
Emergence of a Novel Dengue Virus Serotype-2 Genotype IV Lineage III Strain and Displacement of Dengue Virus Serotype-1 in Central India (2019–2023)

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
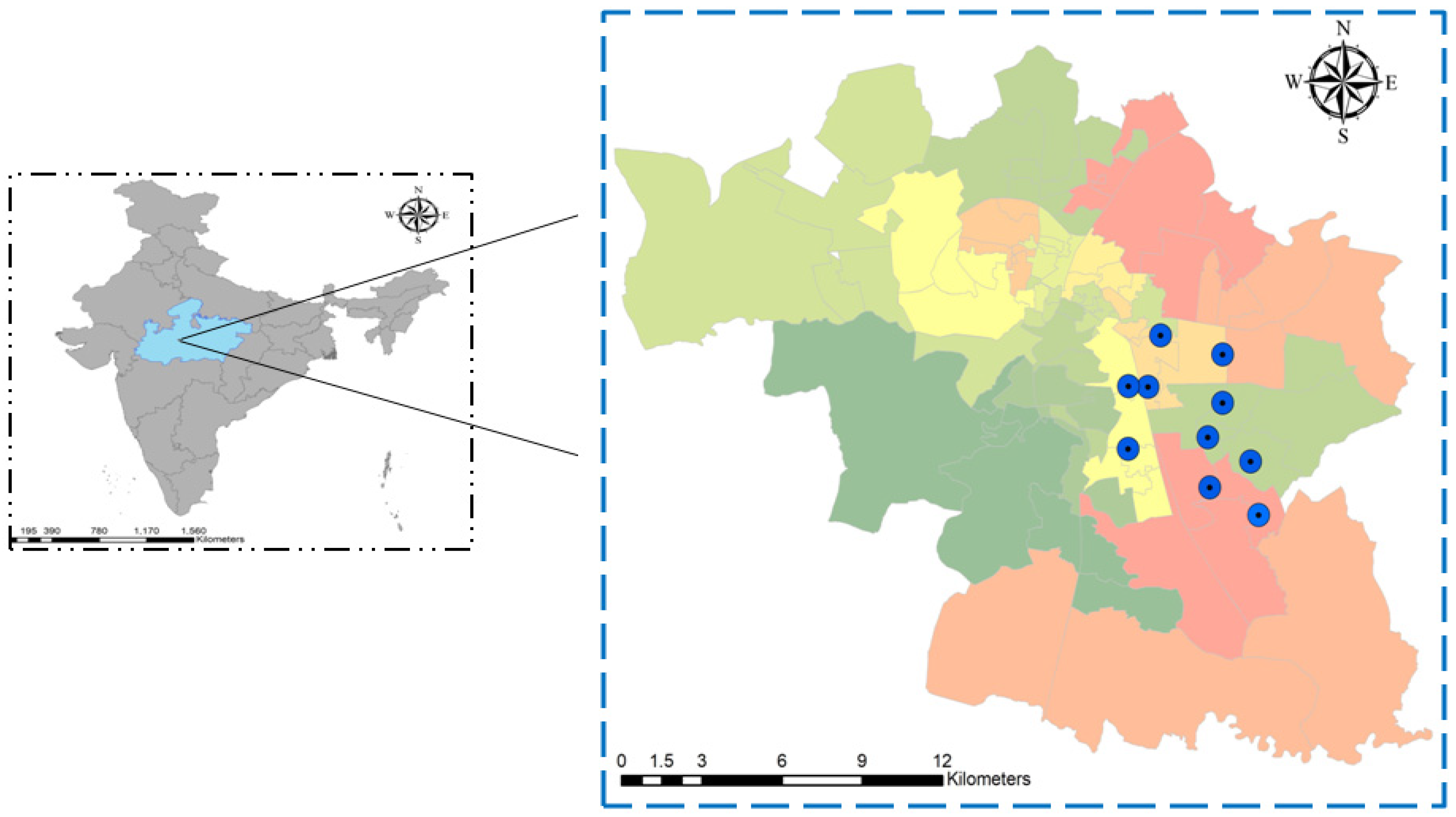
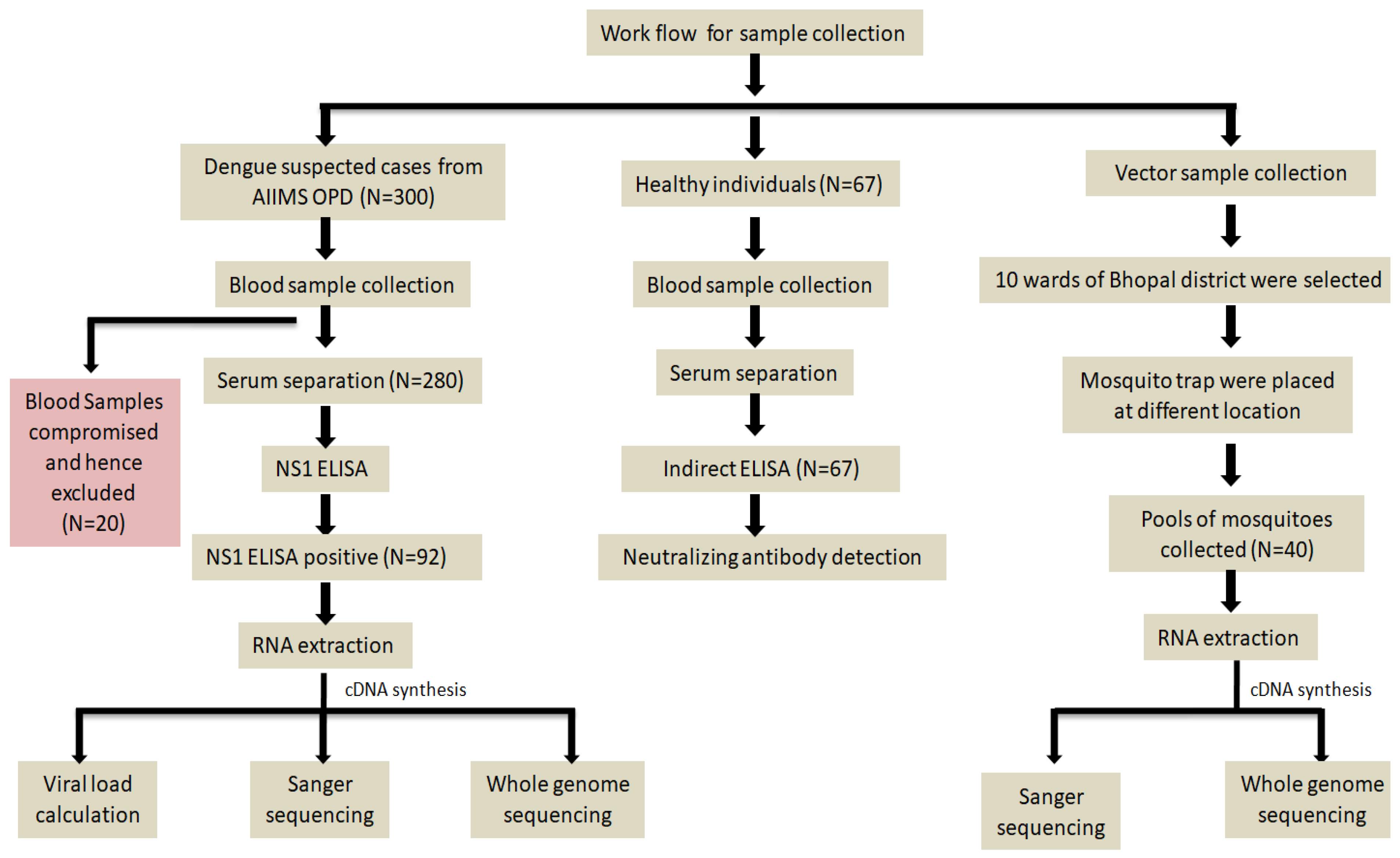
2.1. Study Area, Study Period, and Sample Sources
2.2. Patients Clinical Details
2.3. Serological Diagnosis of Dengue Virus
2.4. Mosquito Sample Collection and Processing
2.5. In-Vitro Culture of Dengue Virus in Vero E6 Cell Line
2.6. Establishing the Positive Control and qRT-PCR for Detection of Dengue Virus
2.7. CPrM Region Sanger Sequencing and Whole Genome Sequencing of DENV Virus
2.8. Phylogenetic Analysis of the CPrM Regions and the Whole Genome Sequences
2.9. Shannon Entropy Analysis for the WGS of DENV Virus
2.10. Molecular Clock Analysis of the WGS Using Bayesian MCMC
2.11. Neutralizing Antibody Assay for Control Samples
3. Results
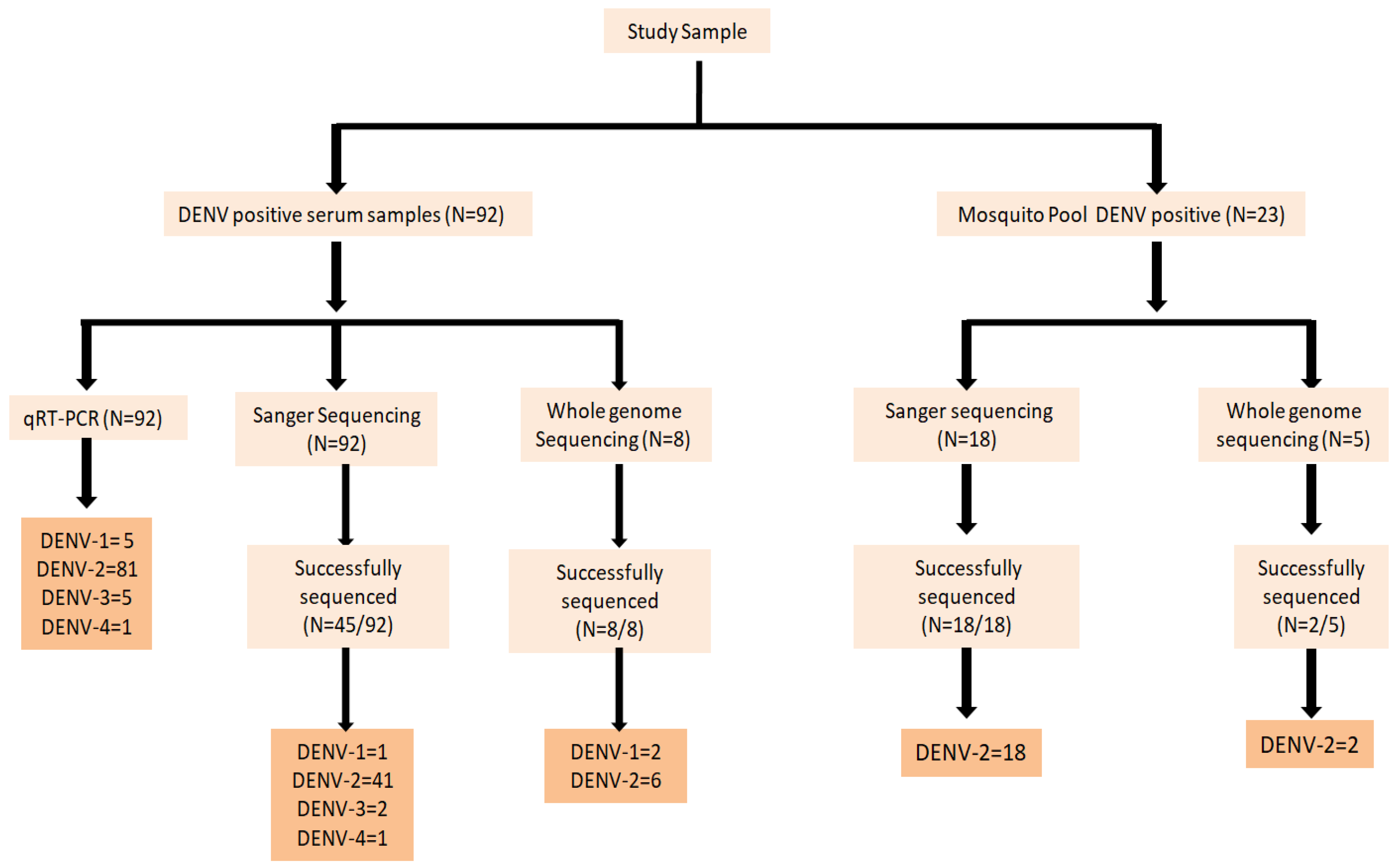
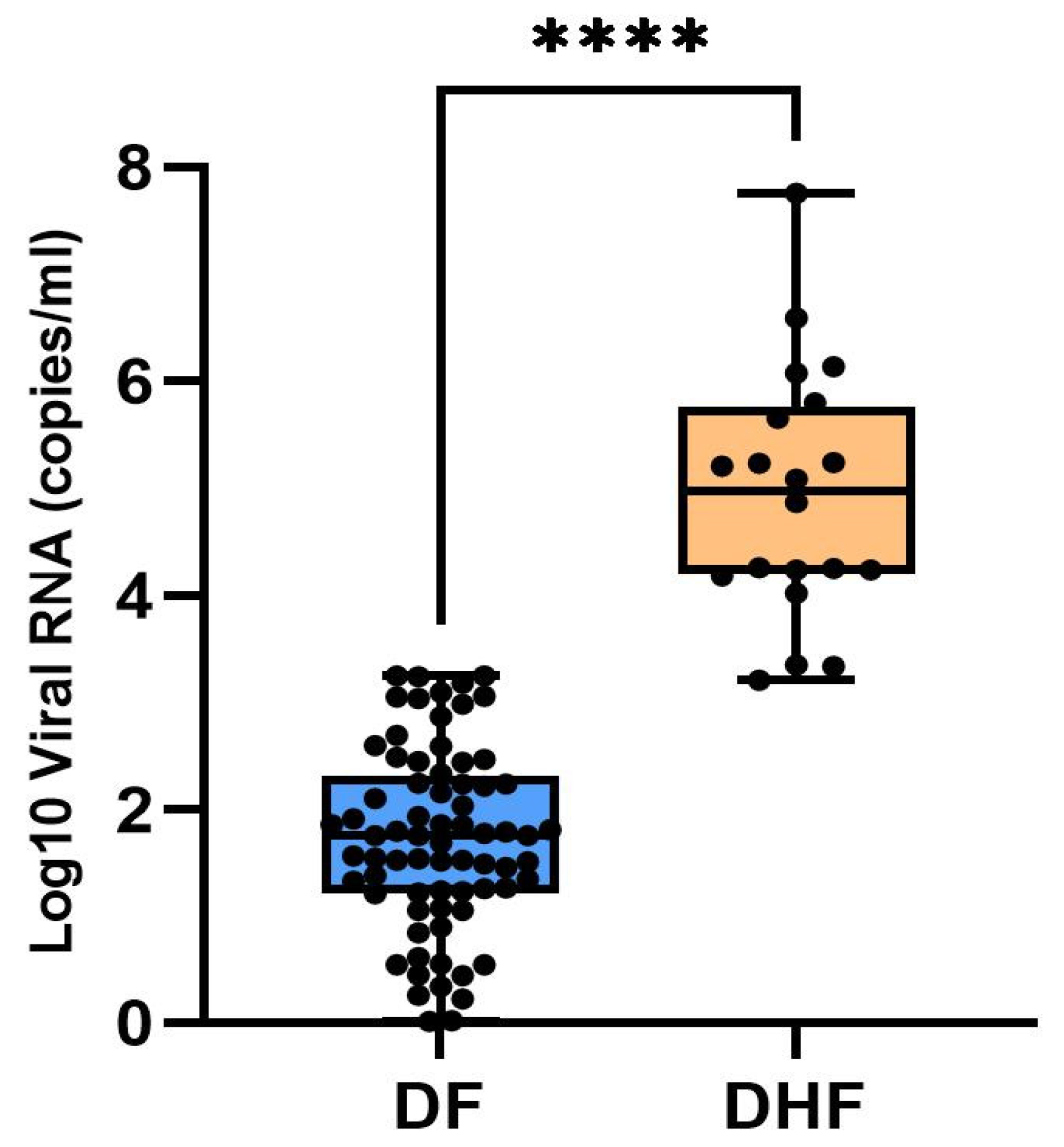
3.1. Serotype Specific qRT-PCR
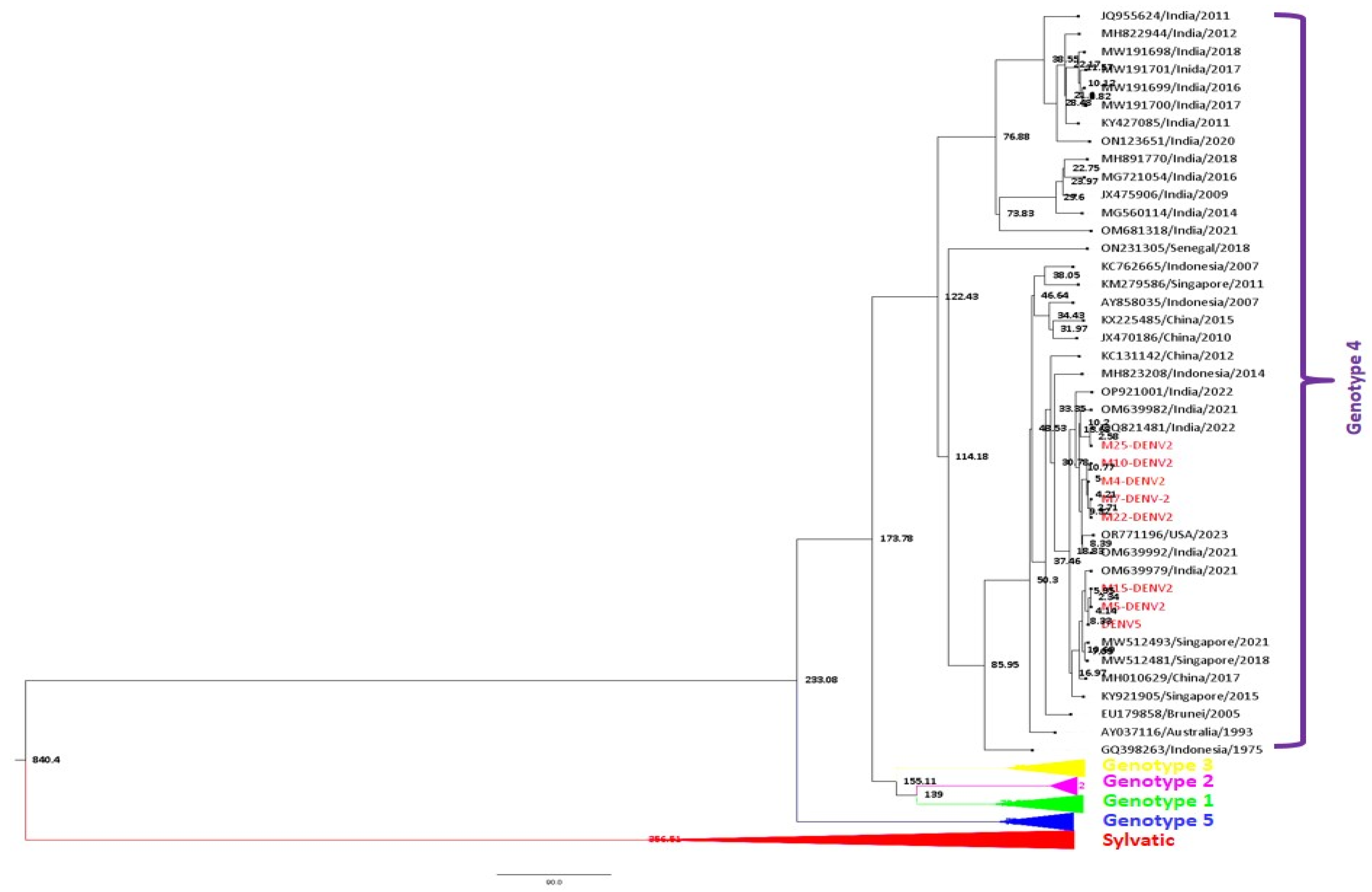
3.2. Phylogenetic Analysis
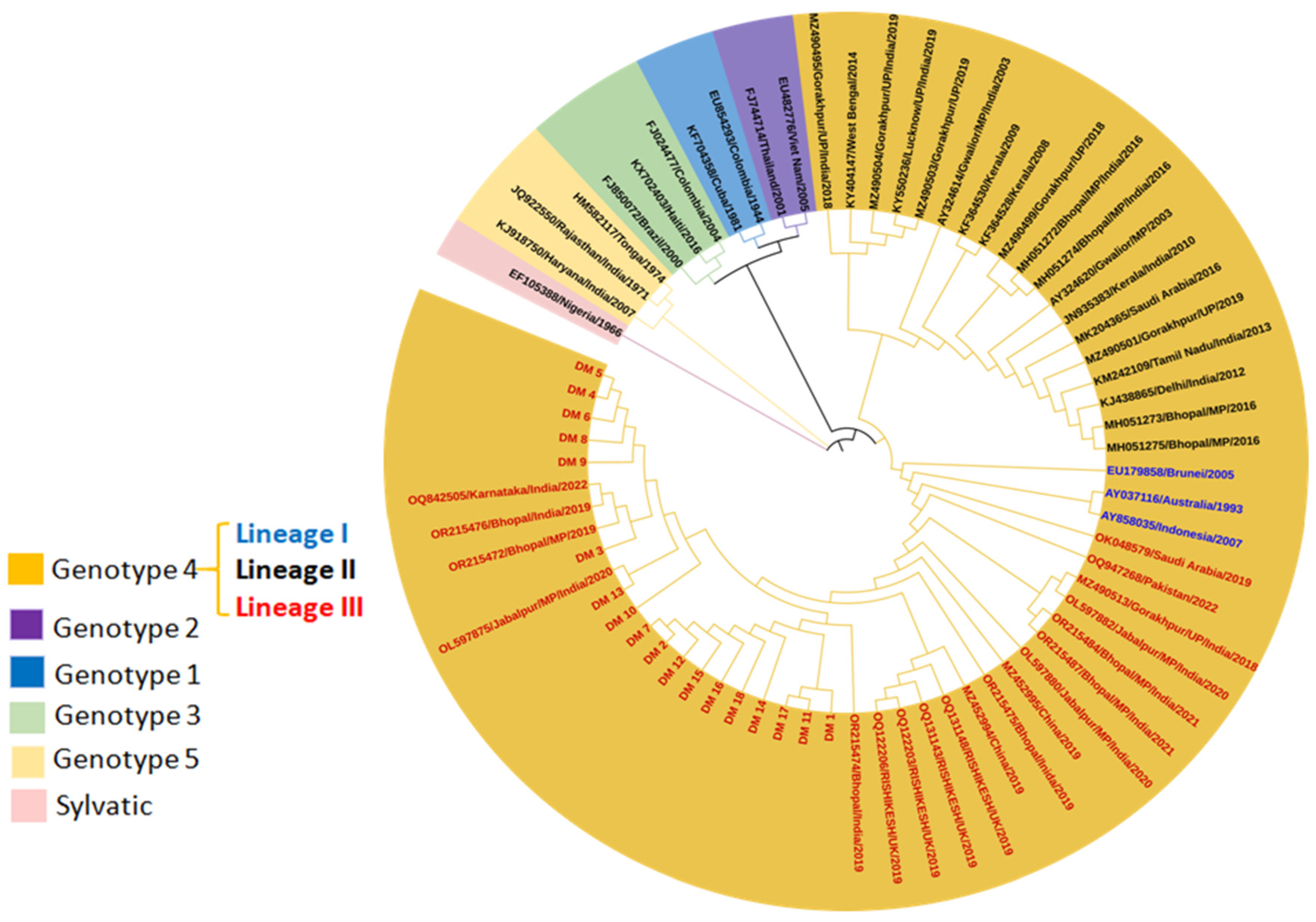
3.2.1. DENV Genotype Distribution for Vector Pool
3.2.2. DENV Genotype Distribution for Host Serum Samples
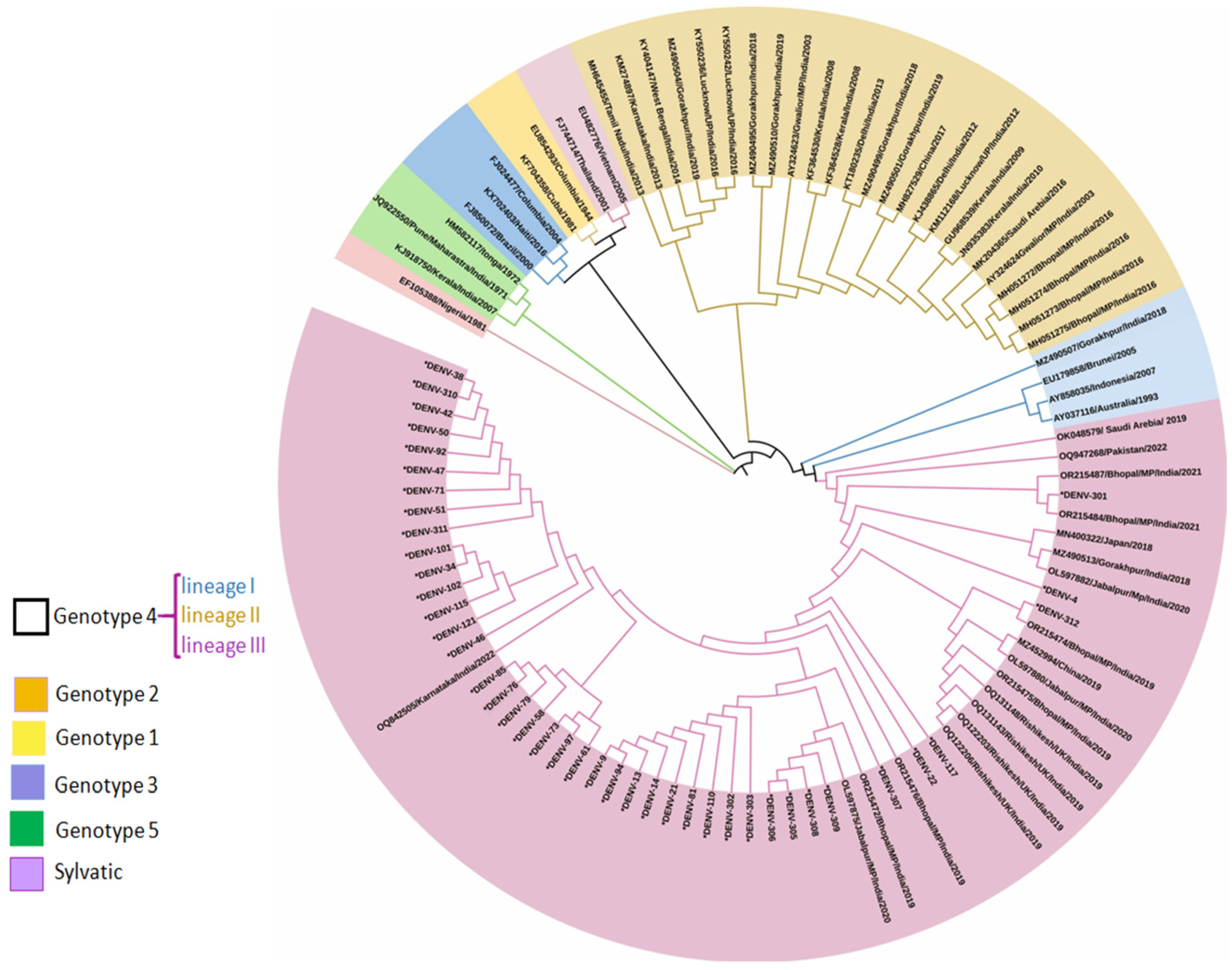
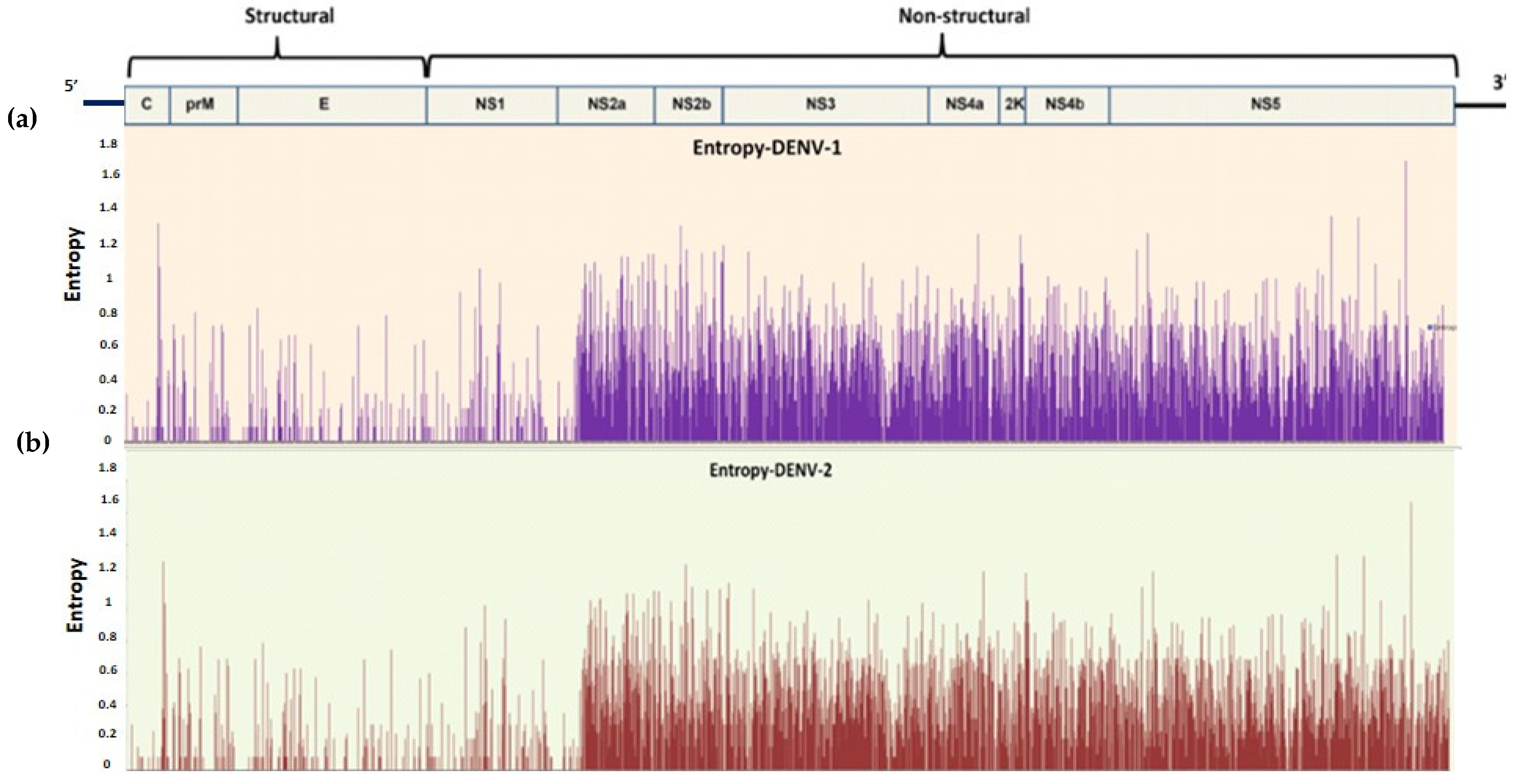
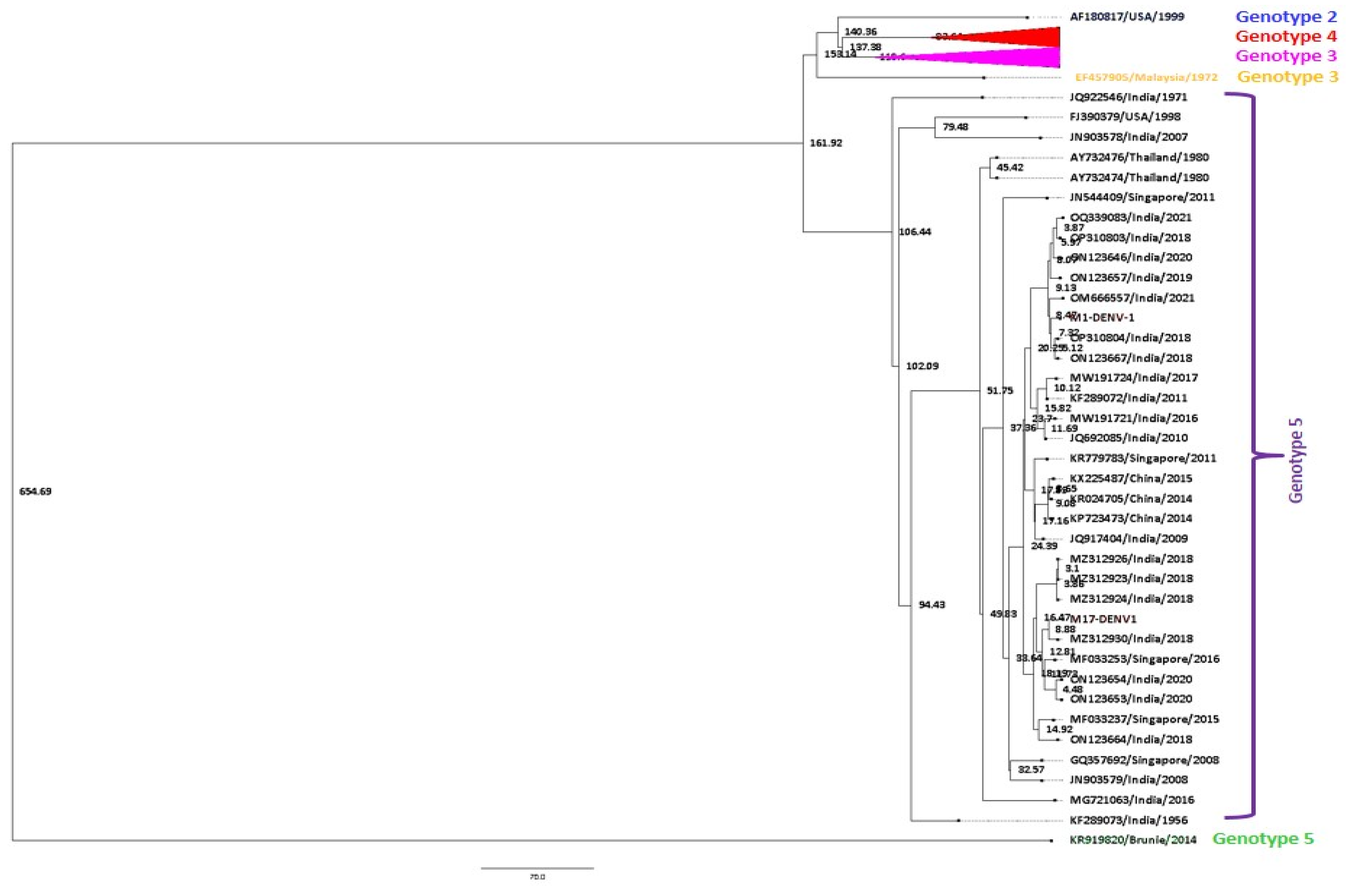
3.2.3. Whole Genome Sequence Analysis
Phylogenetic Analysis
Shannon Entropy Analysis
Bayesian MCMC Analysis
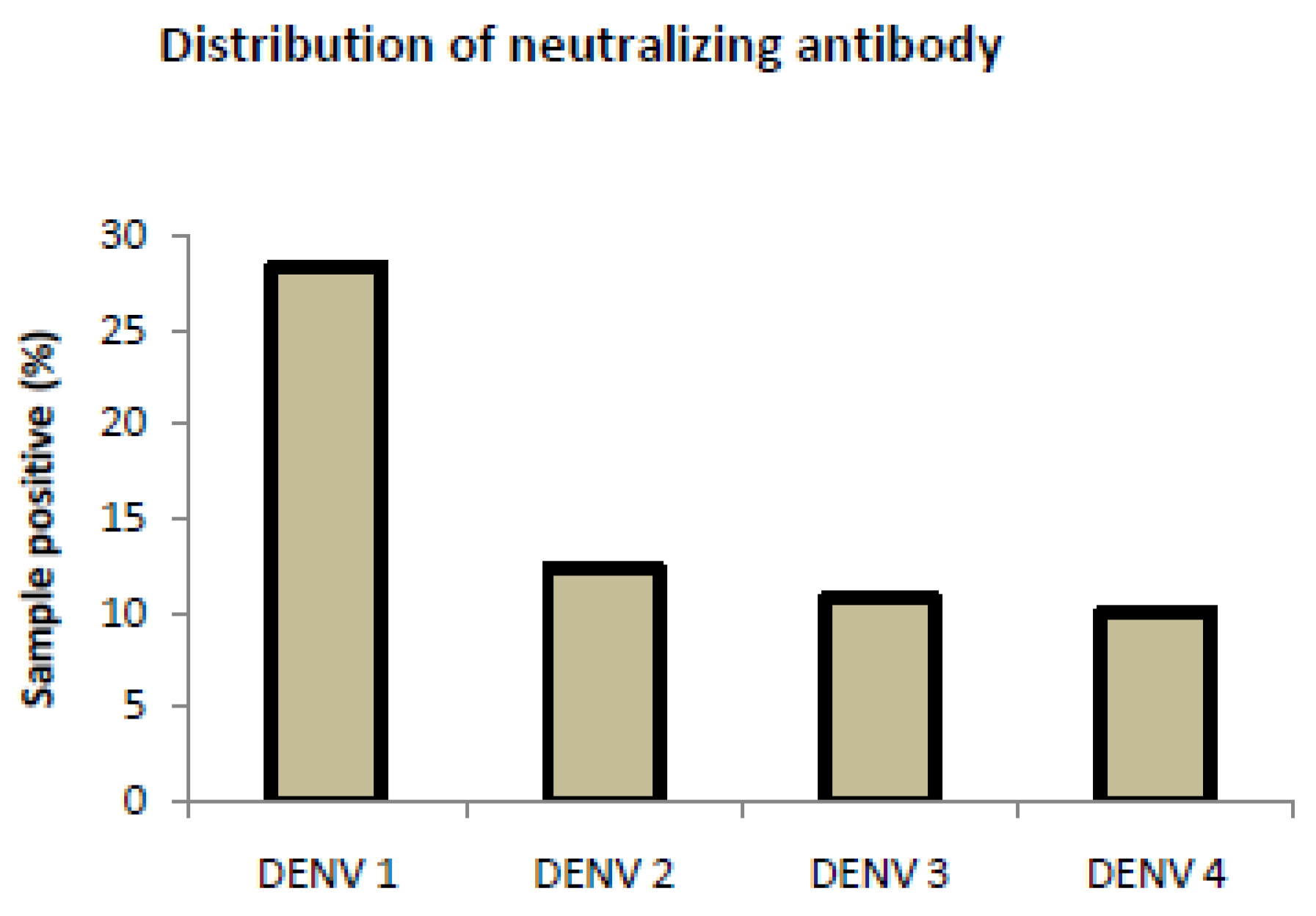
3.3. Analysis of Neutralizing Antibody Assay for Control Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Li, N.-K.; Corander, J.; Grad, Y.H.; Chang, H.-H. Discovering recent selection forces shaping the evolution of dengue viruses based on polymorphism data across geographic scales. Virus Evol. 2022, 8, veac108. [Google Scholar] [CrossRef]
- Rico-Hesse, R. Molecular evolution and distribution of dengue viruses type 1 and 2 in nature. Virology 1990, 174, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C.; Twiddy, S.S. The origin, emergence and evolutionary genetics of dengue virus. Infect. Genet. Evol. 2003, 3, 19–28. [Google Scholar] [CrossRef]
- Putri, D.H.; Sudiro, T.M.; Yunita, R.; Jaya, U.A.; Dewi, B.E.; Sjatha, F.; Konishi, E.; Hotta, H.; Sudarmono, P. Immunogenicity of a Candidate DNA Vaccine Based on the prM/E Genes of a Dengue Type 2 Virus Cosmopolitan Genotype Strain. Jpn. J. Infect. Dis. 2015, 68, 357–363. [Google Scholar] [CrossRef]
- Chen, R.; Vasilakis, N. Dengue—Quo tu et quo vadis? Viruses 2011, 3, 1562–1608. [Google Scholar] [CrossRef] [PubMed]
- Bäck, A.T.; Lundkvist, A. Dengue viruses—An overview. Infect. Ecol. Epidemiol. 2013, 3, 19839. [Google Scholar]
- Heilman, J.M.; De Wolff, J.; Beards, G.M.; Basden, B.J. Dengue fever: A Wikipedia clinical review. Open Med. 2014, 8, e105–e115. [Google Scholar] [PubMed]
- Kularatne, S.M.; Ralapanawa, U.; Dalugama, C.; Jayasinghe, J.; Rupasinghe, S.; Kumarihamy, P. Series of 10 dengue fever cases with unusual presentations and complications in Sri Lanka: A single centre experience in 2016. BMC Infect. Dis. 2018, 18, 674. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Ganvir, R.; Kale, D.; Chaurasia, D.; Kapoor, G. Continued dominance of dengue virus serotype 2 during the recent Central India outbreaks (2019-2021) with evidence of genetic divergence. Pathog. Glob. Health 2024, 118, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Pakhare, A.; Sabde, Y.; Joshi, A.; Jain, R.; Kokane, A.; Joshi, R. A study of spatial and meteorological determinants of dengue outbreak in Bhopal City in 2014. J. Vector Borne Dis. 2016, 53, 225–233. [Google Scholar]
- Baruah, K.; Singh, P.; Mohalia, M.; Dhariwal, A. A study on dengue outbreak during 2009 in Bhopal and Indore districts of Madhya Pradesh, India. J. Commun. Dis. 2010, 42, 273–279. [Google Scholar] [PubMed]
- Jagtap, S.; Pattabiraman, C.; Sankaradoss, A.; Krishna, S.; Roy, R. Evolutionary dynamics of dengue virus in India. PLOS Pathog. 2023, 19, e1010862. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.G.; Alvarez, M.; Rodriguez-Roche, R.; Bernardo, L.; Montes, T.; Vazquez, S.; Morier, L.; Alvarez, A.; Gould, E.A.; Kourí, G.; et al. Neutralizing Antibodies after Infection with Dengue 1 Virus. Emerg. Infect. Dis. 2007, 13, 282–286. [Google Scholar] [CrossRef]
- Vuong, N.L.; Quyen, N.T.; Tien, N.T.; Tuan, N.M.; Kien, D.T.; Lam, P.K.; Tam, D.T.; Van Ngoc, T.; Yacoub, S.; Jaenisch, T.; et al. Higher plasma viremia in the febrile phase is associated with adverse dengue outcomes irrespective of infecting serotype or host immune status: An analysis of 5642 Vietnamese cases. Clin. Infect. Dis. 2021, 72, e1074–e1083. [Google Scholar] [CrossRef] [PubMed]
- de Arruda, T.B.; Bavia, L.; Mosimann, A.L.P.; Aoki, M.N.; Sarzi, M.L.; Conchon-Costa, I.; Wowk, P.F.; Duarte dos Santos, C.N.; Pavanelli, W.R.; Silveira, G.F.; et al. Viremia and Inflammatory Cytokines in Dengue: Interleukin-2 as a Biomarker of Infection, and Interferon-α and -γ as Markers of Primary versus Secondary Infection. Pathogens 2023, 12, 1362. [Google Scholar] [CrossRef]
- Tyagi, B.K.; Munirathinam, A.; Venkatesh, A. A catalogue of Indian mosquitoes. Int. J. Mosq. Res. 2015, 2, 50–97. [Google Scholar]
- de Figueiredo, M.L.; de C Gomes, A.; Amarilla, A.A.; de S Leandro, A.; de S Orrico, A.; de Araujo, R.F.; do SM Castro, J.; Durigon, E.L.; Aquino, V.H.; Figueiredo, L.T. Mosquitoes infected with dengue viruses in Brazil. Virol. J. 2010, 7, 152. [Google Scholar] [CrossRef]
- Armstrong, P.M.; Andreadis, T.G.; Finan, S.L.; Shepard, J.J.; Thomas, M.C. Detection of Infectious Virus from Field-collected Mosquitoes by Vero Cell Culture Assay. J. Vis. Exp. JoVE 2011, 52, 2889. [Google Scholar] [CrossRef]
- Prada-Arismendy, J.; Castellanos, J.E. Real time PCR. Application in dengue studies. Colomb. Medica 2011, 42, 243–258. [Google Scholar] [CrossRef]
- Santiago, G.A.; Vergne, E.; Quiles, Y.; Cosme, J.; Vazquez, J.; Medina, J.F.; Medina, F.; Colón, C.; Margolis, H.; Muñoz-Jordán, J.L. Analytical and Clinical Performance of the CDC Real Time RT-PCR Assay for Detection and Typing of Dengue Virus. PLoS Negl. Trop. Dis. 2013, 7, e2311. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Calisher, C.H.; Gubler, D.J.; Chang, G.J.; Vorndam, A.V. Rapid detection and typing of dengue viruses from clinical samples by using reverse transcriptase-polymerase chain reaction. J. Clin. Microbiol. 1992, 30, 545–551. [Google Scholar] [CrossRef]
- Cruz, C.D.; Torre, A.; Troncos, G.; Lambrechts, L.; Leguia, M. Targeted full-genome amplification and sequencing of dengue virus types 1–4 from South America. J. Virol. Methods 2016, 235, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Pollett, S.; Melendrez, M.; Maljkovic Berry, I.; Duchêne, S.; Salje, H.; Cummings, D.; Jarman, R. Understanding dengue virus evolution to support epidemic surveillance and counter-measure development. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2018, 62, 279–295. [Google Scholar] [CrossRef]
- Papastamoulis, P.; Furukawa, T.; Van Rhijn, N.; Bromley, M.; Bignell, E.; Rattray, M. Bayesian detection of piecewise linear trends in replicated time-series with application to growth data modelling. Int. J. Biostat. 2020, 16, 20180052. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Adam, A.; Schüttoff, T.; Reiche, S.; Jassoy, C. High seroprevalence of dengue virus indicates that dengue virus infections are frequent in central and eastern Sudan. Trop. Med. Int. Health 2018, 23, 960–967. [Google Scholar] [CrossRef]
- Tiraki, D.; Singh, K.; Shrivastava, S.; Mishra, A.C.; Arankalle, V. Complete genome characterization and evolutionary analysis of dengue viruses isolated during 2016–2017 in Pune, India. Infect. Genet. Evol. 2021, 93, 104909. [Google Scholar] [CrossRef]
- Gupta, N.; Srivastava, S.; Jain, A.; Chaturvedi, U.C. Dengue in India. Indian J. Med. Res. 2012, 136, 373–390. [Google Scholar]
- Chaturvedi, U.C.; Mathur, A.; Kapoor, A.K.; Mehrotra, N.K.; Mehrotra, R.M.L. Virological study of an epidemic of febrile illness with haemorrhagic manifestations at Kanpur, India, during 1968. Bull. World Health Organ. 1970, 43, 289–293. [Google Scholar]
- Dash, P.K.; Sharma, S.; Srivastava, A.; Santhosh, S.R.; Parida, M.M.; Neeraja, M.; Subbalaxmi, M.V.S.; Lakshmi, V.; RAO, P.V.L. Emergence of dengue virus type 4 (genotype I) in India. Epidemiol. Infect. 2011, 139, 857–861. [Google Scholar] [CrossRef]
- Islam, A.; Deeba, F.; Tarai, B.; Gupta, E.; Naqvi, I.H.; Abdullah, M.; Dohare, R.; Ahmed, A.; Almajhdi, F.N.; Hussain, T.; et al. Global and local evolutionary dynamics of Dengue virus serotypes 1, 3, and 4. Epidemiol. Infect. 2023, 151, e127. [Google Scholar] [CrossRef]
- Myat Thu, H.; Lowry, K.; Jiang, L.; Hlaing, T.; Holmes, E.C.; Aaskov, J. Lineage extinction and replacement in dengue type 1 virus populations are due to stochastic events rather than to natural selection. Virology 2005, 336, 163–172. [Google Scholar] [CrossRef]
- Vazeille, M.; Gaborit, P.; Mousson, L.; Girod, R.; Failloux, A.B. Competitive advantage of a dengue 4 virus when co-infecting the mosquito Aedes aegypti with a dengue 1 virus. BMC Infect. Dis. 2016, 16, 318. [Google Scholar] [CrossRef]
- O’Connor, O.; Ou, T.P.; Aubry, F.; Dabo, S.; Russet, S.; Girault, D.; In, S.; Minier, M.; Lequime, S.; Home, T.; et al. Potential role of vector-mediated natural selection in dengue virus genotype/lineage replacements in two epidemiologically contrasted settings. Emerg. Microbes Infect. 2021, 10, 1346. [Google Scholar] [CrossRef]
- Stica, C.J.; Barrero, R.A.; Murray, R.Z.; Devine, G.J.; Phillips, M.J.; Frentiu, F.D. Global Evolutionary History and Dynamics of Dengue Viruses Inferred from Whole Genome Sequences. Viruses 2022, 14, 703. [Google Scholar] [CrossRef]
- Khan, A.M.; Hu, Y.; Miotto, O.; Thevasagayam, N.M.; Sukumaran, R.; Abd Raman, H.S.; Brusic, V.; Tan, T.W.; Thomas August, J. Analysis of viral diversity for vaccine target discovery. BMC Med. Genom. 2017, 10, 78. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Pandey, R.K.; Khatoon, N.; Narula, A.; Mishra, A.; Prajapati, V.K. Exploring dengue genome to construct a multi-epitope based subunit vaccine by utilizing immunoinformatics approach to battle against dengue infection. Sci. Rep. 2017, 7, 9232. [Google Scholar] [CrossRef] [PubMed]
- Twiddy, S.S.; Holmes, E.C.; Rambaut, A. Inferring the Rate and Time-Scale of Dengue Virus Evolution. Mol. Biol. Evol. 2003, 20, 122–129. [Google Scholar] [CrossRef]
- Gubler, D.J. Dengue and Dengue Hemorrhagic Fever. Clin. Microbiol. Rev. 1998, 11, 480–496. [Google Scholar] [CrossRef]
- Costa, R.L.; Voloch, C.M.; Schrago, C.G. Comparative evolutionary epidemiology of dengue virus serotypes. Infect. Genet. Evol. 2012, 12, 309–314. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NS1 ELISA Positive | NS-1 Negative (n = 188) | |||
|---|---|---|---|---|
| Clinical and Demographic Characteristics | DF * (n = 72) | DHF * (n = 20) | ||
| Age (years) | 18–30 | 23 | 6 | 88 |
| 30–50 | 34 | 9 | 32 | |
| 50–65 | 15 | 5 | 68 | |
| Sex (Male/female) | 45/27 | 15/5 | 110/78 | |
| Headache | 12 (16%) | 10 (50%) | 56 (29%) | |
| Myalgia | 13 (18%) | 20 (100%) | 40 (21%) | |
| Fever | 65 (90%) | 16 (80%) | 165 (88%) | |
| Arthralgia | 10 (13%) | 16 (80%) | 89 (47%) | |
| Rash | 9 (12.5%) | 4 (20%) | 32 (17%) | |
| Vomiting/Nausea | 6 (8.33%) | 3 (15%) | 23 (12%) | |
| Retro-orbital pain | 5 (6.94%) | 13 (65%) | 15 (8%) | |
| Hematocrit | 14 (19%) | 8 (40%) | 25 (13%) | |
| Thrombocytopenia | 15 (20%) | 18 (90%) | 15 (8%) | |
| Number of patients with increased aminotransferase level | 32 (44%) | 16 (80%) | 8 (4%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yadav, A.K.; Chowdhary, R.; Siddiqui, A.; Malhotra, A.G.; Kanwar, J.R.; Kumar, A.; Biswas, D.; Khadanga, S.; Joshi, R.; Pakhare, A.; et al. Emergence of a Novel Dengue Virus Serotype-2 Genotype IV Lineage III Strain and Displacement of Dengue Virus Serotype-1 in Central India (2019–2023). Viruses 2025, 17, 144. https://doi.org/10.3390/v17020144
Yadav AK, Chowdhary R, Siddiqui A, Malhotra AG, Kanwar JR, Kumar A, Biswas D, Khadanga S, Joshi R, Pakhare A, et al. Emergence of a Novel Dengue Virus Serotype-2 Genotype IV Lineage III Strain and Displacement of Dengue Virus Serotype-1 in Central India (2019–2023). Viruses. 2025; 17(2):144. https://doi.org/10.3390/v17020144
Chicago/Turabian StyleYadav, Ashish Kumar, Rashmi Chowdhary, Arshi Siddiqui, Anvita Gupta Malhotra, Jagat R. Kanwar, Ashok Kumar, Debasis Biswas, Sagar Khadanga, Rajnish Joshi, Abhijit Pakhare, and et al. 2025. "Emergence of a Novel Dengue Virus Serotype-2 Genotype IV Lineage III Strain and Displacement of Dengue Virus Serotype-1 in Central India (2019–2023)" Viruses 17, no. 2: 144. https://doi.org/10.3390/v17020144
APA StyleYadav, A. K., Chowdhary, R., Siddiqui, A., Malhotra, A. G., Kanwar, J. R., Kumar, A., Biswas, D., Khadanga, S., Joshi, R., Pakhare, A., & Goel, S. K. (2025). Emergence of a Novel Dengue Virus Serotype-2 Genotype IV Lineage III Strain and Displacement of Dengue Virus Serotype-1 in Central India (2019–2023). Viruses, 17(2), 144. https://doi.org/10.3390/v17020144