavier, F.; Crepin, T. Advances in Structural Virology via Cryo-EM in 2022. Viruses 2023, 15, 1315. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.E.; Kiessling, V.; Pornillos, O.; White, J.M.; Ganser-Pornillos, B.K.; Tamm, L.K. HIV-cell membrane fusion intermediates are restricted by Serincs as revealed by cryo-electron and TIRF microscopy. J. Biol. Chem. 2020, 295, 15183–15195. [Google Scholar] [CrossRef]
- Zila, V.; Margiotta, E.; Turonova, B.; Muller, T.G.; Zimmerli, C.E.; Mattei, S.; Allegretti, M.; Borner, K.; Rada, J.; Muller, B.; et al. Cone-shaped HIV-1 capsids are transported through intact nuclear pores. Cell 2021, 184, 1032–1046.e18. [Google Scholar] [CrossRef]
- Mangala Prasad, V.; Leaman, D.P.; Lovendahl, K.N.; Croft, J.T.; Benhaim, M.A.; Hodge, E.A.; Zwick, M.B.; Lee, K.K. Cryo-ET of Env on intact HIV virions reveals structural variation and positioning on the Gag lattice. Cell 2022, 185, 641–653.e17. [Google Scholar] [CrossRef]
- Golm, S.K.; Hubner, W.; Muller, K.M. Fluorescence Microscopy in Adeno-Associated Virus Research. Viruses 2023, 15, 1174. [Google Scholar] [CrossRef]
- Huang, B.; Bates, M.; Zhuang, X. Super-resolution fluorescence microscopy. Annu. Rev. Biochem. 2009, 78, 993–1016. [Google Scholar] [CrossRef]
- Chen, G.W.; Song, F.L.; Xiong, X.Q.; Peng, X.J. Fluorescent Nanosensors Based on Fluorescence Resonance Energy Transfer (FRET). Ind. Eng. Chem. Res. 2013, 52, 11228–11245. [Google Scholar] [CrossRef]
- Sekar, R.B.; Periasamy, A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J. Cell Biol. 2003, 160, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Ellis, E.; Veetil, J.V.; Yao, H.; Ye, K. Visualization of human immunodeficiency virus protease inhibition using a novel Forster resonance energy transfer molecular probe. Biotechnol. Prog. 2011, 27, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Sood, C.; Francis, A.C.; Desai, T.M.; Melikyan, G.B. An improved labeling strategy enables automated detection of single-virus fusion and assessment of HIV-1 protease activity in single virions. J. Biol. Chem. 2017, 292, 20196–20207. [Google Scholar] [CrossRef]
- Nagai, T.; Yamada, S.; Tominaga, T.; Ichikawa, M.; Miyawaki, A. Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 10554–10559. [Google Scholar] [CrossRef]
- Chiu, T.Y.; Yang, D.M. Intracellular Pb2+ content monitoring using a protein-based Pb2+ indicator. Toxicol. Sci. 2012, 126, 436–445. [Google Scholar] [CrossRef]
- Hubner, W.; Chen, P.; Del Portillo, A.; Liu, Y.; Gordon, R.E.; Chen, B.K. Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J. Virol. 2007, 81, 12596–12607. [Google Scholar] [CrossRef]
- Albertazzi, L.; Arosio, D.; Marchetti, L.; Ricci, F.; Beltram, F. Quantitative FRET analysis with the EGFP-mCherry fluorescent protein pair. Photochem. Photobiol. 2009, 85, 287–297. [Google Scholar] [CrossRef]
- Bhuckory, S.; Kays, J.C.; Dennis, A.M. In Vivo Biosensing Using Resonance Energy Transfer. Biosensors 2019, 9, 76. [Google Scholar] [CrossRef]
- Fontana, J.; Jurado, K.A.; Cheng, N.; Ly, N.L.; Fuchs, J.R.; Gorelick, R.J.; Engelman, A.N.; Steven, A.C. Distribution and Redistribution of HIV-1 Nucleocapsid Protein in Immature, Mature, and Integrase-Inhibited Virions: A Role for Integrase in Maturation. J. Virol. 2015, 89, 9765–9780. [Google Scholar] [CrossRef] [PubMed]
- Jurado, K.A.; Wang, H.; Slaughter, A.; Feng, L.; Kessl, J.J.; Koh, Y.; Wang, W.; Ballandras-Colas, A.; Patel, P.A.; Fuchs, J.R.; et al. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. USA 2013, 110, 8690–8695. [Google Scholar] [CrossRef]
- Checkley, M.A.; Luttge, B.G.; Soheilian, F.; Nagashima, K.; Freed, E.O. The capsid-spacer peptide 1 Gag processing intermediate is a dominant-negative inhibitor of HIV-1 maturation. Virology 2010, 400, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Tan, E.; Chin, C.S.H.; Lim, Z.F.S.; Ng, S.K. HEK293 Cell Line as a Platform to Produce Recombinant Proteins and Viral Vectors. Front. Bioeng. Biotechnol. 2021, 9, 796991. [Google Scholar] [CrossRef]
- Chen, B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019, 27, 878–891. [Google Scholar] [CrossRef]
- Ma, X.; Lu, M.; Gorman, J.; Terry, D.S.; Hong, X.; Zhou, Z.; Zhao, H.; Altman, R.B.; Arthos, J.; Blanchard, S.C.; et al. HIV-1 Env trimer opens through an asymmetric intermediate in which individual protomers adopt distinct conformations. Elife 2018, 7, e34271. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.B.; Gorman, J.; Ma, X.; Zhou, Z.; Arthos, J.; Burton, D.R.; Koff, W.C.; Courter, J.R.; Smith, A.B., 3rd; Kwong, P.D.; et al. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 2014, 346, 759–763. [Google Scholar] [CrossRef]
- Herschhorn, A.; Ma, X.; Gu, C.; Ventura, J.D.; Castillo-Menendez, L.; Melillo, B.; Terry, D.S.; Smith, A.B., 3rd; Blanchard, S.C.; Munro, J.B.; et al. Release of gp120 Restraints Leads to an Entry-Competent Intermediate State of the HIV-1 Envelope Glycoproteins. mBio 2016, 7, e01598-16. [Google Scholar] [CrossRef]
- Alsahafi, N.; Bakouche, N.; Kazemi, M.; Richard, J.; Ding, S.; Bhattacharyya, S.; Das, D.; Anand, S.P.; Prevost, J.; Tolbert, W.D.; et al. An Asymmetric Opening of HIV-1 Envelope Mediates Antibody-Dependent Cellular Cytotoxicity. Cell Host Microbe 2019, 25, 578–587.e5. [Google Scholar] [CrossRef]
- Checkley, M.A.; Luttge, B.G.; Freed, E.O. HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J. Mol. Biol. 2011, 410, 582–608. [Google Scholar] [CrossRef]
- Hill, C.P.; Worthylake, D.; Bancroft, D.P.; Christensen, A.M.; Sundquist, W.I. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: Implications for membrane association and assembly. Proc. Natl. Acad. Sci. USA 1996, 93, 3099–3104. [Google Scholar] [CrossRef] [PubMed]
- Alfadhli, A.; Barklis, R.L.; Barklis, E. HIV-1 matrix organizes as a hexamer of trimers on membranes containing phosphatidylinositol-(4,5)-bisphosphate. Virology 2009, 387, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Tedbury, P.R.; Ablan, S.D.; Freed, E.O. Global rescue of defects in HIV-1 envelope glycoprotein incorporation: Implications for matrix structure. PLoS Pathog. 2013, 9, e1003739. [Google Scholar] [CrossRef] [PubMed]
- Tedbury, P.R.; Novikova, M.; Ablan, S.D.; Freed, E.O. Biochemical evidence of a role for matrix trimerization in HIV-1 envelope glycoprotein incorporation. Proc. Natl. Acad. Sci. USA 2016, 113, E182–E190. [Google Scholar] [CrossRef] [PubMed]
- Wyma, D.J.; Jiang, J.; Shi, J.; Zhou, J.; Lineberger, J.E.; Miller, M.D.; Aiken, C. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: A novel role of the gp41 cytoplasmic tail. J. Virol. 2004, 78, 3429–3435. [Google Scholar] [CrossRef]
- Murakami, T.; Ablan, S.; Freed, E.O.; Tanaka, Y. Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J. Virol. 2004, 78, 1026–1031. [Google Scholar] [CrossRef]
- Chojnacki, J.; Staudt, T.; Glass, B.; Bingen, P.; Engelhardt, J.; Anders, M.; Schneider, J.; Müller, B.; Hell, S.W.; Kräusslich, H.G. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science 2012, 338, 524–528. [Google Scholar] [CrossRef]
- Li, W.; Qin, Z.; Nand, E.; Grunst, M.W.; Grover, J.R.; Bess, J.W., Jr.; Lifson, J.D.; Zwick, M.B.; Tagare, H.D.; Uchil, P.D.; et al. HIV-1 Env trimers asymmetrically engage CD4 receptors in membranes. Nature 2023, 623, 1026–1033. [Google Scholar] [CrossRef]
- Carlon-Andres, I.; Malinauskas, T.; Padilla-Parra, S. Structure dynamics of HIV-1 Env trimers on native virions engaged with living T cells. Commun. Biol. 2021, 4, 1228. [Google Scholar] [CrossRef]
- Kol, N.; Shi, Y.; Tsvitov, M.; Barlam, D.; Shneck, R.Z.; Kay, M.S.; Rousso, I. A stiffness switch in human immunodeficiency virus. Biophys. J. 2007, 92, 1777–1783. [Google Scholar] [CrossRef]
- Nieto-Garai, J.A.; Arboleya, A.; Otaegi, S.; Chojnacki, J.; Casas, J.; Fabrias, G.; Contreras, F.X.; Krausslich, H.G.; Lorizate, M. Cholesterol in the Viral Membrane is a Molecular Switch Governing HIV-1 Env Clustering. Adv. Sci. 2021, 8, 2003468. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Cimakasky, L.M.; Hampton, R.; Nguyen, D.H.; Hildreth, J.E. Lipid rafts and HIV pathogenesis: Host membrane cholesterol is required for infection by HIV type 1. AIDS Res. Hum. Retroviruses 2001, 17, 1009–1019. [Google Scholar] [CrossRef]
- Waheed, A.A.; Ablan, S.D.; Mankowski, M.K.; Cummins, J.E.; Ptak, R.G.; Schaffner, C.P.; Freed, E.O. Inhibition of HIV-1 replication by amphotericin B methyl ester: Selection for resistant variants. J. Biol. Chem. 2006, 281, 28699–28711. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Chertova, E.; Bess, J., Jr.; Lifson, J.D.; Arthur, L.O.; Liu, J.; Taylor, K.A.; Roux, K.H. Electron tomography analysis of envelope glycoprotein trimers on HIV and simian immunodeficiency virus virions. Proc. Natl. Acad. Sci. USA 2003, 100, 15812–15817. [Google Scholar] [CrossRef]
- Klein, J.S.; Bjorkman, P.J. Few and far between: How HIV may be evading antibody avidity. PLoS Pathog. 2010, 6, e1000908. [Google Scholar] [CrossRef] [PubMed]
- Brandenberg, O.F.; Magnus, C.; Regoes, R.R.; Trkola, A. The HIV-1 Entry Process: A Stoichiometric View. Trends Microbiol. 2015, 23, 763–774. [Google Scholar] [CrossRef]
- Weber, I.T.; Wang, Y.F.; Harrison, R.W. HIV Protease: Historical Perspective and Current Research. Viruses 2021, 13, 839. [Google Scholar] [CrossRef]
- Kohl, N.E.; Emini, E.A.; Schleif, W.A.; Davis, L.J.; Heimbach, J.C.; Dixon, R.A.; Scolnick, E.M.; Sigal, I.S. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. USA 1988, 85, 4686–4690. [Google Scholar] [CrossRef]
- Navia, M.A.; Fitzgerald, P.M.; McKeever, B.M.; Leu, C.T.; Heimbach, J.C.; Herber, W.K.; Sigal, I.S.; Darke, P.L.; Springer, J.P. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature 1989, 337, 615–620. [Google Scholar] [CrossRef]
- Wlodawer, A.; Miller, M.; Jaskolski, M.; Sathyanarayana, B.K.; Baldwin, E.; Weber, I.T.; Selk, L.M.; Clawson, L.; Schneider, J.; Kent, S.B. Conserved folding in retroviral proteases: Crystal structure of a synthetic HIV-1 protease. Science 1989, 245, 616–621. [Google Scholar] [CrossRef]
- Darke, P.L.; Nutt, R.F.; Brady, S.F.; Garsky, V.M.; Ciccarone, T.M.; Leu, C.T.; Lumma, P.K.; Freidinger, R.M.; Veber, D.F.; Sigal, I.S. HIV-1 protease specificity of peptide cleavage is sufficient for processing of gag and pol polyproteins. Biochem. Biophys. Res. Commun. 1988, 156, 297–303. [Google Scholar] [CrossRef]
- Kotler, M.; Katz, R.A.; Danho, W.; Leis, J.; Skalka, A.M. Synthetic peptides as substrates and inhibitors of a retroviral protease. Proc. Natl. Acad. Sci. USA 1988, 85, 4185–4189. [Google Scholar] [CrossRef] [PubMed]
- Atta, M.G.; De Seigneux, S.; Lucas, G.M. Clinical Pharmacology in HIV Therapy. Clin. J. Am. Soc. Nephrol. 2019, 14, 435–444. [Google Scholar] [CrossRef]
- Loos, N.H.C.; Beijnen, J.H.; Schinkel, A.H. The Mechanism-Based Inactivation of CYP3A4 by Ritonavir: What Mechanism? Int. J. Mol. Sci. 2022, 23, 9866. [Google Scholar] [CrossRef] [PubMed]
- Nathan, B.; Bayley, J.; Waters, L.; Post, F.A. Cobicistat: A Novel Pharmacoenhancer for Co-Formulation with HIV Protease and Integrase Inhibitors. Infect. Dis. Ther. 2013, 2, 111–122. [Google Scholar] [CrossRef]
- Koh, Y.; Nakata, H.; Maeda, K.; Ogata, H.; Bilcer, G.; Devasamudram, T.; Kincaid, J.F.; Boross, P.; Wang, Y.F.; Tie, Y.; et al. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 2003, 47, 3123–3129. [Google Scholar] [CrossRef]
- Spielvogel, E.; Lee, S.K.; Zhou, S.; Lockbaum, G.J.; Henes, M.; Sondgeroth, A.; Kosovrasti, K.; Nalivaika, E.A.; Ali, A.; Yilmaz, N.K.; et al. Selection of HIV-1 for resistance to fifth-generation protease inhibitors reveals two independent pathways to high-level resistance. Elife 2023, 12, e80328. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Weber, I.T.; Mitsuya, H. Beyond darunavir: Recent development of next generation HIV-1 protease inhibitors to combat drug resistance. Chem. Commun. 2022, 58, 11762–11782. [Google Scholar] [CrossRef]
- Fujioka, T.; Kashiwada, Y.; Kilkuskie, R.E.; Cosentino, L.M.; Ballas, L.M.; Jiang, J.B.; Janzen, W.P.; Chen, I.S.; Lee, K.H. Anti-AIDS agents, 11. Betulinic acid and platanic acid as anti-HIV principles from Syzigium claviflorum, and the anti-HIV activity of structurally related triterpenoids. J. Nat. Prod. 1994, 57, 243–247. [Google Scholar] [CrossRef]
- Kanamoto, T.; Kashiwada, Y.; Kanbara, K.; Gotoh, K.; Yoshimori, M.; Goto, T.; Sano, K.; Nakashima, H. Anti-human immunodeficiency virus activity of YK-FH312 (a betulinic acid derivative), a novel compound blocking viral maturation. Antimicrob. Agents Chemother. 2001, 45, 1225–1230. [Google Scholar] [CrossRef]
- Kashiwada, Y.; Hashimoto, F.; Cosentino, L.M.; Chen, C.H.; Garrett, P.E.; Lee, K.H. Betulinic acid and dihydrobetulinic acid derivatives as potent anti-HIV agents. J. Med. Chem. 1996, 39, 1016–1017. [Google Scholar] [CrossRef]
- Li, F.; Goila-Gaur, R.; Salzwedel, K.; Kilgore, N.R.; Reddick, M.; Matallana, C.; Castillo, A.; Zoumplis, D.; Martin, D.E.; Orenstein, J.M.; et al. PA-457: A potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc. Natl. Acad. Sci. USA 2003, 100, 13555–13560. [Google Scholar] [CrossRef] [PubMed]
- Schur, F.K.; Obr, M.; Hagen, W.J.; Wan, W.; Jakobi, A.J.; Kirkpatrick, J.M.; Sachse, C.; Krausslich, H.G.; Briggs, J.A. An atomic model of HIV-1 capsid-SP1 reveals structures regulating assembly and maturation. Science 2016, 353, 506–508. [Google Scholar] [CrossRef]
- Wagner, J.M.; Zadrozny, K.K.; Chrustowicz, J.; Purdy, M.D.; Yeager, M.; Ganser-Pornillos, B.K.; Pornillos, O. Crystal structure of an HIV assembly and maturation switch. Elife 2016, 5, e17063. [Google Scholar] [CrossRef]
- Purdy, M.D.; Shi, D.; Chrustowicz, J.; Hattne, J.; Gonen, T.; Yeager, M. MicroED structures of HIV-1 Gag CTD-SP1 reveal binding interactions with the maturation inhibitor bevirimat. Proc. Natl. Acad. Sci. USA 2018, 115, 13258–13263. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Louis, J.M.; Tycko, R. Effects of an HIV-1 maturation inhibitor on the structure and dynamics of CA-SP1 junction helices in virus-like particles. Proc. Natl. Acad. Sci. USA 2020, 117, 10286–10293. [Google Scholar] [CrossRef]
- Adamson, C.S.; Ablan, S.D.; Boeras, I.; Goila-Gaur, R.; Soheilian, F.; Nagashima, K.; Li, F.; Salzwedel, K.; Sakalian, M.; Wild, C.T.; et al. In vitro resistance to the human immunodeficiency virus type 1 maturation inhibitor PA-457 (Bevirimat). J. Virol. 2006, 80, 10957–10971. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zoumplis, D.; Matallana, C.; Kilgore, N.R.; Reddick, M.; Yunus, A.S.; Adamson, C.S.; Salzwedel, K.; Martin, D.E.; Allaway, G.P.; et al. Determinants of activity of the HIV-1 maturation inhibitor PA-457. Virology 2006, 356, 217–224. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, C.H.; Aiken, C. Human immunodeficiency virus type 1 resistance to the small molecule maturation inhibitor 3-O-(3′,3′-dimethylsuccinyl)-betulinic acid is conferred by a variety of single amino acid substitutions at the CA-SP1 cleavage site in Gag. J Virol 2006, 80, 12095–12101. [Google Scholar] [CrossRef]
- Zhou, J.; Yuan, X.; Dismuke, D.; Forshey, B.M.; Lundquist, C.; Lee, K.H.; Aiken, C.; Chen, C.H. Small-molecule inhibition of human immunodeficiency virus type 1 replication by specific targeting of the final step of virion maturation. J. Virol. 2004, 78, 922–929. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Feasley, C.L.; Jackson, K.W.; Nitz, T.J.; Salzwedel, K.; Air, G.M.; Sakalian, M. The prototype HIV-1 maturation inhibitor, bevirimat, binds to the CA-SP1 cleavage site in immature Gag particles. Retrovirology 2011, 8, 101. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, C.H.; Aiken, C. The sequence of the CA-SP1 junction accounts for the differential sensitivity of HIV-1 and SIV to the small molecule maturation inhibitor 3-O-3′,3′-dimethylsuccinyl-betulinic acid. Retrovirology 2004, 1, 15. [Google Scholar] [CrossRef]
- Zhou, J.; Huang, L.; Hachey, D.L.; Chen, C.H.; Aiken, C. Inhibition of HIV-1 maturation via drug association with the viral Gag protein in immature HIV-1 particles. J. Biol. Chem. 2005, 280, 42149–42155. [Google Scholar] [CrossRef]
- Sarkar, S.; Zadrozny, K.K.; Zadorozhnyi, R.; Russell, R.W.; Quinn, C.M.; Kleinpeter, A.; Ablan, S.; Meshkin, H.; Perilla, J.R.; Freed, E.O.; et al. Structural basis of HIV-1 maturation inhibitor binding and activity. Nat. Commun. 2023, 14, 1237. [Google Scholar] [CrossRef]
- Prabu-Jeyabalan, M.; Nalivaika, E.; Schiffer, C.A. Substrate shape determines specificity of recognition for HIV-1 protease: Analysis of crystal structures of six substrate complexes. Structure 2002, 10, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.F.; Ogundele, A.; Forrest, A.; Wilton, J.; Salzwedel, K.; Doto, J.; Allaway, G.P.; Martin, D.E. Phase I and II study of the safety, virologic effect, and pharmacokinetics/pharmacodynamics of single-dose 3-o-(3′,3′-dimethylsuccinyl)betulinic acid (bevirimat) against human immunodeficiency virus infection. Antimicrob. Agents Chemother. 2007, 51, 3574–3581. [Google Scholar] [CrossRef]
- DeJesus, E.; Harward, S.; Jewell, R.C.; Johnson, M.; Dumont, E.; Wilches, V.; Halliday, F.; Talarico, C.L.; Jeffrey, J.; Gan, J.; et al. A Phase IIa Study Evaluating Safety, Pharmacokinetics, and Antiviral Activity of GSK2838232, a Novel, Second-generation Maturation Inhibitor, in Participants With Human Immunodeficiency Virus Type 1 Infection. Clin. Infect. Dis. 2020, 71, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Schurmann, D.; Sobotha, C.; Boffito, M.; Sevinsky, H.; Ray, N.; Ravindran, P.; Xiao, H.; Keicher, C.; Huser, A.; et al. Antiviral Activity, Safety, and Exposure-Response Relationships of GSK3532795, a Second-Generation Human Immunodeficiency Virus Type 1 Maturation Inhibitor, Administered as Monotherapy or in Combination With Atazanavir With or Without Ritonavir in a Phase 2a Randomized, Dose-Ranging, Controlled Trial (AI468002). Clin. Infect. Dis. 2017, 65, 442–452. [Google Scholar]
- Morales-Ramirez, J.; Bogner, J.R.; Molina, J.M.; Lombaard, J.; Dicker, I.B.; Stock, D.A.; DeGrosky, M.; Gartland, M.; Pene Dumitrescu, T.; Min, S.; et al. Safety, efficacy, and dose response of the maturation inhibitor GSK3532795 (formerly known as BMS-955176) plus tenofovir/emtricitabine once daily in treatment-naive HIV-1-infected adults: Week 24 primary analysis from a randomized Phase IIb trial. PLoS ONE 2018, 13, e0205368. [Google Scholar] [CrossRef]
- Abstracts of the 19th European AIDS Conference (#EACS2023), October 18–21, 2023, Warsaw, Poland. HIV Med. 2023, 24 (Suppl. 5), 3–788.
- Benn, P.D.; Zhang, Y.; Kahl, L.; Greene, T.J.; Bainbridge, V.; Fishman, C.; Wen, B.; Gartland, M. A phase I, first-in-human study investigating the safety, tolerability, and pharmacokinetics of the maturation inhibitor GSK3739937. Pharmacol. Res. Perspect. 2023, 11, e01093. [Google Scholar] [CrossRef] [PubMed]
- Yant, S.R.; Mulato, A.; Hansen, D.; Tse, W.C.; Niedziela-Majka, A.; Zhang, J.R.; Stepan, G.J.; Jin, D.; Wong, M.H.; Perreira, J.M.; et al. A highly potent long-acting small-molecule HIV-1 capsid inhibitor with efficacy in a humanized mouse model. Nat. Med. 2019, 25, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Faysal, K.M.R.; Walsh, J.C.; Renner, N.; Marquez, C.L.; Shah, V.B.; Tuckwell, A.J.; Christie, M.P.; Parker, M.W.; Turville, S.G.; Towers, G.J.; et al. Pharmacologic hyperstabilisation of the HIV-1 capsid lattice induces capsid failure. Elife 2024, 13, e83605. [Google Scholar] [PubMed]
- Marquis, K.A.; Everett, J.; Cantu, A.; McFarland, A.; Sherrill-Mix, S.; Krystal, M.; Parcella, K.; Gillis, E.; Fridell, R.A.; Bushman, F.D. The HIV-1 Capsid-Targeted Inhibitor GSK878 Alters Selection of Target Sites for HIV DNA Integration. AIDS Res. Hum. Retroviruses 2024, 40, 114–126. [Google Scholar] [CrossRef]
- McFadden, W.M.; Snyder, A.A.; Kirby, K.A.; Tedbury, P.R.; Raj, M.; Wang, Z.; Sarafianos, S.G. Rotten to the core: Antivirals targeting the HIV-1 capsid core. Retrovirology 2021, 18, 41. [Google Scholar] [CrossRef]
- Zhou, J.; Price, A.J.; Halambage, U.D.; James, L.C.; Aiken, C. HIV-1 Resistance to the Capsid-Targeting Inhibitor PF74 Results in Altered Dependence on Host Factors Required for Virus Nuclear Entry. J. Virol. 2015, 89, 9068–9079. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Alam, S.L.; Fricke, T.; Zadrozny, K.; Sedzicki, J.; Taylor, A.B.; Demeler, B.; Pornillos, O.; Ganser-Pornillos, B.K.; Diaz-Griffero, F.; et al. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc. Natl. Acad. Sci. USA 2014, 111, 18625–18630. [Google Scholar] [CrossRef]
- Lamorte, L.; Titolo, S.; Lemke, C.T.; Goudreau, N.; Mercier, J.F.; Wardrop, E.; Shah, V.B.; von Schwedler, U.K.; Langelier, C.; Banik, S.S.; et al. Discovery of novel small-molecule HIV-1 replication inhibitors that stabilize capsid complexes. Antimicrob. Agents Chemother. 2013, 57, 4622–4631. [Google Scholar] [CrossRef]
- Blair, W.S.; Pickford, C.; Irving, S.L.; Brown, D.G.; Anderson, M.; Bazin, R.; Cao, J.; Ciaramella, G.; Isaacson, J.; Jackson, L.; et al. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 2010, 6, e1001220. [Google Scholar] [CrossRef]
- Wang, C.; Huang, H.; Mallon, K.; Valera, L.; Parcella, K.; Cockett, M.I.; Kadow, J.F.; Gillis, E.P.; Krystal, M.; Fridell, R.A. Antiviral Properties of HIV-1 Capsid Inhibitor GSK878. Antimicrob. Agents Chemother. 2023, 67, e0169422. [Google Scholar] [CrossRef]
- Gillis, E.P.; Parcella, K.; Bowsher, M.; Cook, J.H.; Iwuagwu, C.; Naidu, B.N.; Patel, M.; Peese, K.; Huang, H.; Valera, L.; et al. Potent Long-Acting Inhibitors Targeting the HIV-1 Capsid Based on a Versatile Quinazolin-4-one Scaffold. J. Med. Chem. 2023, 66, 1941–1954. [Google Scholar] [CrossRef]
- Gupta, S.K.; Berhe, M.; Crofoot, G.; Benson, P.; Ramgopal, M.; Sims, J.; McDonald, C.; Ruane, P.; Sanchez, W.E.; Scribner, A.; et al. Lenacapavir administered every 26 weeks or daily in combination with oral daily antiretroviral therapy for initial treatment of HIV: A randomised, open-label, active-controlled, phase 2 trial. Lancet HIV 2023, 10, e15–e23. [Google Scholar] [CrossRef] [PubMed]
- Margot, N.; Pennetzdorfer, N.; Naik, V.; Rhee, M.; Callebaut, C. Cross-resistance to entry inhibitors and lenacapavir resistance through Week 52 in study CAPELLA. Antivir. Ther. 2023, 28, 13596535231220754. [Google Scholar] [CrossRef] [PubMed]
- Margot, N.A.; Naik, V.; VanderVeen, L.; Anoshchenko, O.; Singh, R.; Dvory-Sobol, H.; Rhee, M.S.; Callebaut, C. Resistance Analyses in Highly Treatment-Experienced People with Human Immunodeficiency Virus (HIV) Treated with the Novel Capsid HIV Inhibitor Lenacapavir. J. Infect. Dis. 2022, 226, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Segal-Maurer, S.; DeJesus, E.; Stellbrink, H.J.; Castagna, A.; Richmond, G.J.; Sinclair, G.I.; Siripassorn, K.; Ruane, P.J.; Berhe, M.; Wang, H.; et al. Capsid Inhibition with Lenacapavir in Multidrug-Resistant HIV-1 Infection. N. Engl. J. Med. 2022, 386, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Orkin, C.; Schapiro, J.M.; Perno, C.F.; Kuritzkes, D.R.; Patel, P.; DeMoor, R.; Dorey, D.; Wang, Y.; Han, K.; Van Eygen, V.; et al. Expanded Multivariable Models to Assist Patient Selection for Long-Acting Cabotegravir + Rilpivirine Treatment: Clinical Utility of a Combination of Patient, Drug Concentration, and Viral Factors Associated With Virologic Failure. Clin. Infect. Dis. 2023, 77, 1423–1431. [Google Scholar] [CrossRef]
- Cutrell, A.G.; Schapiro, J.M.; Perno, C.F.; Kuritzkes, D.R.; Quercia, R.; Patel, P.; Polli, J.W.; Dorey, D.; Wang, Y.; Wu, S.; et al. Exploring predictors of HIV-1 virologic failure to long-acting cabotegravir and rilpivirine: A multivariable analysis. AIDS 2021, 35, 1333–1342. [Google Scholar] [CrossRef]
- Bekker, L.G.; Das, M.; Abdool Karim, Q.; Ahmed, K.; Batting, J.; Brumskine, W.; Gill, K.; Harkoo, I.; Jaggernath, M.; Kigozi, G.; et al. Twice-Yearly Lenacapavir or Daily F/TAF for HIV Prevention in Cisgender Women. N. Engl. J. Med. 2024; ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Landovitz, R.J.; Donnell, D.; Clement, M.E.; Hanscom, B.; Cottle, L.; Coelho, L.; Cabello, R.; Chariyalertsak, S.; Dunne, E.F.; Frank, I.; et al. Cabotegravir for HIV Prevention in Cisgender Men and Transgender Women. N. Engl. J. Med. 2021, 385, 595–608. [Google Scholar] [CrossRef]
- Delany-Moretlwe, S.; Hughes, J.P.; Bock, P.; Ouma, S.G.; Hunidzarira, P.; Kalonji, D.; Kayange, N.; Makhema, J.; Mandima, P.; Mathew, C.; et al. Cabotegravir for the prevention of HIV-1 in women: Results from HPTN 084, a phase 3, randomised clinical trial. Lancet 2022, 399, 1779–1789. [Google Scholar] [CrossRef]
- Fujiwara, T.; Mizuuchi, K. Retroviral DNA integration: Structure of an integration intermediate. Cell 1988, 54, 497–504. [Google Scholar] [CrossRef]
- Brown, P.O.; Bowerman, B.; Varmus, H.E.; Bishop, J.M. Retroviral integration: Structure of the initial covalent product and its precursor, and a role for the viral IN protein. Proc. Natl. Acad. Sci. USA 1989, 86, 2525–2529. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.J.; Schwartzberg, P.L.; Goff, S.P. Structure of the termini of DNA intermediates in the integration of retroviral DNA: Dependence on IN function and terminal DNA sequence. Cell 1989, 58, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Sherman, P.A.; Fyfe, J.A. Human immunodeficiency virus integration protein expressed in Escherichia coli possesses selective DNA cleaving activity. Proc. Natl. Acad. Sci. USA 1990, 87, 5119–5123. [Google Scholar] [CrossRef]
- Zhao, A.V.; Crutchley, R.D.; Guduru, R.C.; Ton, K.; Lam, T.; Min, A.C. A clinical review of HIV integrase strand transfer inhibitors (INSTIs) for the prevention and treatment of HIV-1 infection. Retrovirology 2022, 19, 22. [Google Scholar] [CrossRef]
- Kessl, J.J.; Kutluay, S.B.; Townsend, D.; Rebensburg, S.; Slaughter, A.; Larue, R.C.; Shkriabai, N.; Bakouche, N.; Fuchs, J.R.; Bieniasz, P.D.; et al. HIV-1 Integrase Binds the Viral RNA Genome and Is Essential during Virion Morphogenesis. Cell 2016, 166, 1257–1268.e12. [Google Scholar] [CrossRef]
- Allen, P.; Worland, S.; Gold, L. Isolation of high-affinity RNA ligands to HIV-1 integrase from a random pool. Virology 1995, 209, 327–336. [Google Scholar] [CrossRef]
- Engelman, A. In vivo analysis of retroviral integrase structure and function. Adv. Virus. Res. 1999, 52, 411–426. [Google Scholar] [PubMed]
- Engelman, A.; Englund, G.; Orenstein, J.M.; Martin, M.A.; Craigie, R. Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication. J. Virol. 1995, 69, 2729–2736. [Google Scholar] [CrossRef]
- Llano, M.; Vanegas, M.; Hutchins, N.; Thompson, D.; Delgado, S.; Poeschla, E.M. Identification and characterization of the chromatin-binding domains of the HIV-1 integrase interactor LEDGF/p75. J. Mol. Biol. 2006, 360, 760–773. [Google Scholar] [CrossRef]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar] [CrossRef] [PubMed]
- Maertens, G.; Cherepanov, P.; Pluymers, W.; Busschots, K.; De Clercq, E.; Debyser, Z.; Engelborghs, Y. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J. Biol. Chem. 2003, 278, 33528–33539. [Google Scholar] [CrossRef]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; De Clercq, E.; Debyser, Z. HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells. J. Biol. Chem. 2003, 278, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Desimmie, B.A.; Demeulemeester, J.; Christ, F.; Debyser, Z. Rational design of LEDGINs as first allosteric integrase inhibitors for the treatment of HIV infection. Drug Discov. Today Technol. 2013, 10, e517–e522. [Google Scholar] [CrossRef]
- Le Rouzic, E.; Bonnard, D.; Chasset, S.; Bruneau, J.M.; Chevreuil, F.; Le Strat, F.; Nguyen, J.; Beauvoir, R.; Amadori, C.; Brias, J.; et al. Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology 2013, 10, 144. [Google Scholar] [CrossRef]
- Sharma, A.; Slaughter, A.; Jena, N.; Feng, L.; Kessl, J.J.; Fadel, H.J.; Malani, N.; Male, F.; Wu, L.; Poeschla, E.; et al. A new class of multimerization selective inhibitors of HIV-1 integrase. PLoS Pathog. 2014, 10, e1004171. [Google Scholar] [CrossRef]
- Fader, L.D.; Malenfant, E.; Parisien, M.; Carson, R.; Bilodeau, F.; Landry, S.; Pesant, M.; Brochu, C.; Morin, S.; Chabot, C.; et al. Discovery of BI 224436, a Noncatalytic Site Integrase Inhibitor (NCINI) of HIV-1. ACS Med. Chem. Lett. 2014, 5, 422–427. [Google Scholar] [CrossRef]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; Van der Veken, N.J.; Van Remoortel, B.; Strelkov, S.V.; et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Turkki, V.; Sherrill-Mix, S.; Hwang, Y.; Eilers, G.; Taylor, L.; McDanal, C.; Wang, P.; Temelkoff, D.; Nolte, R.T.; et al. Structural Basis for Inhibitor-Induced Aggregation of HIV Integrase. PLoS Biol. 2016, 14, e1002584. [Google Scholar] [CrossRef]
- Gupta, K.; Brady, T.; Dyer, B.M.; Malani, N.; Hwang, Y.; Male, F.; Nolte, R.T.; Wang, L.; Velthuisen, E.; Jeffrey, J.; et al. Allosteric inhibition of human immunodeficiency virus integrase: Late block during viral replication and abnormal multimerization involving specific protein domains. J. Biol. Chem. 2014, 289, 20477–20488. [Google Scholar] [CrossRef]
- Balakrishnan, M.; Yant, S.R.; Tsai, L.; O’Sullivan, C.; Bam, R.A.; Tsai, A.; Niedziela-Majka, A.; Stray, K.M.; Sakowicz, R.; Cihlar, T. Non-catalytic site HIV-1 integrase inhibitors disrupt core maturation and induce a reverse transcription block in target cells. PLoS ONE 2013, 8, e74163. [Google Scholar]
- Eilers, G.; Gupta, K.; Allen, A.; Montermoso, S.; Murali, H.; Sharp, R.; Hwang, Y.; Bushman, F.D.; Van Duyne, G. Structure of a HIV-1 IN-Allosteric inhibitor complex at 2.93 A resolution: Routes to inhibitor optimization. PLoS Pathog. 2023, 19, e1011097. [Google Scholar] [CrossRef]
- Singer, M.R.; Dinh, T.; Levintov, L.; Annamalai, A.S.; Rey, J.S.; Briganti, L.; Cook, N.J.; Pye, V.E.; Taylor, I.A.; Kim, K.; et al. The Drug-Induced Interface That Drives HIV-1 Integrase Hypermultimerization and Loss of Function. mBio 2023, 14, e0356022. [Google Scholar] [CrossRef] [PubMed]
- Bonnard, D.; Le Rouzic, E.; Singer, M.R.; Yu, Z.; Le Strat, F.; Batisse, C.; Batisse, J.; Amadori, C.; Chasset, S.; Pye, V.E.; et al. Biological and Structural Analyses of New Potent Allosteric Inhibitors of HIV-1 Integrase. Antimicrob. Agents Chemother. 2023, 67, e0046223. [Google Scholar] [CrossRef]
- Maehigashi, T.; Ahn, S.; Kim, U.I.; Lindenberger, J.; Oo, A.; Koneru, P.C.; Mahboubi, B.; Engelman, A.N.; Kvaratskhelia, M.; Kim, K.; et al. A highly potent and safe pyrrolopyridine-based allosteric HIV-1 integrase inhibitor targeting host LEDGF/p75-integrase interaction site. PLoS Pathog. 2021, 17, e1009671. [Google Scholar] [CrossRef]
- Hitchcock, A.M.; Kufel, W.D.; Dwyer, K.A.M.; Sidman, E.F. Lenacapavir: A novel injectable HIV-1 capsid inhibitor. Int. J. Antimicrob. Agents 2024, 63, 107009. [Google Scholar] [CrossRef] [PubMed]
- Cihlar, T.; Fordyce, M. Current status and prospects of HIV treatment. Curr. Opin. Virol. 2016, 18, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Barthold, D.; Saldarriaga, E.M.; Brah, A.T.; Hauber, B.; Banerjee, P.; Fuller, S.M.; McCaslin, D.; Moldoveanu, A.M.; Marconi, V.C.; Simoni, J.M.; et al. Preference for daily oral pills over long-acting antiretroviral therapy options among people with HIV. AIDS 2023, 37, 1545–1553. [Google Scholar] [CrossRef]
- Swanstrom, A.E.; Gorelick, R.J.; Welker, J.L.; Schmidt, F.; Lu, B.; Wang, K.; Rowe, W.; Breed, M.W.; Killoran, K.E.; Kramer, J.A.; et al. Long-acting lenacapavir protects macaques against intravenous challenge with simian-tropic HIV. EBioMedicine 2023, 95, 104764. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Structure | Stage of Development |
|---|---|---|
| Protease Inhibitors (PIs): | ||
| Saquinavir (SQV) |  | FDA approved, 1995 |
| Darunavir (DRV) |  | FDA approved, 2006 |
| Maturation Inhibitors (MIs): | ||
| Bevirimat (BVM) |  | Clinical, terminated |
| GSK2838232 | 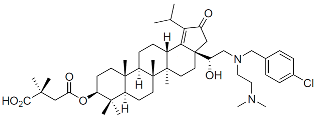 | Clinical, terminated |
| GSK3532795 |  | Clinical, terminated |
| GSK3640254 |  | Clinical, terminated |
| Capsid Inhibitors (CAIs): | ||
| PF-3450074 (PF74) |  | Discovery |
| BI-1 |  | Discovery |
| BI-2 |  | Discovery |
| GSK878 |  | Pre-clinical |
| GS-CA1 |  | Pre-clinical |
| Lenacapavir (LEN) |  | FDA approved, 2022 |
| Allosteric Integrase Inhibitors (ALLINIs): | ||
| Pirmitegravir (PIR) |  | Clinical, Phase 2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGraw, A.; Hillmer, G.; Medehincu, S.M.; Hikichi, Y.; Gagliardi, S.; Narayan, K.; Tibebe, H.; Marquez, D.; Mei Bose, L.; Keeting, A.; et al. Exploring HIV-1 Maturation: A New Frontier in Antiviral Development. Viruses 2024, 16, 1423. https://doi.org/10.3390/v16091423
McGraw A, Hillmer G, Medehincu SM, Hikichi Y, Gagliardi S, Narayan K, Tibebe H, Marquez D, Mei Bose L, Keeting A, et al. Exploring HIV-1 Maturation: A New Frontier in Antiviral Development. Viruses. 2024; 16(9):1423. https://doi.org/10.3390/v16091423
Chicago/Turabian StyleMcGraw, Aidan, Grace Hillmer, Stefania M. Medehincu, Yuta Hikichi, Sophia Gagliardi, Kedhar Narayan, Hasset Tibebe, Dacia Marquez, Lilia Mei Bose, Adleigh Keeting, and et al. 2024. "Exploring HIV-1 Maturation: A New Frontier in Antiviral Development" Viruses 16, no. 9: 1423. https://doi.org/10.3390/v16091423